
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable polymer brushes with effectively varied grafting density synthesized from highly crosslinked random copolymer thin films†
Hyungoo Kang‡
a,
Sol An‡a,
Woo Jung Leeb,
Ga Ryang Kangc,
Sangwon Kimd,
Su-Mi Hur*c,
Keewook Paeng*b and
Myungwoong Kim *a
*a
aDepartment of Chemistry and Chemical Engineering, Inha University, Incheon 22212, Republic of Korea. E-mail: mkim233@inha.ac.kr
bDepartment of Chemistry, Sungkyunkwan University, Suwon 16419, Republic of Korea. E-mail: paeng@skku.edu
cSchool of Polymer Science and Engineering, Chonnam National University, Gwangju 61186, Republic of Korea. E-mail: shur@chonnam.ac.kr
dDepartment of Polymer Science and Engineering, Inha University, Incheon 22212, Republic of Korea
First published on 3rd July 2018
Abstract
We demonstrate the synthesis of poly(methyl methacrylate) (PMMA) and poly(styrene-b-methyl methacrylate) (P(S-b-MMA)) brushes on crosslinked random copolymer thin films, compositionally varied poly(styrene-r-glycidyl methacrylate) (P(S-r-GMA)), which can be further functionalized with a molecule featuring an initiator group upon crosslinking to form highly stable thin films. With careful optimizations, PMMA brushes were successfully grown from the surfaces of initiator functionalized P(S-r-GMA) via surface-initiated atom transfer radical polymerization. The grafting densities of the PMMA and P(S-b-MMA) brushes were effectively controlled to be in different density regimes by controlling the composition of P(S-r-GMA) and post-crosslinking functionalization methods. Synthesized BCP brushes were stable upon repetitive washing and thermal annealing processes even at high grafting density, highlighting that the outstanding stability of crosslinked P(S-r-GMA) thin films enables close examination of the morphology of thermally annealed P(S-b-MMA) brushes in different grafting density regimes.
Introduction
Surface coatings consisting of polymer chains tethered to a surface, namely polymer brushes, have been of great academic and industrial interest owing to their high stabilities and versatility for tailoring surface properties and functionalities.1–3 Hence, they are widely applicable as functional materials for applications such as external stimuli responsive surfaces,4,5 coatings for colloid and surface stabilizations,6 friction reducing agents to achieve semi-permanent lubrication coatings,7 surface active and passive materials (e.g. for cell adhesion control and fouling release control),8–11 and functional freestanding membranes such as polymer carpets.12,13 In particular, the ability of polymer brushes to be used for the fabrication of complex surface structures has attracted attention. For example, in block copolymer (BCP) lithography, homopolymer, random copolymer or BCP brushes, formed by grafting reaction between hydroxyl group at the chain end to a silicon oxide surface by thermal annealing at elevated temperature, can be utilized for control of surface energy even in the nanoscale, which is considerably beneficial for rational fabrication of highly aligned BCP domains.14–17 More importantly, BCP brushes themselves offer unique routes toward complex structures; Paik et al. showed that e-beam patterning of BCP brushes can be used to fabricate sub-100 nm channels on a surface.18,19 Furthermore, it has been known that BCP brushes themselves can micro-phase separate, forming periodic nanodomains in thin films, which does not involve any dewetting of copolymers owing to the anchoring.20For the expansion of the applications of complex polymer brushes, it is of great importance to control grafting density. In case of BCP brushes, previous studies by O'Driscoll et al. explored the phase behaviors of BCP brushes by both self-consistent field theory (SCFT) calculations and experiments involving grafting pre-defined end-functionalized poly(styrene-b-methyl methacrylate) (P(S-b-MMA)) to a surface, namely the “grafting-to” method.20 They observed clear microphase separations in BCP brushes; however, the grafting density was fixed at ∼0.05 chains per nm2, which is in the mushroom regime typically attained when high molecular weight polymers are grafted.1,2,21 Grafting density is an important parameter that governs the physical and chemical properties of brushes;22,23 the phase behaviors are also expected to be significantly affected by the grafting density, and systematic studies are required to explore the correlation.20 The challenge of increasing the grafting density has been overcome by changing the synthetic strategy. For example, Sparnacci et al. reported the grafting of hydroxyl terminated P(S-r-MMA)s (1.7–69.0 kg mol−1) by rapid thermal annealing method, achieving the grafting density in range of 0.19–0.87 chains per nm2.24 However, since the grafting-to process is diffusion-limited, high grafting density is difficult to attain when brush molecular weight increases.2 A “grafting-from” method has addressed this issue; it employs a variety of surface-initiated (SI) polymerization techniques, such as atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain-transfer (RAFT) polymerization, and nitroxide-mediated polymerization (NMP) using immobilized initiators on surfaces.21,25–28 The most widely used method to define surface initiators are utilizing initiator molecules containing silane or thiol groups, which can form self-assembled monolayers.29–32 However, there are several significant challenges. First, controlling the initiator concentration has been quite challenging. Second, the covalent bonds often fail to deliver the sufficient stability of the resulting brush films against thermal annealing at elevated temperatures to induce thermally equilibrated phase separation in complex brush systems.33 Furthermore, the synthetic routes for various thiol- and silane-based initiators involve multiple steps, and often they suffer from side reactions such as hydrolysis.34 Therefore, facilitating scale-up with high throughput can be quite challenging.
In this study, to address these challenges, we demonstrate the use of a very simple and easily synthesizable reactive copolymer, poly(styrene-random-glycidyl methacrylate) (P(S-r-GMA)), to produce a highly stable self-crosslinked copolymer thin film,35 which additionally offers the control of the degree of functionality, since its composition can be tuned in synthesis (Fig. 1). A post-crosslinking modification involving the reaction of an ATRP initiator containing acyl bromide groups with hydroxyl groups in the thin film, which was formed through the thermal self-crosslinking of glycidyl groups, allows us to control the concentration of the ATRP initiator, which was closely examined using a variety of characterization tools. Poly(methyl methacrylate) (PMMA) brushes were grown from the surfaces and characterized, and it was found that different brush density regimes were achieved effectively. In the course of optimization of the post-crosslinking modification, we found that the initiator functionalization relied heavily on the processing conditions, namely, whether it was conducted in the solution phase or vapor phase. With the conditions optimized, P(S-b-MMA) brushes were synthesized through the ATRP of styrene from PMMA brushes grown from the crosslinked thin film. The grafting density was controlled to range in different grafting density regimes. The resulting brush films were thermally stable enough against repetitive washing and thermal annealing at the elevated temperature. This thermal stability enabled the close examination of phase separation of the BCP brushes, confirming the effectiveness of the simple and readily scalable synthetic route with epoxy containing random copolymers in the brush film formation.
