
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe theoretical chemical calculations clarify the mechanism of beta-alkylation of 1-phenylethanol with benzyl alcohol catalyzed by iron(II) acetylacetonate methods†
Wei Fenga,
Junfeng Zhaoc,
Aiwen Weia,
Dandan Zhanga,
Huiling Liu *a,
Xuri Huang*a and
Kai Sun*b
*a,
Xuri Huang*a and
Kai Sun*b
aLaboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Technology Building of Jilin University, Liutiao Road 2, Changchun 130023, China. E-mail: huiling@jlu.edu.cn; huangxr@jlu.edu.cn
bSchool of Pharmaceutical Sciences, Jilin University, Changchun 130021, China. E-mail: thomassk@jlu.edu.cn
cDepartment of Materials, School of Technology, Yantai Nanshan University, Longkou, Shandong Province 265713, China
First published on 3rd July 2018
Abstract
Iron(II) acetylacetonate was suggested to be a better catalyst of the β-alkylation of 1-phenylethanol with benzyl alcohol to form 1,3-diphenyl-1-propanol. DFT calculations have been performed to study the internal mechanism, the structures of intermediates and transition states, and the exchange of electronic density in detail. The energetic results show that this β-alkylation reaction proceeds via the hydrogen autotransfer mechanism and the catalytic cycle includes three sequential stages: (1) alcohol oxidation to produce aldehyde associated with hydride anion transfer, (2) cross-aldol condensation to form a chalcone and (3) chalcone reduction with multi-step hydrogenation. In order to study whether the only by-product, water, has clearly influenced the reaction, eight catalyst hydrogenation pathways and four catalyst dehydrogenation pathways have been studied. We are delighted to find that the presence of the only by-product, water, can significantly increase the reduction energy barrier of dihydrochalcone. The energy barrier of the catalyst's hydrogenation is less than 6 kcal mol−1. Our calculation results are fundamentally coincident with the experimental detections, and suggest that the crossing-coupling reaction occurs through a reliable mechanism. Two dihydrochalcone catalysts were designed on the basis of how the β-alkylation reaction proceeds.
Introduction
The alkylation reaction is one of the most studied reactions in organic synthesis and catalysis due to its importance in carbon–carbon bond creation and long chain compound formation. Various alkylating agents are used in this process such as halohydrocarbon, sulfonate, and carbonyl compounds.1–5 However, most reactions generate considerable amounts of side products or waste. How to increase the atom-efficiency of the creation of carbon–carbon bonds is a key step of modern green chemistry. Alcohol, as one of the most common reagents, has been used in carbon–carbon bond formation reactions. In 2015, Jian and MacMillan developed a dual catalyst system that directly installs alkyl group fragments containing singly bonded carbon and hydrogen atoms. They showed that it is possible to accomplish mild, non-traditional alkylation using alcohols as radical precursors.6 This environment-friendly reaction aroused great interest, and more investigations using alcohols as the alkylating agents were reported.7–9The catalyst is another important factor of alkylation reaction. Most catalysts used in the alkylation reaction are mainly based on Ru, Pt, and Ir.10–13 However, these noble metals catalysts are expensive and are not easy to obtain. The use of more eco-friendly and widely abundant metals for the synthesis should be more beneficial for chemical research and industry manufacture. Iron, as the most productive metallic element on Earth, has been wildly used in alkylation reactions.14–16 Furthermore, iron as a catalyst has also been used in the alkylation reaction with alcohol as the alkylating agent.17,18
In 2012, Sun et al. found that the ferrocenecarboxaldehyde catalyst has a very good catalytic effect on the β-alkylation reaction of secondary alcohols with primary alcohols.19 We have conducted detailed theoretical calculations on the catalytic mechanism of ferrocenecarboxaldehyde. Sun et al. are always looking for cheaper, more stable, efficient and environmentally friendly catalysts. Recently, they found that under the same reaction conditions, the inexpensive acetylacetonate catalyst can also achieve very high yields (>95%) compared to the ferrocenecarboxaldehyde catalyst with an order of magnitude reduction.20
This microscale reaction mechanism has not been previously studied or explained. In this paper, we will use density functional method to analyze the reaction process in detail. According to our previous studies,21 a simplified plausibility process for the β-alkylation of water-involved secondary and primary alcohols is proposed in this paper (see Scheme 1).
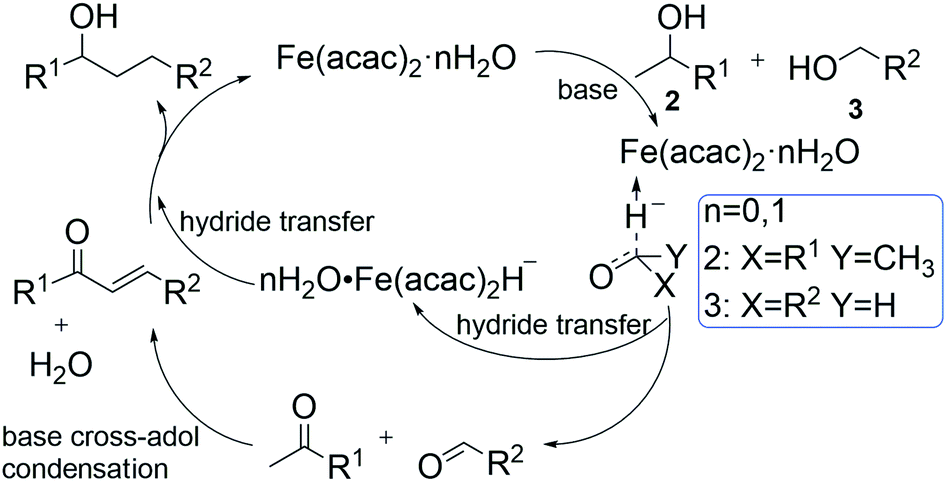 | ||
| Scheme 1 Mechanistic proposed for the iron-catalyzed crossing-coupling reaction. | ||
We conducted the elementarily theoretical calculation of the mechanism of the Fe(acac)2 catalyzed cross-coupling reaction. It also provides inspiration and reference for the synthesis of new cheap catalysts. At the end of the article, two ideal catalysts have also been suggested to control the product selectivity. We hope that this study could provide useful information for the industrial production.
Computational details
All geometrical optimization calculations were performed using the GAUSSIAN 09 revision D.01 (ref. 22) at the level M06-L23/BSI, BSI represents the basis set combination of SDD pseudopotential for Fe and 6-31g(d,p) for C, O and H atoms. At the same level of theory, frequency calculations were performed to identify the stationary points as real minima or transition states and to provide thermal corrections to the enthalpies and Gibbs free energies at 463.15 K. Intrinsic reaction coordinate (IRC) calculations24 were carried out to confirm the correct connections between a transition state and its forward and backward reactive intermediates. ORCA 3.0.3 (ref. 25) was used to perform the single-point calculations on previously optimized geometries. In order to obtain a better estimation of total electronic energies, the double-hybrid PWPB95 functional,26 including the D3 dispersion correction27,28 due to Grimme and the Becke–Johnson damping function, and the large basis set def2-TZVPP29 were adopted. We have verified the energies of the key species including diffuse functions in the basis set, and the results didn't changed greatly. In order to reduce the computational time, diffuse functions didn't included in and the resolution-of-identity (RI) technique is used for accelerating both self-consistent field (SCF) process and second-order perturbative correction part. The loss of accuracy due to above two factors is negligible. Schematic frontier molecular orbital (FMO) diagrams were represented by quasi-restricted orbitals, and were plotted using Chimera.30,31 Natural population analysis (NPA) charges were calculated by the NBO 3.1 module embedded in Gaussian 09 program.The solvation free energies of the optimized geometries were calculated at the M05-2X32/BSII (SMD, p-xylene) level (BSII designates the basis set combination of SDD pseudopotential for the Fe atom and 6-31G(d) for the other atoms; SMD corresponds to Truhlar and coworkers' solvation model which does an IEFPCM calculation with radii and non-electrostatic terms). All the energy that appears in this article is Gibbs free energy in kcal mol−1.