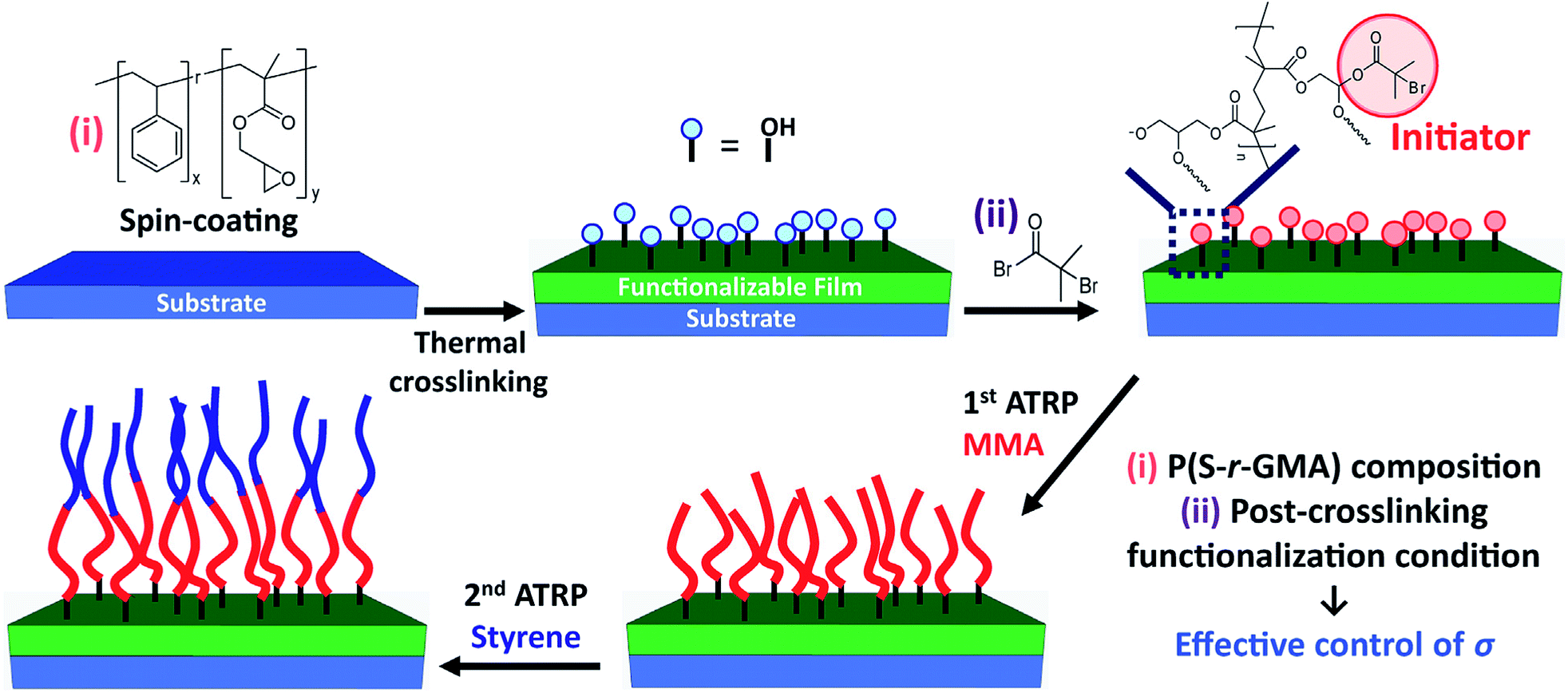 | ||
| Fig. 1 Schematics showing fabrication of P(S-b-MMA) brushes on crosslinked random copolymer thin film with an effective controlled grafting density via adjusting the composition of random copolymer and different post-crosslinking functionalization methods. | ||
Experimental section
Materials and methods
Methyl methacrylate (MMA), styrene, and glycidyl methacrylate (GMA) were purchased from Samchun Chemicals, TCI Chemicals, and Sigma-Aldrich Co., Ltd., respectively. The monomers were passed through neutral alumina columns to remove inhibitors prior to use for polymerization. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was obtained from Junsei Chemicals and was recrystallized from methanol, and dried under vacuum. 2-Bromoisobutyryl bromide (BiBB) was purchased from Alfa Aesar and used without purification. Other chemicals were purchased from Sigma-Aldrich Co., Ltd. and used without purification.Synthesis of crosslinkable polymers
A homopolymer of GMA and a series of random copolymers of styrene and GMA were synthesized through conventional free radical polymerization.35,36 In a typical preparation of P(S-r-GMA), the desired amounts of styrene, GMA, and AIBN ([M]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]0 ∼ 150) were mixed in toluene (100 wt% to monomer) in a Schlenk flask containing a magnetic stirring bar. The flask was placed in an oil bath and stirred at 80 °C overnight. For the homopolymerization of GMA, methyl ethyl ketone was used as the solvent. [M]0:[I]0 was fixed at 290, and the polymerization temperature and time were 60 °C and 6 hours, respectively. The polymerization mixture was cooled to room temperature and then exposed to air. The mixture was diluted with tetrahydrofuran (THF), and then precipitated into methanol. The polymer sample was collected as a white powder through vacuum filtration, followed by drying under vacuum at room temperature.
:[I]0 ∼ 150) were mixed in toluene (100 wt% to monomer) in a Schlenk flask containing a magnetic stirring bar. The flask was placed in an oil bath and stirred at 80 °C overnight. For the homopolymerization of GMA, methyl ethyl ketone was used as the solvent. [M]0:[I]0 was fixed at 290, and the polymerization temperature and time were 60 °C and 6 hours, respectively. The polymerization mixture was cooled to room temperature and then exposed to air. The mixture was diluted with tetrahydrofuran (THF), and then precipitated into methanol. The polymer sample was collected as a white powder through vacuum filtration, followed by drying under vacuum at room temperature.
Crosslinked polymer film formation and surface functionalization
A solution of the synthesized random copolymer (0.3 wt%) in toluene was spin-coated onto pre-cleaned silicon wafers with piranha solution (H2SO4/H2O2 = 7/3 v/v. Caution: the solution reacts violently with organic compounds). The samples were thermally annealed at 160 °C under vacuum for 3 hours, generating crosslinked thin films. After annealing, the substrates were washed with copious amounts of THF and toluene to remove uncrosslinked polymer. The substrates were then treated with BiBB using two methods: (i) immersion in a 0.40 M solution of BiBB in anhydrous dichloromethane for an hour, or (ii) exposure to BiBB vapor under reduced pressure at 35 °C for an hour. After functionalization using either method, the samples were thoroughly rinsed with THF to remove any residues.SI-ATRP of MMA
MMA (10.3 g, 102.9 mmol), ethyl 2-bromoisobutyrate (ebib) (10.0 mg, 0.05 mmol), CuBr (14.7 mg, 0.10 mmol), CuBr2 (11.5 mg, 0.05 mmol), pentamethyldiethylenetriamine (PMDETA) (20.0 mg, 0.12 mmol), and anisole (5.0 g) were added to a Schlenk flask containing a magnetic stirring bar and the substrate coated with a crosslinked polymeric thin film. The flask was then degassed via three freeze-pump-thaw cycles, warmed to room temperature, and then immersed in a 60 °C oil bath for the desired time. The resulting sample was removed from the flask and then rinsed with THF and deionized water. The substrate was further soaked in THF overnight, sonicated in THF for 5 min, thoroughly rinsed with THF for complete removal of ungrafted polymer chains, and then dried with a stream of nitrogen.Chain extension of PMMA brush through the SI-ATRP of styrene
Styrene (10.7 g, 102.7 mmol), ebib (10.0 mg, 0.05 mmol), CuBr (41.9 mg, 0.29 mmol), PMDETA (50.6 mg, 0.29 mmol), and anisole (5.0 g) were mixed in a Schlenk flask containing a magnetic stirring bar and the sample coated with the PMMA brush film grown in the first SI-ATRP. The flask was degassed via three freeze-pump-thaw cycles, warmed to room temperature, and then immersed in a 90 °C oil bath for the desired time. The resulting sample was removed from the solution, then rinsed with THF and deionized water. The substrate was further immersed in THF overnight, sonicated in THF for 5 min, rinsed with THF for complete removal of remaining residues, and then dried with a stream of nitrogen.Characterization
Size exclusion chromatography (SEC) was conducted using a Thermo Scientific UltiMate 3000 chromatography system employing THF as the eluent at 35 °C with a flow rate of 1 mL min−1. Obtained chromatograms were analyzed with the calibration using nine standard polystyrene (PS) samples with molecular weights of 1–1400 kg mol−1). 1H-NMR spectra were obtained using a JEOL JNM-ECZ400S 400 MHz spectrometer with CDCl3 containing TMS as an internal reference. AFM was conducted to examine the surfaces of crosslinked mats and brush films with noncontact tapping mode using a Nanoscope Multimode Iva (Bruker) and NX-10 AFM (Park systems). The film thicknesses were measured using a spectroscopic ellipsometer (SE MG-1000, Nano-view Co., Ltd.) in the 350–840 nm wavelength range. X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific K-Alpha. Water contact angles were measured using an SEO Phoenix 300.Results and discussion
Synthesis of crosslinkable polymers and the formation of crosslinked films
We synthesized one PGMA and five P(S-r-GMA) samples with the composition, FGMA, ranging from 0.08 to 1.00 (measured with quantitative 1H-NMR spectroscopy, Fig. S1–S6†) by varying the feeding composition, fGMA. Classical free radical polymerization using AIBN as an initiator led to typical homopolymers or copolymers with number average molecular weights (Mn) ranging from 20 to 72 kg mol−1 with relatively broad dispersity ranging from 1.67 to 3.48 (Table 1). In free radical copolymerization of GMA and styrene, it was found that the reactivity ratio values of rGMA and rstyrene were found to be 0.53 and 0.45.37 These values are comparable to a