Results and discussion
Catalyst initiation
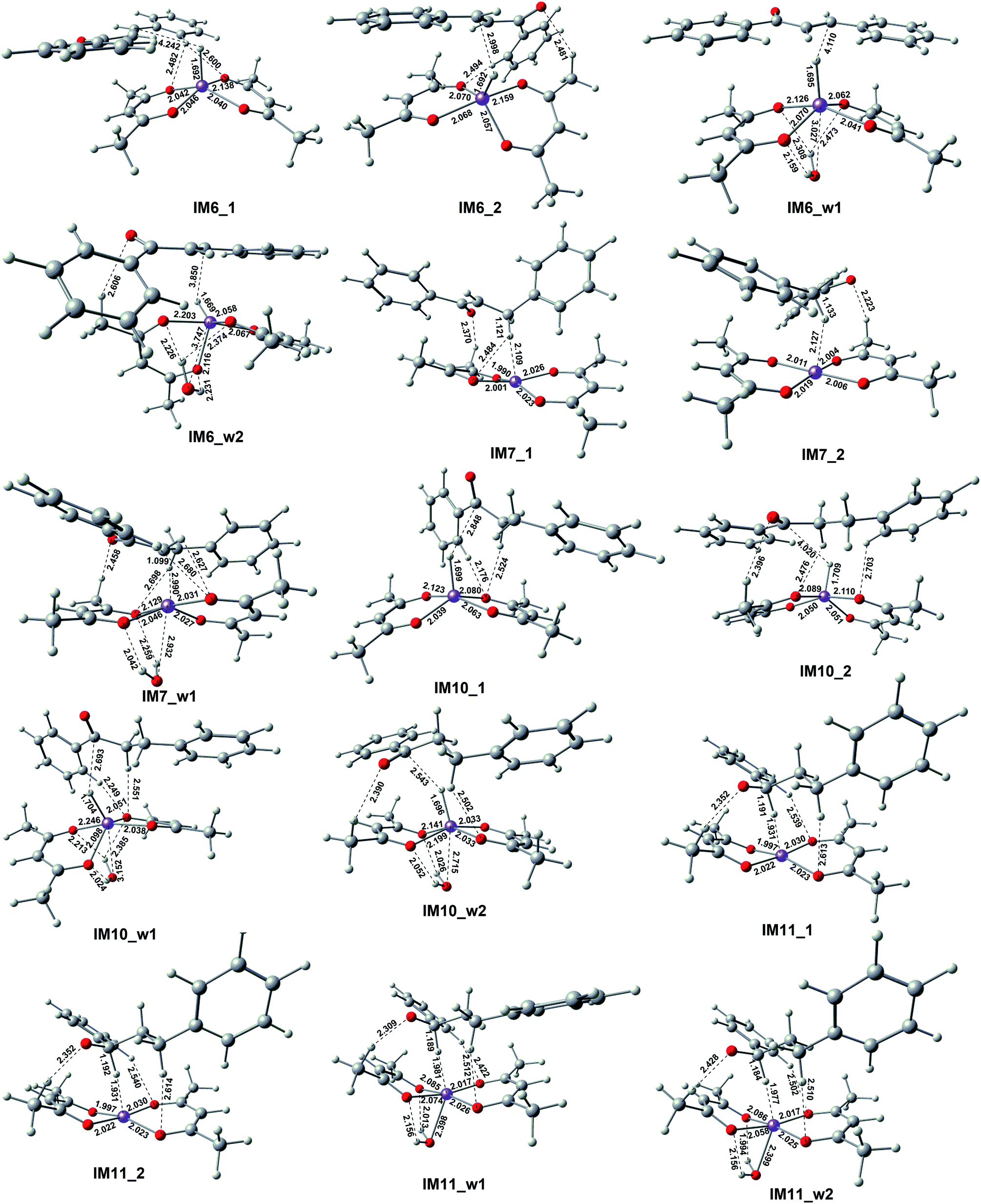
This reactant system includes 1-phenylethanol, benzyl alcohol, Fe(acac)2 and base (NaOH). First, the base accelerates the ionization of the 1-phenylethanol and benzyl alcohol, which give the 1-phenylethanol anion (R1) and benzyl alcohol anion (R2) respectively. Then, iron-catalyst, come into effect with 1-phenylethanol anion and benzyl alcohol anion to form the substrate–catalyst complexes by sigma complexation between the H and Fe. The optimized geometries of the intermediates and transition with selected pivotal parameters are illustrated in Fig. 2. As shown in Fig. 2, the phenethyl alcohol anion forms a complex with Fe(acac)2 in two different conformations, IM1_1 and IM1_2, followed by the corresponding two kinds of significant differences in the transition state TS1_1 and TS1_2. For TS1_1, the two hydrogen atoms of the alcohol anion points to the oxygen atom of the ligand. While for TS1_2, the benzene ring is almost parallel to the ligand plane. We refer to these two conformations as type I and type II conformations, respectively, and apply them to the discussion of the conformation of compounds appearing below. The distance of Fe–H in type II (1.728 Å) is shorter than that of type I (1.742 Å). This situation also applies to benzyl alcohol anions. As a control, reactions with water participation are also shown in Fig. 2. As an abbreviation of water, if letter “w” appears in the name of the compound, it means that there is reaction water involved, and this applies to the nomenclature of all the compounds herein. When water participates, the Fe–H distance in the transition state is shortened, and the Fe–H distance in type II is shorter. For TS1_w4, the distance of H2O–Fe is the shortest, and the strong effect between the water and Fe causing the effects of Fe and H to be greatly weakened, and the distance of the Fe–O (2.686 Å) is the longest among all transition states.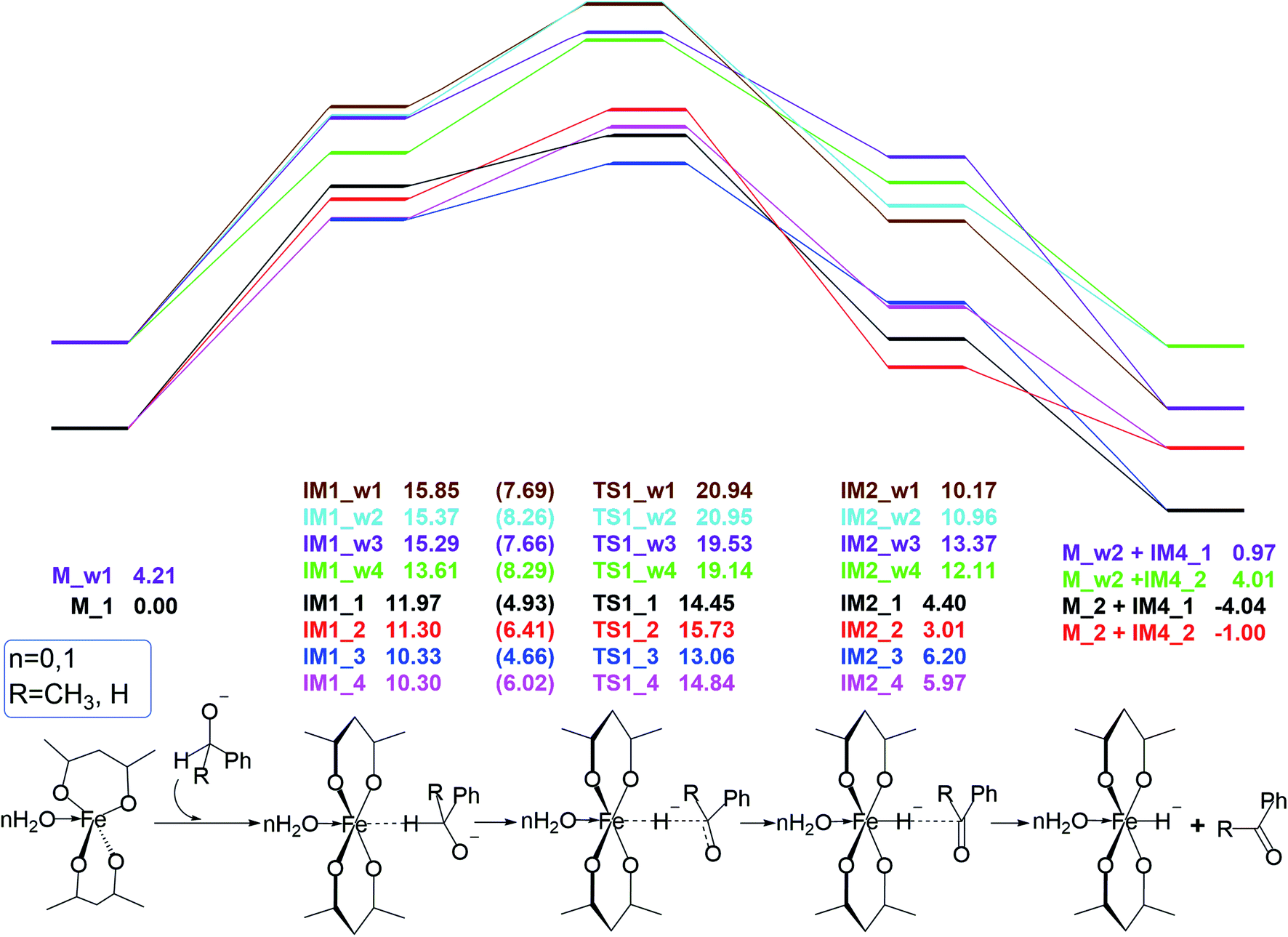 | ||
| Fig. 1 Eight reaction pathways and corresponding free energy profiles of H-adsorption step in catalyst initiation. The numbers in parentheses refer to the activation barriers. | ||
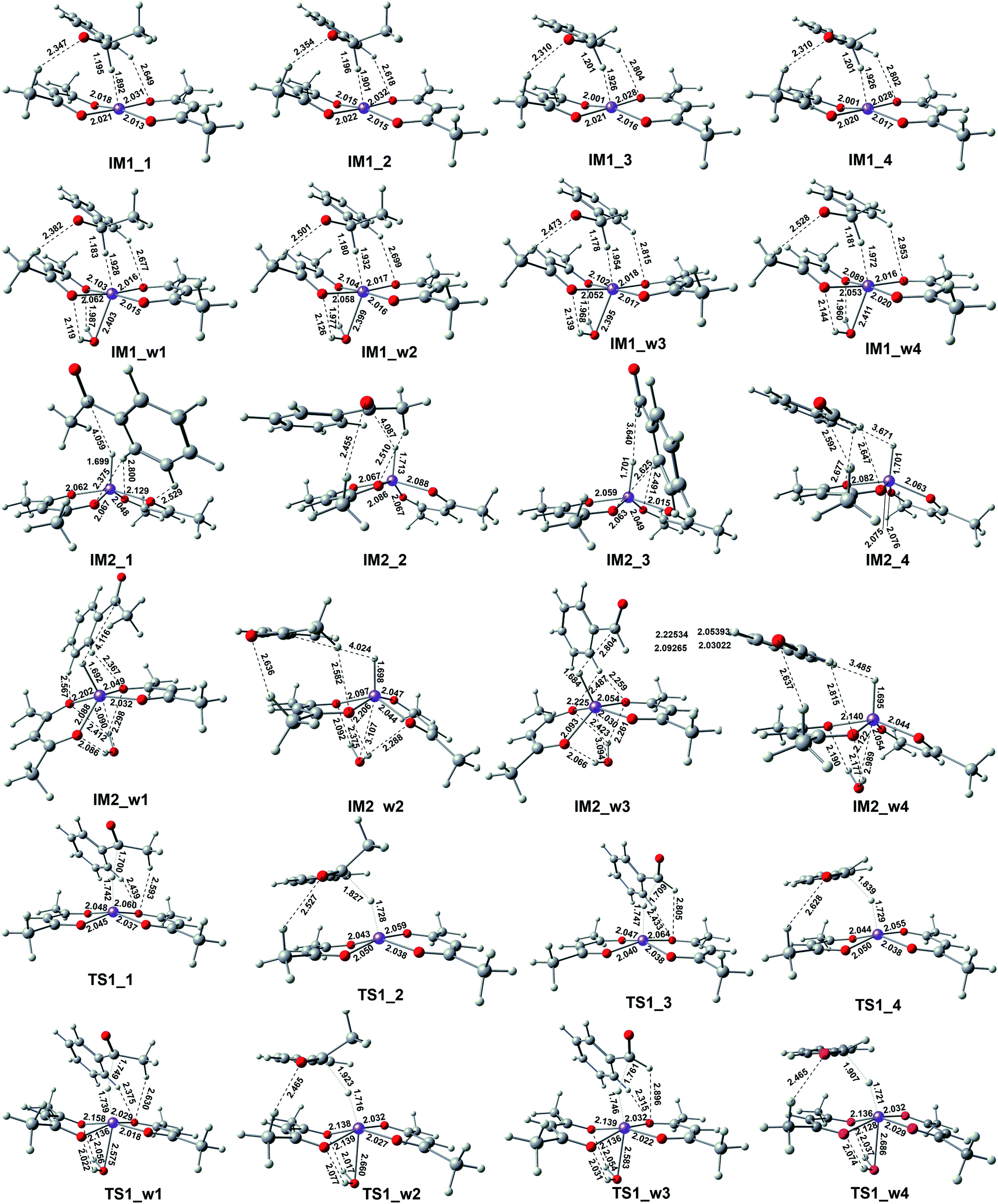 | ||
| Fig. 2 Optimized geometries of the complexes with selected bond distances (in Å). | ||
Free energy profiles for the 1-phenylethanol anion (R1) and benzyl alcohol anion (R2) oxidation via the inner-sphere hydrogen transfer pathways are illustrated in Fig. 1. In the absence of water, the barrier of reaction energy in Fig. 1 is in the range of 2.48 to 4.53 kcal mol−1. As a control, the reaction energy barrier for water participation is in the range of 4.24 to 5.53 kcal mol−1. It can be seen that the presence of water hardly affects the hydrogen atoms transfer from alcohol anion to the Fe of catalyst. This is because the reaction core areas of the two cases are very similar to each other.
Cross-aldol condensation