monomer pair or methyl methacrylate and styrene (0.46 and 0.52);38 and therefore, P(S-r-GMA) likely exhibits a similar sequence to P(S-r-MMA), i.e. a sequence between alternating copolymer and random copolymer. The synthesized samples were utilized to fabricate functionalizable crosslinked polymer thin films on substrates. Solutions of the polymer samples in toluene (0.3 wt%) were spin-coated onto silicon wafers, producing ∼8–10 nm thick films. Thermal annealing was performed to fully crosslink the coated films under the conditions established for full self-crosslinking of the copolymers with FGMA higher than 0.04 without any additional crosslinkers.27,35,36 The thicknesses of the crosslinked films after washing with toluene were almost identical, further confirming the successful formation of crosslinked stable polymer films on substrates.Post-crosslinking functionalization to immobilize initiator for SI-ATRP
First, the water contact angles of the resulting crosslinked thin films were measured. Since we aim to change the amount of reactive methacrylate groups in the thin film, the water contact angle is expected to vary systematically as a function of the composition of the copolymers. Fig. 2a shows that the contact angle of crosslinked thin films increased linearly from approximately 50° to 90° as the amount of GMA decreased, indicating that the surface became more hydrophobic. The surfaces were functionalized with BiBB in both solution phase and vapor phase to create surface initiators for ATRP. The byproduct of the functionalization reaction is HBr, which can penetrate into crosslinked thin films or be formed and trapped inside the film. Also, the physical state of a crosslinked film under the reaction conditions may affect the functionalization; the swelling behaviors of thin films in contact with solution and vapor molecules are different.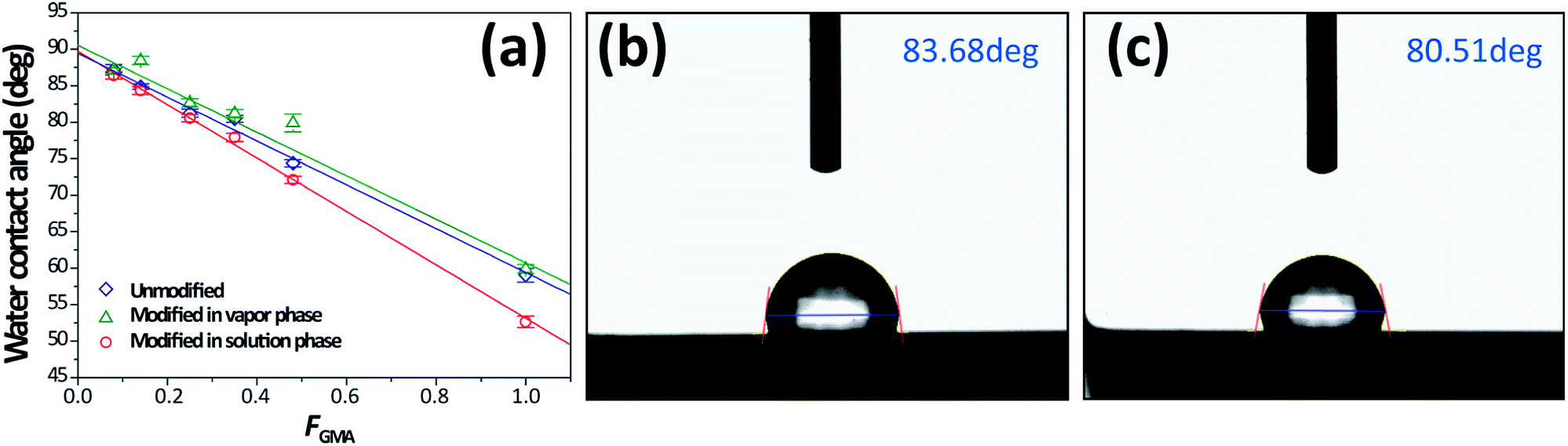 | ||
| Fig. 2 (a) Water contact angles on crosslinked copolymer films with varying FGMA, the solid lines are guides to the eye, (b) a representative water contact angle image of initiator-incorporated thin film fabricated from PSG0.35 through vapor functionalization, observed to be higher than (c) solution functionalization. | ||
In order to fully understand the resulting surfaces, we carefully examined them with a variety of surface characterization techniques: water contact angle measurement, XPS, and AFM. The water contact angle of a surface that was functionalized using BiBB vapor is generally higher than those of pristine thin films. However, treatment of the samples with a BiBB solution led to lower contact angles (Fig. 2). From this result, we suppose that it is due to byproduct, HBr, which can penetrate and hydrolyze polymer thin films.
Fig. 3 shows the AFM images of the surfaces of PGMA, PSG0.35, and PSG0.08 thin film samples before and after functionalization. The surfaces were quite even and homogeneous before functionalization, with root mean square (rms) roughness (Rq) in the 0.8–1.3 nm range. Vapor functionalization did not lead to significant changes in the roughness (0.9–1.0 nm); however, solution-phase functionalization resulted in higher Rq values (0.8–2.4 nm). In the XPS spectra of treated samples, the emergence of a Br(3d) peak at ∼70 eV indicates that the reaction of BiBB with the surface occurred (Fig. 4a). As FGMA decreases, the integrated intensity of the Br(3d) peak clearly decreases (Fig. 4b). It is noteworthy that the Br(3d) peak intensity for surfaces that were functionalized in solution was approximately two times higher than that for a surface treated with BiBB vapor. This could be attributed to initiator groups and bromine ions being trapped inside the film during the reaction in solution phase; this is in good agreement with contact angle measurements and AFM studies. On the other hand, functionalization occurs only near the surface during the vapor-phase reaction, resulting in lower-intensity Br(3d) signals in the XPS spectra.
 | ||
| Fig. 3 Height AFM images of unfunctionalized and functionalized crosslinked P(S-r-GMA) thin films (image size = 2.7 μm × 2.4 μm). | ||
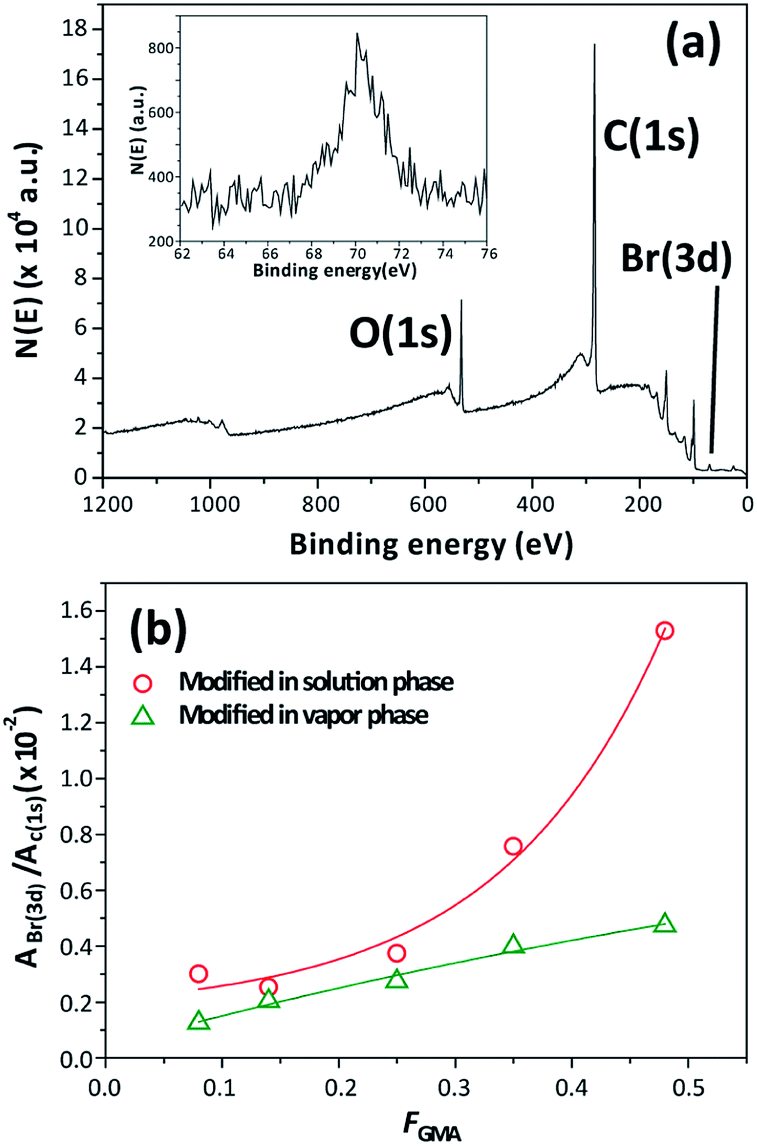 | ||
| Fig. 4 (a) Representative XPS spectra of initiator-incorporated thin film fabricated from PSG0.35 and through solution functionalization, and (b) integrated intensity ratios of the Br(3d) and C(1s) peaks as functions of the composition. The solid lines are guides to the eye. | ||
SI-ATRP of MMA from functionalized thin films: controlled grafting density
Towards obtaining P(S-b-MMA) BCP brushes, we first examined the ATRP of MMA from the surfaces that were created through the solution-phase and vapor-phase post-crosslinking functionalization of PGMA, PSG0.35, and PSG0.08 thin films. The solution growth of PMMA under standard ATRP conditions using CuBr, CuBr2, and PMDETA was first conducted with the addition of a small amount of sacrificial initiator, ebib. The kinetic studies for the solution growth showed that Mn increased linearly as a function of time (Fig. 5a). Also, the film thicknesses of grown brushes increased linearly with polymerization time for all of the films, confirming that the ATRP from the surfaces was well-controlled (Fig. 5a and c). The grafting density, which is given by σ = hNavρ/Mn (where σ is the grafting density, h is the brush thickness, Nav is Avogadro's number, ρ is the bulk density of the polymer, and Mn is the number average molecular weight), is calculated with the molecular weights measured from a polymer grown using a sacrificial initiator in the same polymerization solution.26–28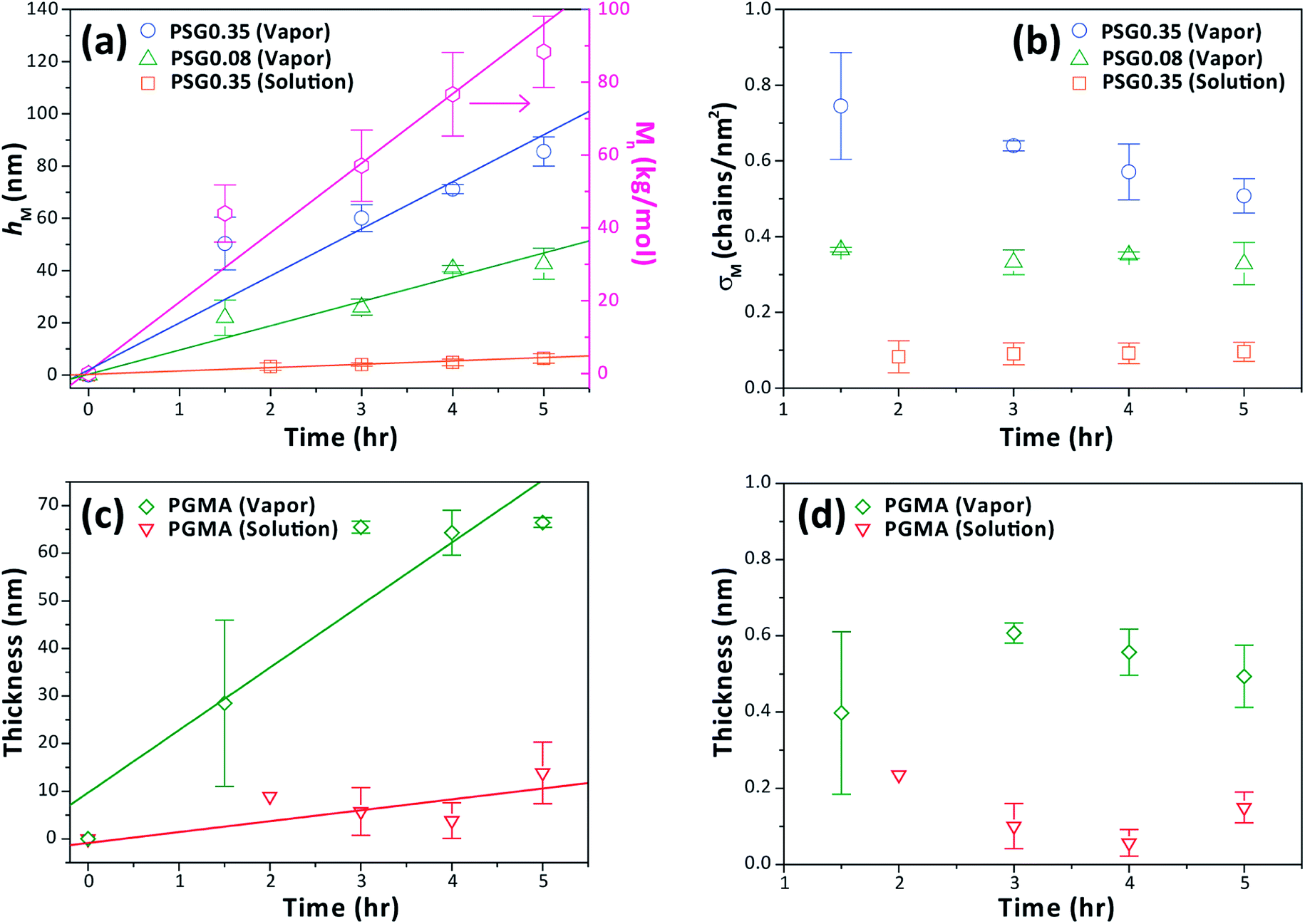 | ||
| Fig. 5 (a and c) Kinetic plots showing the linear increase of the Mn of PMMA grown from the additional sacrificial initiator (pink trace and right y-axis) and the thickness of PMMA brush film (hM) over polymerization time (left y-axis), and (b and d) corresponding grafting density of PMMA brush (σM) estimated from measured hM as a function of polymerization time. The solid lines are guides to the eye. | ||
Fig. 5b shows the σM values at various polymerization times calculated by plugging the measured Mn and hM values into the equation. In the case of vapor-phase functionalization of P(S-r-GMA), σM remains close to constant as the regime changes from moderate to high density (0.3–0.8 chains per nm2), indicating nearly homogeneous growth of the brushes from the initiating sites that are effectively controlled by varying the composition of the copolymer. Interestingly, the σM of brushes grown from the crosslinked PGMA film shown in Fig. 5d was similar to that of the brushes grown from crosslinked PSG0.35, possibly because the amount of hydroxyl groups formed upon self-crosslinking does not increase linearly as the amount of GMA increases. In the case of solution-phase immobilization of BiBB, σM was calculated to be significantly lower than the σM values of brushes grown from the substrates prepared using the vapor-phase functionalization (Fig. 5b and d). As discussed in the previous section, functionalized surfaces in solution gave different results than expected; brush growth studies further showed that the density of initiating groups was not effectively controlled owing to side effects. One possible scenario is that acid generated during the reaction triggers partial hydrolysis of a given surface, as reflected in the characterization results, namely, the anomalously high intensity of the Br(3d) peak in the XPS spectra and the unexpectedly low water contact angle upon functionalization. This scenario is visualized in Fig. S7,† higher bromine intensity in solution-processed samples can be due to residual bromide ions or covalently bound bromine in the network. In addition, polar groups such as alcohol and carboxylic acid by the hydrolysis with an acid catalyst can lead to lower water contact angle. As a consequence, the low degree of immobilization led to brushes in the low-density regime. Therefore, by controlling both process parameters and copolymer composition, desired brush density regimes can be effectively achieved.