The intermediate products acetophenone (IM4_1) and benzaldehyde (IM4_2) can undergo aldol condensation without the catalyst in an alkaline environment to form chalcone. The mechanism has been detailed studied in our previous study,21 so in this article, we will not focus on this process.In our previous study, we illuminate the details of the mechanism aspects of the ferrocenecarboxaldehyde-catalyzed cross-coupling reaction of 1-phenylethanol with benzyl alcohol based on the density functional theory (DFT) at the M06-2X level of the theory. It should be noted that this reaction system only required inexpensive and commercially available ferrocenecarboxaldehyde and a catalytic amount of NaOH without sacrificial agent (hydrogen acceptors or hydrogen donors) and complex nitrogen or phosphorus ligands. The intermediate products acetophenone and benzaldehyde can undergo aldol condensation without catalyst in an alkaline environment to form chalcone. The formation of new C–C bond from cross-coupling reaction has been studied in detail. On account of the similar feature of benzyl alcohol anion and 1-phenylethanol anion, we use the benzyl alcohol anion as the research object to participate in reaction. This part begins with acetophenone transfers a hydrogen proton to benzyl alcohol anion to form the acetophenone anion via the first transition state with the barrier of 0.3 kcal mol−1. Then the acetophenone anion will attack benzaldehyde to form of a new anion intermediate via the second transition state with the barrier of 0.9 kcal mol−1. Then a hydrogen proton transfer from benzyl alcohol to this anion intermediate to form a new electrically neutral intermediate via the third transition state with the barrier of −0.7 kcal mol−1. After the generation of this electrically neutral intermediate, benzyl alcohol anion enters this reaction process again to capture a hydrogen proton to form another new anion intermediate and benzyl alcohol via the fourth transition state with the barrier of 0.8 kcal mol−1. Then benzyl alcohol will provide a hydrogen proton to this anion intermediate to form intermediate chalcone with fifth transition state with the barrier of 11.5 kcal mol−1.
Chalcone reduction
After the generation of chalcone (IM5), which is a conjugated planar macromolecule containing both C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC double bonds, iron hydride species (M_2 or M_w2), benzyl alcohol and phenylethyl alcohol enter the hydrogen transfer process. The reaction mechanism and the energy are depicted in Fig. 3. First, the H atom on Fe attacks the unsaturated C to form the first step hydrogen reduction intermediate product. Then this reduction product can take hydrogen protons from the benzyl alcohol or phenyl ethanol of the reactants, and realize the second step hydrogen reduction.
O and CC double bonds, iron hydride species (M_2 or M_w2), benzyl alcohol and phenylethyl alcohol enter the hydrogen transfer process. The reaction mechanism and the energy are depicted in Fig. 3. First, the H atom on Fe attacks the unsaturated C to form the first step hydrogen reduction intermediate product. Then this reduction product can take hydrogen protons from the benzyl alcohol or phenyl ethanol of the reactants, and realize the second step hydrogen reduction.