Formation of BCP brushes to examine the stability of copolymer brush films
Towards obtaining P(S-b-MMA) BCP brushes, we first examined the ATRP of MMA from the surfaces that were created through solution-phase and vapor-phase post-crosslinking functionalization of crosslinked films fabricated using PSG0.35 and PSG0.08. Then, we carried out the second ATRP of styrene, under standard polymerization conditions using CuBr and PMDETA, for chain extension from PMMA brushes to obtain tethered P(S-b-MMA).18,39 The Mn of both PMMA and PS blocks were estimated by adding ebib as a sacrificial initiator in each polymerization. The increases in Mn and film thickness as functions of polymerization time confirmed that the polymerization was controlled well (Fig. 6a and b). With the optimized conditions, we have synthesized P(S-b-MMA) brushes having different grafting density to examine morphologies upon thermal annealing. Representative results created by the chain extension from various PMMA brushes synthesized from the functionalized surfaces are tabulated in Table 2. Different regimes were achieved with symmetric (FMMA ∼ 0.43) compositions with various Mn values from 67 K to 240 K. σBCP was estimated from measured Mn values from polymer samples grown using a sacrificial initiator in both polymerizations, hBCP of resulting BCP brush films and the bulk density of P(S-b-MMA) weight-averaged from the bulk densities of PS and PMMA.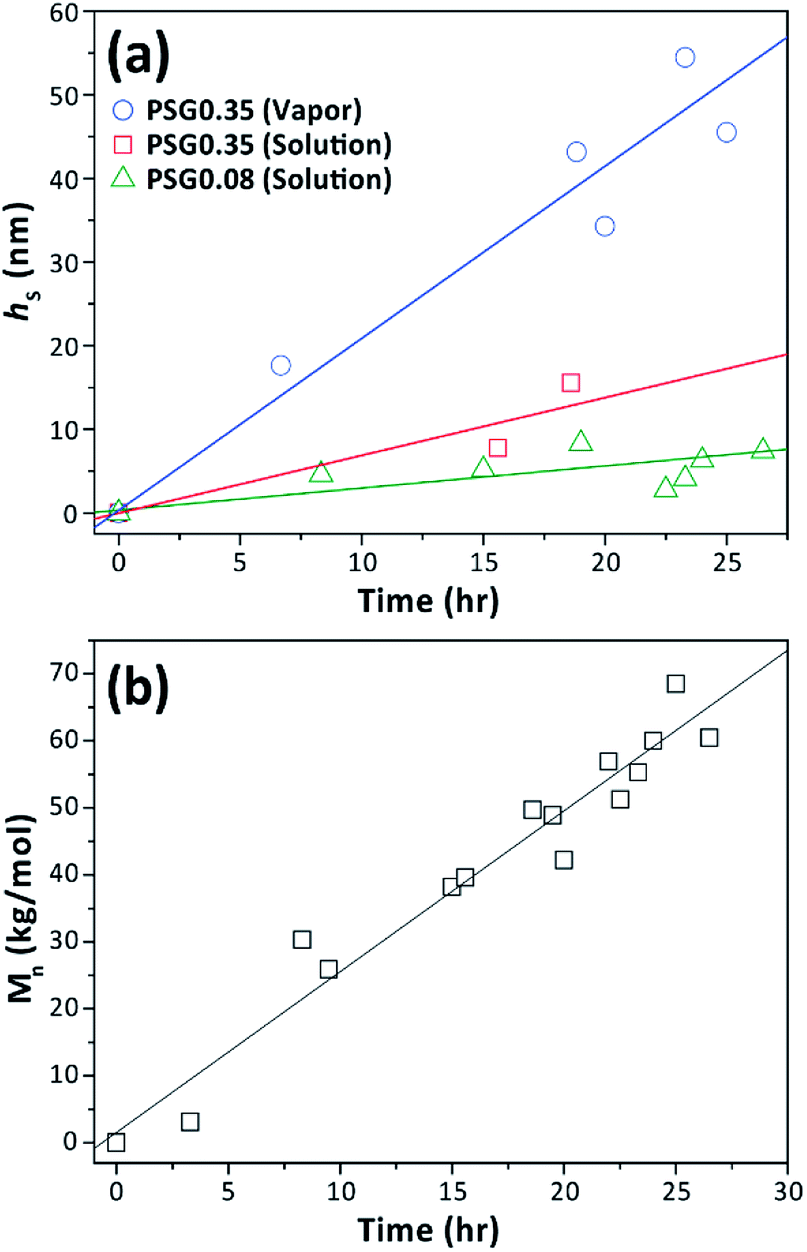 | ||
| Fig. 6 Kinetic plots showing (a) increased brush thickness (hS) during the chain extension with styrene from PMMA brushes prepared from various surfaces, (b) Mn of the PS grown from a sacrificial initiator as a function of polymerization time. | ||
Fig. 7 shows AFM height and phase images of as-prepared samples of representative symmetric P(S-b-MMA) block copolymer brushes (Fig. 7a, high grafting density, 27–40 K; Fig. 7b, moderate grafting density, 46–56 K; Fig. 7c, low grafting density, 101–137 K). In order to investigate the stability of BCP brushes, resulting samples were subjected to thermal annealing at 140 °C for 72 hours under vacuum, followed by washing with THF (Fig. 8a–c). The samples did not show any damages in the films with no significant changes in film thickness. If polymer brushes are damaged, the surface morphology is disrupted with the changes in the thickness and grafting density due to the detachment of brushes.33,40,41 Price et al. reported that PS and PMMA brushes at 0.1–0.3 chains per nm2 (moderate to high density regime) synthesized from the initiator tethered to the surface through Si–O bond were not stable enough at elevated temperature; a detachment of brushes and a significant decrease of film thickness were observed.33 In addition, the thiol–Au bond is classified as a semi-covalent bond: its bond energy is found to be ∼170 kJ mol−1,42,43 which is lower than that of the Si–O bond (∼452 kJ mol−1),44 suggesting the possibility to have a similar issue.