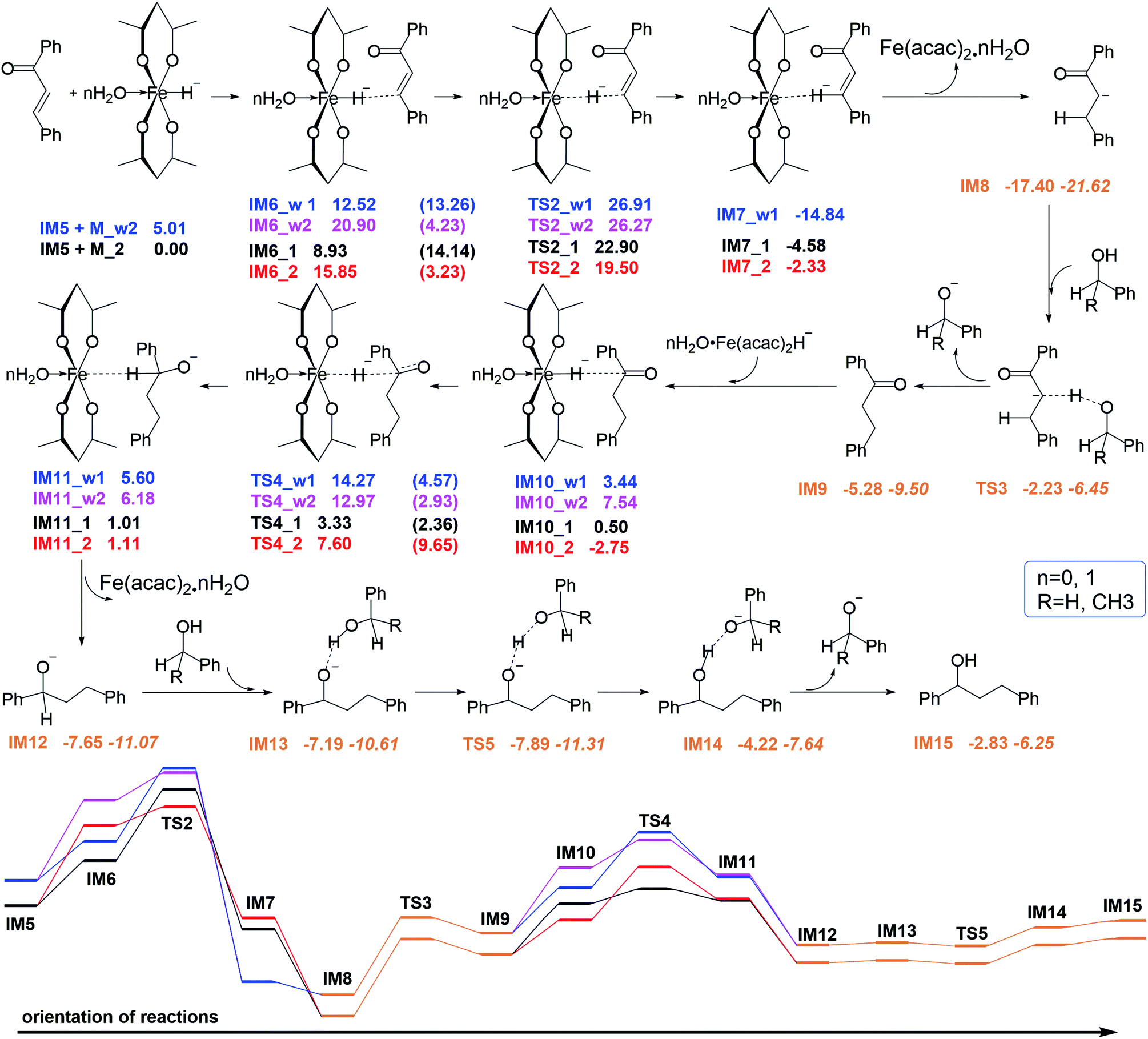 | ||
| Fig. 3 Four reaction pathways and corresponding free energy profiles of chalcone reduction processes. The orange line represents the process by which the intermediate product abstracts hydrogen protons, regardless of the catalyst, and the energy in italics corresponds to the path without water. The numbers in parentheses refer to the activation barriers. | ||
If the reaction stops at this step, dihydrochalcone will accumulate. In fact, dihydrochalcone was indeed experimentally detected from the product. As the reaction continues, the carbonyl will be reduced. The reduction process is also a hydrogenation catalyst that first provides a hydride ion to carbon, and then this hydrogenation intermediate product abstracts hydrogen protons from benzyl alcohol and phenylethanol. The two sets of corresponding transition states with slightly different energy barrier (TS3_1 (17.25 kcal mol−1), TS3_2 (15.17 kcal mol−1), TS5_1 (−0.70 kcal mol−1) and TS5_2 (−2.84 kcal mol−1)) when benzyl alcohol and phenylethyl alcohol were used as donors for hydrogen protons. Thus we selected one set to make the energy change line graph of Fig. 3 for more concise. The intermediate products and transition state structures are shown in Fig. 4 and 5.
 | ||
| Fig. 4 The geometries of the optimized intermediates correspond to those in Fig. 3 with selected bond distances (in Å). | ||
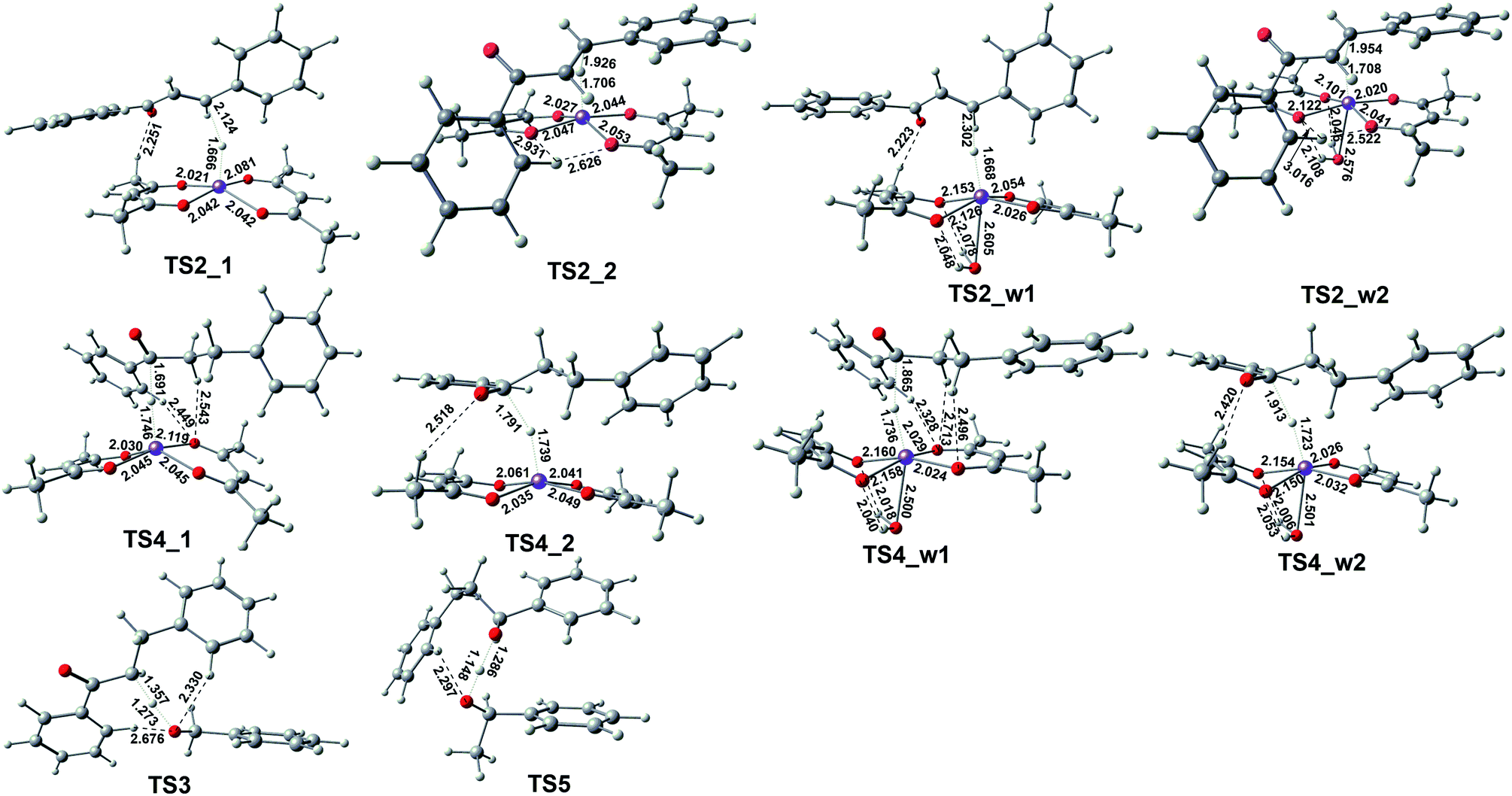 | ||
| Fig. 5 The geometries of the optimized transition states correspond to those in Fig. 3 with selected bond distances (in Å). | ||
As shown in Fig. 5, the first step hydrogenation transition configurations of chalcone catalytic belong to type II, with the difference that the benzene ring parallel to the catalyst is close to the carbonyl group. The distance of H2O–Fe in the transition state TS2_w1 (2.605 Å) and the transition state TS2_w2 (2.576 Å) indicates that the water and Fe have a stronger effect. As a result, the distance of Fe–H and H–C is longer than that without water. It is worth noting that these two transitional states generate the same intermediate IM7_w1. We are pleased to find that the presence of water also hardly affects the reaction energy barrier (about 14 kcal mol−1 and 5 kcal mol−1) of the first step hydrogenation of chalcone. Then through the transition state TS3, the chalcone is reduced to dihydrochalcone, and the original conjugated macrosystem is destroyed (Fig. 5). The transitional state of catalytic hydrogenation of dihydrochalcone simultaneously contains both type I and type II structures. The distance of H2O–Fe in the type I transition state TS4_w1 (2.500 Å) and the type II transition state TS2_w2 (2.501 Å) indicates that the water and Fe have a stronger effect. The distance of H2O–Fe in the type I transition state TS4_w1 and the type II transition state TS4_w2 is quite equal to 2.5 Å, which is obviously shorter than that of other aqueous transition states. This shows that the water–catalyst interaction is stronger in this step hydrogenation reduction reaction, and can most significantly affect the reaction. The type I transition state without water is the lowest energy barrier (2.83 kcal mol−1), and the energy barrier is raised to 10.83 kcal mol−1 when water is present. On the contrary, the type II transition energy barrier (10.35 kcal mol−1) was reduced to 5.43 kcal mol−1. In summary, the presence of water improves the reaction energy barrier for the catalytic hydrogenation of dihydrochalcone, and the dominant reaction pathways have also been interchanged (from type I to type II). The final step of the reaction is a hydrogen proton of the hydroxyl on the phenethyl alcohol transfer process to create 1,3-diphenyl-1-propanol. This procedure proceeds by transition state TS5 to produce the product. The barrier height via TS5 is measured to be −0.7 kcal mol−1. Since the product of all the reaction is mainly alcohol, it is quite fit with the experimental result.