 | ||
| Fig. 7 AFM height images of as-prepared P(S-b-MMA) block copolymer brushes of (a) 27–40 K (rms roughness (Rq) = 0.8 nm), (b) 46–56 K (Rq = 0.9 nm), and (c) 101–137 K (Rq = 2.9 nm). The size of images is 1.5 μm × 1.5 μm. | ||
 | ||
| Fig. 8 AFM height (left) and phase (right) images of (a) 27–40 K (Rq = 1.0 nm), (b) 46–56 K (Rq = 0.5 nm), and (c) 101–137 K (Rq = 2.5 nm), after THF washing. (d) (Rq = 1.0 nm), (e) (Rq = 0.8 nm), and (f) (Rq = 2.0 nm) are after annealing of (a), (b), and (c), respectively. The size of images is 0.5 μm × 0.5 μm. | ||
The surface exhibited quite low surface roughness values and microphase separations which are interpreted as kinetically trapped morphologies during solvent evaporation in the washing step (Fig. 8a–c). These BCP brush samples were further subjected to second thermal annealing under the same conditions to the first annealing. As shown in the AFM images presented in Fig. 8d–f, it should be emphasized that any detachment of polymer brushes upon the second thermal annealing was not observed in all samples; also, the imparted stability of the brush samples enabled us to observe the surface morphologies upon thermal annealing, which are different from those before the annealing. In 27–40 K, mixed morphology of dot and stripe domains are observed (Fig. 8d). In case of 46–56 K, the majority is stripes with the small number of dots (Fig. 8e). The 101–137 K showed mostly stripe patterns with the largest domain size (Fig. 8f). It has been shown that a BCP brush with a symmetric composition exhibited poorly ordered stripe pattern at σBCP of 0.04 chains per nm2.20 The 101–137 K having a symmetric composition and σBCP in a mushroom regime also exhibited mostly stripe pattern, in good agreement with the previous studies. However, the morphologies at higher grafting density have not been systematically studied to the best of our knowledge; our results provide the BCP brushes at moderate and high grafting density regimes also can exhibit clear segregation behaviors, suggesting a possible grafting density effect.
Further experimental and theoretical studies are ongoing to provide a complete phase diagram including the effect of grafting density. These results highlight that synthesized BCP brushes are highly stable upon repetitive washing with the organic solvent and thermal annealing at the elevated temperature even at high grafting density, elucidating outstanding stability of epoxy-based crosslinked random copolymer thin films.
Conclusions
We report the synthesis of polymer brushes with effectively varied grafting densities using highly stable crosslinked P(S-r-GMA) thin films functionalized with BiBB, which can initiate SI-ATRP. Functionalized surfaces prepared under different processing conditions were carefully characterized with a variety of techniques in order to further optimize the brush growth step. With fully optimized SI-ATRP conditions, PMMA and P(S-b-MMA) brushes were successfully synthesized. The grafting densities of the homopolymer and the BCP brushes were controlled by the composition of the random copolymer and post-crosslinking functionalization methods. In this study, we achieved P(S-b-MMA) brushes in three different grafting density regimes. These BCP brush systems were highly stable upon several times of solvent washing and thermal annealing processes. The imparted stability of the brush samples enabled to observe the surface morphologies of BCP brushes with different grafting density. The simple, readily scalable, and versatile random copolymers provide highly stable functional films which make close examinations of assembled morphologies of the copolymer brushes upon thermal annealing possible.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Basic Science Research Program through the National Research Foundation of Korea (2015R1C1A1A01053266). We also acknowledge the financial support from the National Research Foundation of Korea (2015R1C1A2A01054713). S. K. acknowledges support from Inha University Research Grant (Grant No. INHA-52556).Notes and references
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef PubMed.
- M. Kim, S. K. Schmitt, J. W. Choi, J. D. Krutty and P. Gopalan, Polymers, 2015, 7, 1346–1378 CrossRef.
- W.-L. Chen, R. Cordero, H. Tran and C. K. Ober, Macromolecules, 2017, 50, 4089–4113 CrossRef.
- B. P. Crulhas, J. R. Sempionatto, M. F. Cabral, S. Minko and V. A. Pedrosa, Electroanalysis, 2014, 26, 815–822 CrossRef.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- Y. Chen, Z. Xu, D. Zhu, X. Tao, Y. Gao, H. Zhu, Z. Mao and J. Ling, J. Colloid Interface Sci., 2016, 483, 201–210 CrossRef PubMed.
- H. Arafune, T. Kamijo, T. Morinaga, S. Honma, T. Sato and Y. Tsujii, Adv. Mater. Interfaces, 2015, 2, 1500187 CrossRef.