Electronic structure analysis
In the process of dihydrochalcone reduction, the critical step is the catalytic hydrogenation of carbonyl (the process from IM10 to IM11 involving TS4 shown in Fig. 3). Detailed electronic structure analysis can provide deep insight for the reactivity of hydrogen transfer. The schematic frontier molecular orbitals (FMOs) corresponding to the process from IM10 to IM11 are shown in Fig. 6–9. In order to concise, the Fig. 6–9 only list the molecular orbitals that are most closely related to the reaction core.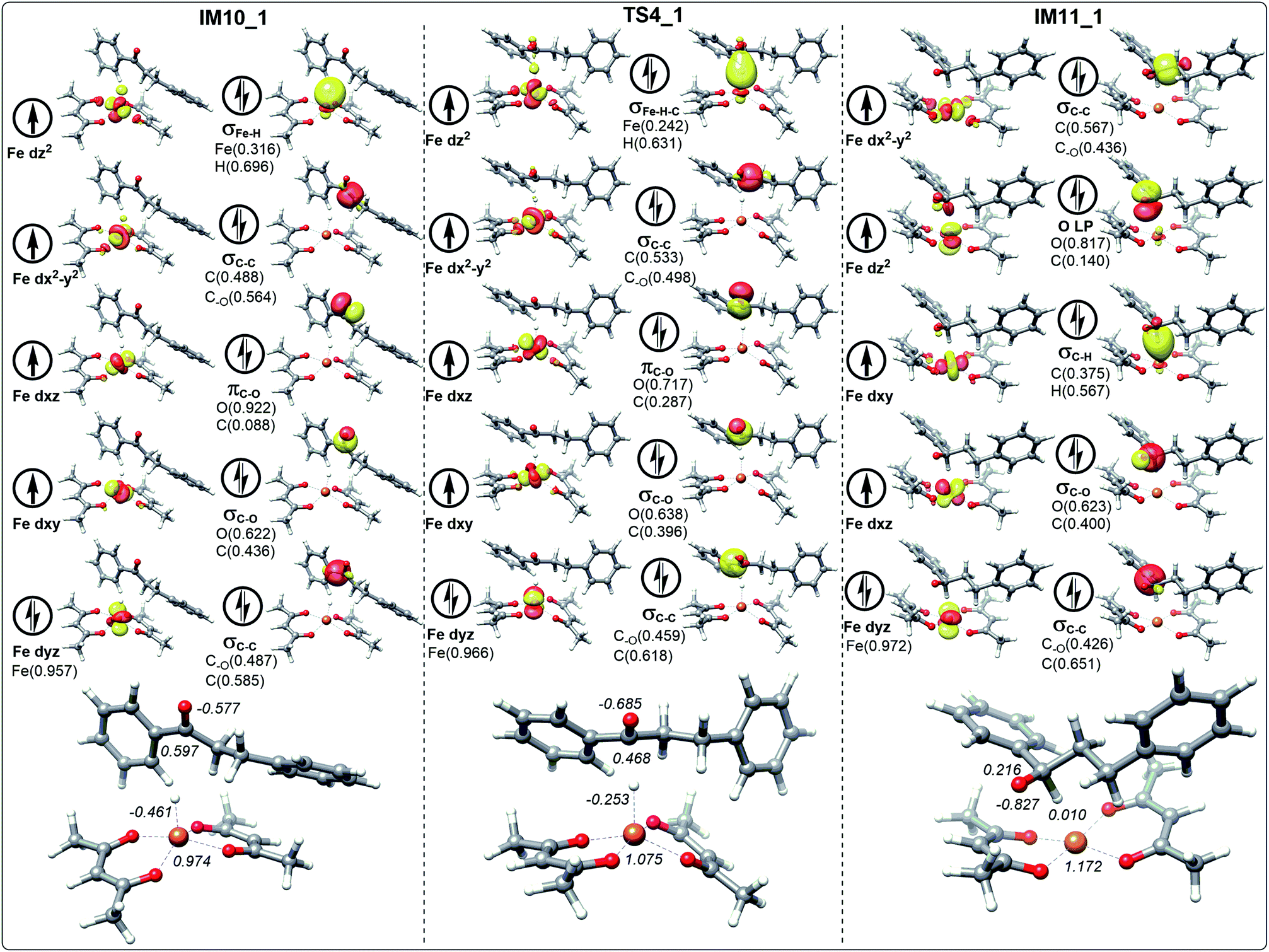 | ||
| Fig. 6 The schematic frontier molecular orbitals (including the composition) of hydrogen transfer corresponding to IM10_1, TS4_1 and IM11_1 involving the path without water. The values in italic represent the NPA charges of Fe, H, C and O. | ||
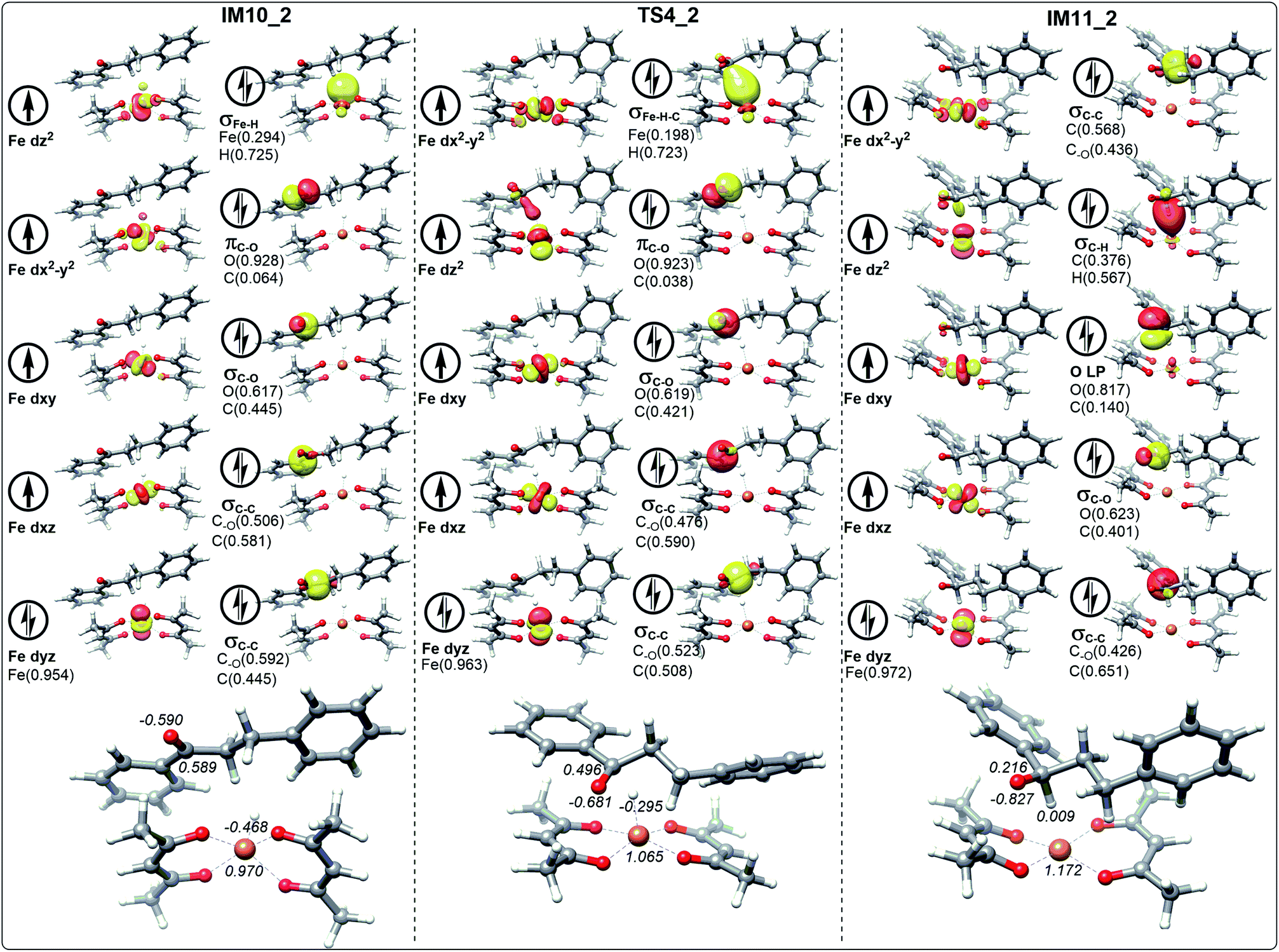 | ||
| Fig. 7 The schematic frontier molecular orbitals (including the composition) of hydrogen transfer corresponding to IM10_2, TS4_2 and IM11_2 involving the path without water. The values in italic represent the NPA charges of Fe, H, C and O. | ||
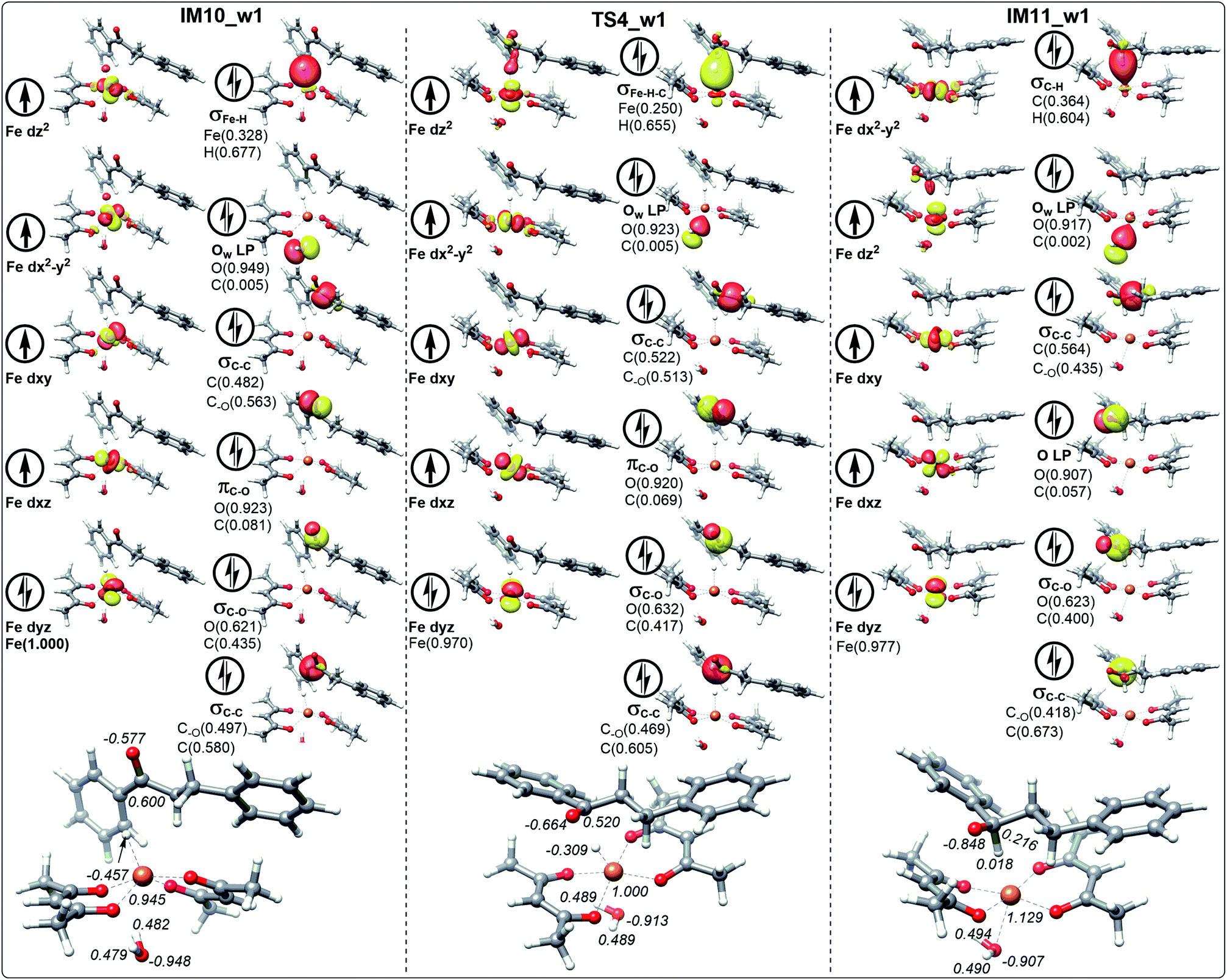 | ||
| Fig. 8 The schematic frontier molecular orbitals (including the composition) of hydrogen transfer corresponding to IM10_w1, TS4_w1 and IM11_w1 involving the path with water. The values in italic represent the NPA charges of Fe, H, C and O. | ||
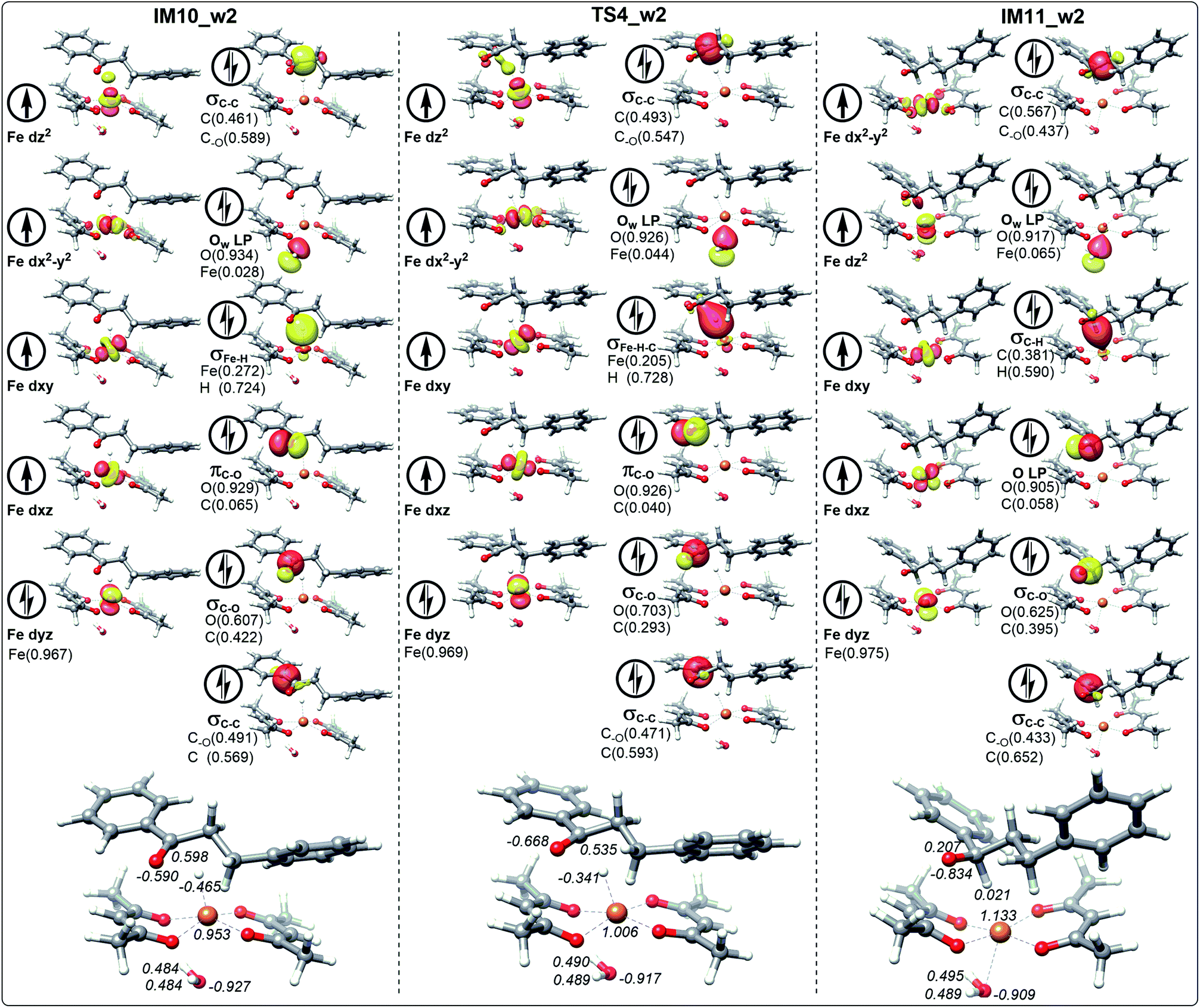 | ||
| Fig. 9 The schematic frontier molecular orbitals (including the composition) of hydrogen transfer corresponding to IM10_w2, TS4_w2 and IM11_w2 involving the path with water. The values in italic represent the NPA charges of Fe, H, C and O. | ||
For the ten molecular orbitals of IM10_1 in Fig. 6, the four single-occupied orbitals dz2, dx2−y2, dxz, and dxy, are the highest occupied orbitals. The remaining six orbitals are the lower-energy localized dual-occupant molecular orbitals and are ranked in the right-hand column in terms of energy from high to low. As we known, the s-orbitals and the p-orbitals of iron center are double occupancy. The composition of hydrogen in σFe–H orbital is 0.696, which means that the σFe–H bond is mainly composed of a pair of electrons of hydrogen. This is very helpful to understand that hydrogen has a negative NPA charge of −0.461. With chalcone close to the catalyst, hydrogen gradually moves away from the Fe center and approaches the carbonyl carbon. The NPA charge of Fe in transition state TS4_1 increases to 1.075. In contrast, the charge of carbonyl carbon decreases to 0.468 and increases to 0.287 in the composition of πC–O orbital. We also found that the composition of carbonyl carbon in the σC–O and σC–C orbitals is decreasing, which indicates that the electron delocalization of carbonyl carbon is decreasing. Subsequently, the electrons transfer from σFe–H–C to σC–H intensifies to the transition of carbonyl carbon from sp2 hybrid to sp3 hybrid. In IM11-1, the lone electron pairs of carbonyl oxygen have been concentrated on O p-orbital, resulting in the disappearance of πC–O bond and the decrease of its NPA charge to −0.827. It is found that in the newly formed σC–H orbital, the composition of hydrogen is higher than that of carbon, but the NPA charge of carbon decreases to 0.216, and the charge of hydrogen increases to a positive 0.010. This is because the electronegativity of carbon is stronger than that of hydrogen, causing the σC–H orbital to move toward carbon. In Fig. 7, 8 and 9, the process of hydrogen transfer to carbonyl carbon is very similar.
It is worth noting that the NPA charge of Fe in Fig. 8 (Fig. 9) is smaller than that in Fig. 6 (Fig. 7) due to water presence. The ten molecular orbitals of IM10_2 (TS4_2) are very similar to those of IM10_1 (TS4_1) except that the order of σC–C orbital becomes lower than the πC–O and σC–O orbitals. This may be due to the interaction of the oxygen atoms of dihydrochalcone with the hydrogen atoms on the catalyst.
Fig. 8 and 9 show the schematic frontier molecular orbitals of the reactions with water participation. It can be observed that O atom of water molecular interacts with Fe center (2.500 Å) to form a coordination bond through O p-orbital (Ow LP) in TS4_w1 and TS4_w2. Similarly, in the reactions of hydrogen transfer corresponding to the path with water participation also proceeds via hydrogen anion transfer process. The orbital energy of Ow LP is only lower than that of σFe–H of IM10_w1 in Fig. 8. By comparing TS4_w1 and TS4_1 with their pre-complexes IM10_w1 and IM10_1, it can be seen that the orbital order does not change when water is present. It is worth noting that in contrast to Fig. 6, 7 and 8, the σFe–H orbital in Fig. 9 is no longer the energy highest among double occupancy molecular orbitals, which results in a lower energy barrier.
Design of catalysts for the production of dihydrochalcone
To our knowledge, dihydrochalcone has been established as effective compounds for a number of pharmaceutical applications, such as antidiabetic, antimalaria, antibacterial, antifungal, anti-inflammatory and anti-tumor drugs.33–39 It is desirable if the reaction can stay at the dihydrochalcone stage. Therefore we focus on the catalysts for the production of dihydrochalcone. Through all the discussions above, three important points can be found:(1) Although the adsorption energy of water and catalyst is positive and thermodynamically unfavorable, the value of about 5 kcal mol−1 indicates that water can be easily adsorbed to the catalyst. When the hydride ion is transferred to the center iron of the catalyst, the H2O–Fe bond is significantly elongated (from 2.219 to 3.048 Å). At this point, the interaction between water and catalyst is almost entirely a weak hydrogen bond between water's hydrogen and ligand oxygen (it can be seen from the water's orientation and O–H bond length). This case also applies to the catalytic process.
(2) The interaction between water and iron center in transition state is stronger than that in the pre-complexes. It shows that the H2O–Fe bond length is shorter, and the water's p orbital clearly points to the iron center in transition states. Compared to other transition states, the interaction in TS4_w1 and TS4_w2 are enhancive since their H2O–Fe bond lengths are shorter (very consistent equals to 2.500 Å).
(3) In the presence of water, the reduction energy barriers of dihydrochalcone, IM9, are affected in the three groups containing H-anion transfer processes (IM1 → TS1, IM6 → TS2 and IM10 → TS4). It is obviously that the energy barriers in the first two groups are barely affected, and the last group is the critical step. In this step, the reduction energy barrier of type I is increased to 10.83 kcal mol−1, and it is greater than the inverse reaction energy barrier of 8.67 kcal mol−1. While the reduction energy barrier of type II is reduced to 5.53 kcal mol−1, which is less than the inverse reaction energy barrier of 6.79 kcal mol−1. It can be seen that the type I reaction favors to stay in the dihydrochalcone.
Based on the above three points of discussion, we can modify the acetylacetonate ferrous catalyst to make the reaction stay at the dihydrochalcone stage. The theoretically designed catalysts, M_3 and M_4, based on acetylacetonate ferrous are shown in Fig. 10. They have three outstanding advantages: (1) the methyl group on the catalyst ligand is replaced with a CC double bond, which avoids the type II reaction and expands the electron delocalization of the ligand to stabilize the catalyst; (2) the change of O–Fe distance can be easily achieved by flexible alkanes twisting. When the hydride ion is transferred to the center iron of M_3 or M_4, the O–Fe distance is as long as 3.800 Å or 3.663 Å. While there is no H anion, the O–Fe distance is only 2.607 Å or 2.283 Å; (3) the reaction core Fe is in semi-enveloping state, which makes the catalyst's applicability and anti-interference stronger.
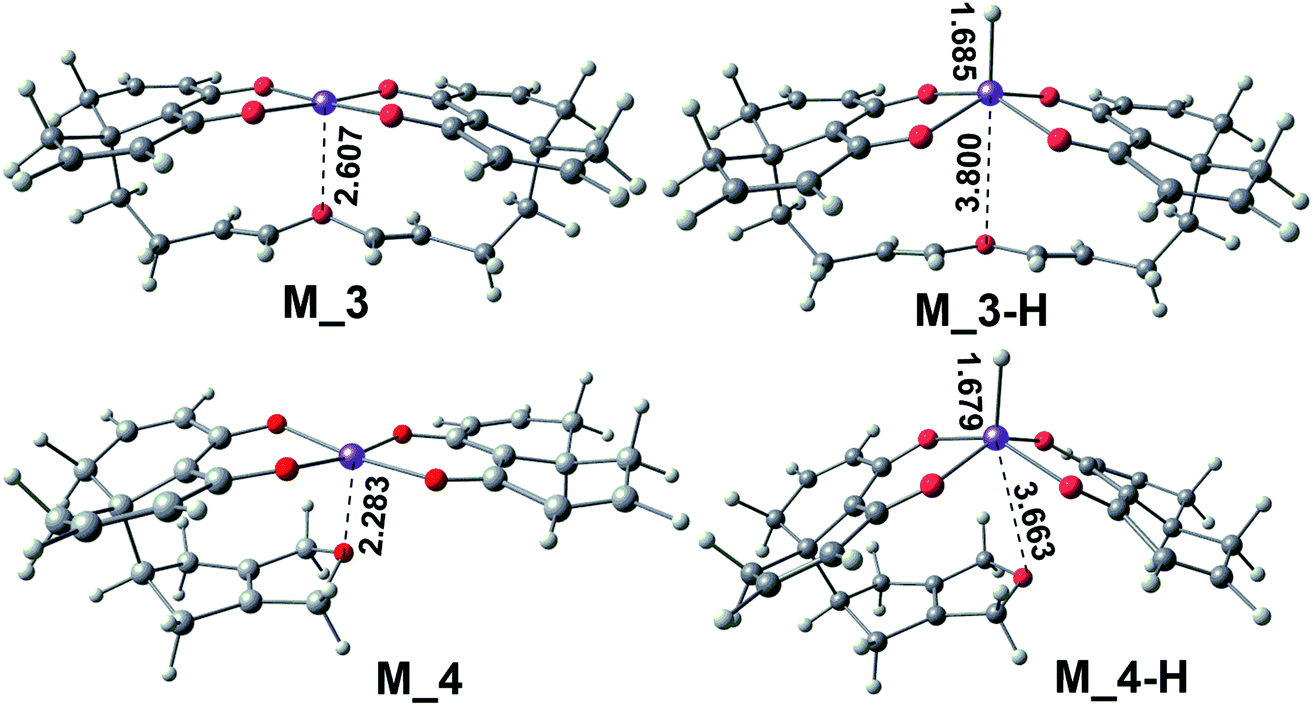 | ||
| Fig. 10 Two dihydrochalcone catalysts were designed on the basis of chemical reactions in β-alkylation reaction proceeds. | ||
Finally, we also found that the benzene ring is far from the reaction core (the metal atom in the catalyst center) and is also almost unaffected by the catalyst. This is also the reason why acetylacetonate catalysts can catalyze the alkylation of alcohols with substituents (such as halogen and alkoxy groups) on the benzene ring. The catalysts we design should also have good catalytic effects on the derivatives of such alcohols.
Conclusions
In this calculation, we illuminate the details of the mechanism aspects of the Fe(acac)2-catalyzed cross-coupling reaction of 1-phenylethanol with benzyl alcohol based on the density functional theory (DFT) includes the effect of paraxylene solvent effect. It should be noted that this reaction system only required inexpensive and commercially available Fe(acac)2 and a catalytic amount of NaOH without sacrificial agent (hydrogen acceptors or hydrogen donors) and complex nitrogen or phosphorus ligands. The following findings can be revealed by our calculation. (1) The catalyzed reaction consists of three steps: hydride transfer from α-C of benzyl alcohol and 1-phenylethanol accompanied by the electron transfer; cross-coupling reaction to generate new C–C bond; and the reduction step with the migration of hydrogen from metal hydride (M_1 or M_w1) and benzyl alcohol, leading to the generation of product. (2) The energy of the entire cycle indicates that the activation barriers for the hydrogen abstraction are a very low, under 5 kcal mol−1, but it is quite fit for the experiment conditions. (3) The only by-product water has only clearly influenced the reduction of the intermediate dihydrochalcone. Detailed electronic structure analysis was performed to achieve a deep insight for this step. (4) Based on these results, two dihydrochalcone catalysts were designed on the basis of chemical reactions in β-alkylation reaction proceeds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 21573090, No. 21403086), and Scientific Research Fund of Jilin Provincial Education Department (2015437) for financial support of this research. Project of Scientific Research Development of Shandong Universities (Grant No. J17KB076). Science and Technology Research Project of Jilin Provincial Department of Education in 12th Five-Year Plan (No. 388[2011]).References
- A. Gradillas, E. Belmonte, R. F. da Silva and J. Pérez-Castells, Eur. J. Org. Chem., 2014, 2014, 1935–1941 CrossRef.
- S. O. Poelma, G. L. Burnett, E. H. Discekici, K. M. Mattson, N. J. Treat, Y. Luo, Z. M. Hudson, S. L. Shankel, P. G. Clark and J. W. Kramer, J. Org. Chem., 2016, 81, 7155–7160 CrossRef PubMed.
- J. Yang, J. Zhang, T. T. Chen, D. M. Sun, J. Li and X. F. Wu, Chin. Chem. Lett., 2011, 22, 1391–1394 CrossRef.
- J.-H. Fan, J. Yang, R.-J. Song and J.-H. Li, Org. Lett., 2015, 17, 836–839 CrossRef PubMed.
- A. Nakatani, K. Hirano, T. Satoh and M. Miura, Chem.–Eur. J., 2013, 19, 7691–7695 CrossRef PubMed.
- J. Jin and D. W. MacMillan, Nature, 2015, 525, 87–90 CrossRef PubMed.
- P. Liu, R. Liang, L. Lu, Z. Yu and F. Li, J. Org. Chem., 2017, 82, 1943–1950 CrossRef PubMed.
- P. Paul, P. Bhanja, N. Salam, U. Mandi, A. Bhaumik, S. M. Alam and S. M. Islam, J. Colloid Interface Sci., 2017, 493, 206–217 CrossRef PubMed.
- W. Bai and G. Jia, Inorg. Chim. Acta, 2015, 431, 234–241 CrossRef.
- J. s. Campos, J. n. López-Serrano, E. Álvarez and E. Carmona, J. Am. Chem. Soc., 2012, 134, 7165–7175 CrossRef PubMed.
- T. Matsuda and I. Yuihara, Chem. Commun., 2015, 51, 7393–7396 RSC.
- G. Li, X. Ma, C. Jia, Q. Han, Y. Wang, J. Wang, L. Yu and S. Yang, Chem. Commun., 2017, 53, 1261–1264 RSC.
- Z.-Z. Zhou, F.-S. Liu, D.-S. Shen, C. Tan and L.-Y. Luo, Inorg. Chem. Commun., 2011, 14, 659–662 CrossRef.
- L. D. Tran and O. Daugulis, Org. Lett., 2010, 12, 4277–4279 CrossRef PubMed.
- E. R. Fruchey, B. M. Monks and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 13130–13133 CrossRef PubMed.
- Q. Yang, P. Wu, J. Chen and Z. Yu, Chem. Commun., 2014, 50, 6337–6339 RSC.
- T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef PubMed.
- S. Elangovan, J. B. Sortais, M. Beller and C. Darcel, Angew. Chem., Int. Ed., 2015, 54, 14483–14486 CrossRef PubMed.
- J. Yang, X. Liu, D. L. Meng, H. Y. Chen, Z. H. Zong, T. T. Feng and K. Sun, Adv. Synth. Catal., 2012, 354, 328–334 CrossRef.
- H. Xu, Master thesis, Jilin University, 2016.
- P. Y. Qin, W. Feng, R. Wang, H. Y. Zhao, H. L. Liu, X. R. Huang and K. Sun, Polyhedron, 2016, 111, 179–184 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 Search PubMed.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, J. Am. Chem. Soc., 2006, 128, 10213–10222 CrossRef PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef PubMed.
- Y. Zhao, N. Mahaja and J. Y. Fang, Small, 2006, 2, 364–367 CrossRef PubMed.
- W. Lesse, Gynecol. Obstet. Invest., 1902, 15, 680–693 CrossRef.
- J. Nelson and R. Falk, Anticancer Res., 1993, 13, 2287–2292 Search PubMed.
- J. Nelson and R. Falk, Anticancer Res., 1993, 13, 2293–2299 Search PubMed.
- Y. Nakamura, S. Watanabe, N. Miyake, H. Kohno and T. Osawa, J. Agric. Food Chem., 2003, 51, 3309–3312 CrossRef PubMed.
- C. Echeverria, J. F. Santibanez, O. Donoso-Tauda, C. A. Escobar and R. Ramirez-Tagle, Int. J. Mol. Sci., 2009, 10, 221–231 CrossRef PubMed.
- L. Norton, R. A. DeFronzo and M. A. Abdul-Ghani, US Endocrinology, 2010, 6, 42–47 CrossRef.
- M. V. B. Reddy, H. Y. Hung, P. C. Kuo, G. J. Huang, Y. Y. Chan, S. C. Huang, S. J. Wu, S. L. Morris-Natschke, K. H. Lee and T. S. Wu, Bioorg. Med. Chem. Lett., 2017, 27, 1547–1550 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The atomic coordinates, DFT-based single-point energy, thermodynamics and solvation corrections of all optimized compounds studied. See DOI: 10.1039/c8ra03735c |
| This journal is © The Royal Society of Chemistry 2018 |