- Y. Deng, X. Zhang, X. Zhao, Q. Li, Z. Ye, Z. Li, Y. Liu, Y. Zhou, H. Ma, G. Pan, D. Pei, J. Fang and S. Wei, Acta Biomater., 2013, 9, 8840–8850 CrossRef PubMed.
- L. Moroni, M. K. Gunnewiek and E. M. Benetti, Acta Biomater., 2014, 10, 2367–2378 CrossRef PubMed.
- S. Tugulu and H.-A. Klok, Biomacromolecules, 2008, 9, 906–912 CrossRef PubMed.
- B. R. Coad, Y. Lua and L. Meagher, Acta Biomater., 2012, 8, 608–618 CrossRef PubMed.
- I. Amin, M. Steenackers, N. Zhang, A. Beyer, X. Zhang, T. Pirzer, T. Hugel, R. Jordan and A. Gölzhäuser, Small, 2010, 6, 1623–1630 CrossRef PubMed.
- I. Amin, M. Steenackers, N. Zhang, R. Schubel, A. Beyer, A. Gölzhäuser and R. Jordan, Small, 2011, 7, 683–687 CrossRef PubMed.
- E. Han, M. Kim and P. Gopalan, ACS Nano, 2012, 6, 1823–1829 CrossRef PubMed.
- E. Han, M. Leolukman, M. Kim and P. Gopalan, ACS Nano, 2010, 4, 6527–6534 CrossRef PubMed.
- P. Mansky, Y. Liu, E. Huang, T. Russell and C. Hawker, Science, 1997, 275, 1458–1460 CrossRef.
- W. Lee, S. Park, Y. Kim, V. Sethuraman, N. Rebello, V. Ganesan and D. Y. Ryu, Macromolecules, 2017, 50, 5858–5866 CrossRef.
- M. Y. Paik, Y. Xu, A. Rastogi, M. Tanaka, Y. Yi and C. K. Ober, Nano Lett., 2010, 10, 3873–3879 CrossRef PubMed.
- A. Rastogi, M. Y. Park, M. Tanaka and C. K. Ober, ACS Nano, 2010, 4, 771–780 CrossRef PubMed.
- B. M. D. O'Driscoll, G. H. Griffiths, M. W. Matsen, S. Perrier, V. Ladmiral and I. W. Hamley, Macromolecules, 2010, 43, 8177–8184 CrossRef.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC.
- L. C. H. Moh, M. D. Losego and P. V. Braun, Langmuir, 2011, 27, 3698–3702 CrossRef PubMed.
- C. Zhao, L. Li and J. Zheng, Langmuir, 2010, 26, 17375–17382 CrossRef PubMed.
- K. Sparnacci, D. Antonioli, V. Gianotti, M. Laus, F. Ferrarese Lupi, T. J. Giammaria, G. Seguini and M. Perego, ACS Appl. Mater. Interfaces, 2015, 7, 10944–10951 CrossRef PubMed.
- O. Azzaroni, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3225–3258 CrossRef.
- K. Matyjaszewski, H. Dong, W. Jakubowski, J. Pietrasik and A. Kusumo, Langmuir, 2007, 23, 4528–4531 CrossRef PubMed.
- D. P. Sweat, M. Kim, X. Yu and P. Gopalan, Langmuir, 2013, 29, 3805–3812 CrossRef PubMed.
- D. P. Sweat, M. Kim, X. Yu, S. K. Schmitt, E. Han, J. W. Choi and P. Gopalan, Langmuir, 2013, 29, 12858–12865 CrossRef PubMed.
- D. R. Calabrese, D. Ditter, C. Liedel, A. Blumfield, R. Zentel and C. K. Ober, ACS Macro Lett., 2015, 4, 606–610 CrossRef.
- M. E. Welch and C. K. Ober, ACS Macro Lett., 2013, 2, 241–245 CrossRef.
- M. E. Welch, Y. Xu, H. Chen, N. Smith, M. E. Tague, H. D. Abruña, B. Baird and C. K. Ober, J. Photopolym. Sci. Technol., 2013, 25, 53–56 CrossRef PubMed.
- H. Ma, M. Wells, T. P. Beebe and A. Chilkoti, Adv. Funct. Mater., 2006, 16, 640–648 CrossRef.
- A. D. Price, S.-M. Hur, G. H. Fredrickson, A. L. Frischknecht and D. L. Huber, Macromolecules, 2012, 45, 510–524 CrossRef.
- S. Edmondson, C.-D. Vo, S. P. Armes, G.-F. Unali and M. P. Weir, Langmuir, 2008, 24, 7208–7215 CrossRef PubMed.
- P. Gopalan, E. Han and M. Kim, US Pat., US9115255, 2015.
- E. Han and P. Gopalan, Langmuir, 2010, 26, 1311–1315 CrossRef PubMed.
- S. Soundararajan, B. Reddy and S. Rajadurai, Polymer, 1990, 31, 366–370 CrossRef.
- P. C. Hiemenz and T. P. Lodge, Polymer Chemistry, CRC Press, Boca Raton, edn 2nd, 2007 Search PubMed.
- J. D. Jeyaprakash, S. Samuel, R. Dhamodharan and J. Rühe, Macromol. Rapid Commun., 2002, 23, 277–281 CrossRef.
- V. Gianotti, D. Antonioli, K. Sparnacci, M. Laus, T. J. Giammaria, F. Ferrarese Lupi, G. Seguini and M. Perego, Macromolecules, 2013, 46, 8224–8234 CrossRef.
- K. Sparnacci, D. Antonioli, V. Gianotti, M. Laus, G. Zuccheri, F. Ferrarese Lupi, T. J. Giammaria, G. Seguini, M. Ceresoli and M. Perego, ACS Appl. Mater. Interfaces, 2015, 7, 3920–3930 CrossRef PubMed.
- H. McNally, M. Pingle, S. W. Lee, D. Guo, D. E. Bergstrom and R. Bashira, Appl. Surf. Sci., 2003, 214, 109–119 CrossRef.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef PubMed.
- S. S. Zumdahl and S. A. Zumdahl, Chemistry, Cengage Learning, Boston, edn 9th, 2013 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04480e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |