
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-specific anchoring aptamer C2NP on DNA origami nanostructures for cancer treatment†
Pengchao Sun‡
abc,
Nan Zhang‡abd,
Yafang Tangab,
Yanan Yangab,
Jie Zhou*ab and
Yongxing Zhao *abd
*abd
aSchool of Pharmaceutical Science, Zhengzhou University, Zhengzhou, Henan 450001, P. R China. E-mail: zhaoyx@zzu.edu.cn; Fax: +86 0371 67739546; Tel: +86 0371 67739165
bKey Laboratory of Targeting Therapy and Diagnosis for Critical Diseases, Henan Province, Zhengzhou, Henan 450001, P. R China
cInstitute for Biological Interfaces 1, Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, Germany
dKey Laboratory of Advanced Pharmaceutical Technology, Ministry of Education of China, Zhengzhou, Henan 450001, P. R China
First published on 23rd July 2018
Abstract
Because of the remarkable features, including biocompatibility and biodegradability, DNA origami nanostructures have drawn much attention as ideal carriers for drug delivery. However, the cellular uptake of DNA origami nanostructures was a passive targeting process, resulting in limited therapeutic effect. To address this problem, we anchored the aptamer C2NP (Apt) on rectangular DNA origami nanostructures (RE) to enhance the tumor targeting properties and anticancer effects of doxorubicin (DOX). Apt was anchored onto RE with low or high density (RE-4Apt, RE-16Apt), followed by incubation with DOX to obtain DOX@RE-4Apt and DOX@RE-16Apt. The results showed that DOX@RE-4Apt and DOX@RE-16Apt exhibited excellent biocompatibility and targeting ability, as well as a synergic biological effect with chemotherapy on cancer therapy. More importantly, after conjugation with RE, the bioactivity of Apt was significantly increased. These results revealed that Apt anchored DNA nanostructures not only are potential carriers for precise therapy, but also supply a strategy to enhance the bioactivity of aptamers.
1. Introduction
Recently, DNA origami nanostructures have witnessed a rapid development since the concept was first proposed by Seeman.1 With a versatile and “one-pot” method, Rothemund made another contribution to DNA nanotechnology. In his work, different two-dimensional shapes of DNA nanostructures, such as rectangles and pentagrams, have been constructed through rational design.2 The scaffold DNA (long single stranded DNA) can be easily folded into a nanoscale by hundreds of staples (short single stranded DNA). This simple single-step method makes fantastic nanomaterials via DNA origami.Up to now, proteins, peptides, and nanoparticles have been site-specifically anchored on DNA origami based on non-covalent or covalent bonds, demonstrating that DNA origami nanostructures exhibit a powerful role in microarrays.3–7,22 Furthermore, DNA nanostructures as a type of biomaterial show natural properties, including superb stability, excellent biocompatibility, and biodegradability. These remarkable features have made the DNA nanostructure an ideal carrier for cell culture study and in vivo study.8 For example, Liedl's group used DNA origami nanostructures as carriers for CpG oligonucleotides to study immunostimulation in mammalian cells.9 In Mikkila's study, DNA origami nanostructures were coated with virus capsid proteins by electrostatic interaction for enhancing intracellular delivery.10 Church's group designed a hollow DNA nanorobot with a smart gate keeper system to deliver antibodies for tissue culture.11
Instead of such complex nanorobots, DNA base-pairs based simple nanostructures are seen as alternative carriers for doxorubicin (DOX) since DOX could insert into the DNA double helix by hydrogen bonds between the amino group of DOX and the cytosine of DNA.12–14 Recently, several groups have used DNA origami nanostructures to deliver DOX for anticancer therapy.15–17 However, since they lack ligands, DNA origami nanostructures showed a passive tumor targeting process, the result of which was that little DOX was effectively delivered to the therapeutic target sites. To address this problem, a possible solution is to modify DNA nanostructures with some ligands which can specifically recognize cancer cell markers.18
C2NP (Apt), a novel DNA aptamer, was selected by systematic evolution of ligands by exponential enrichment (SELEX) and exhibited high affinity to the CD30 cell marker.19 Apt has been utilized as a probe for tumor imaging, with high sensitivity and a long-lasting signal.20 After binding to CD30 receptors, the downstream signalling will be activated, inducing the apoptosis of T-cell lymphoma cells.21 Based on the fact that Apt is a DNA aptamer with 31 nucleotides in length, Apt could easily be conjugated to DNA origami nanostructures by hybridization method or designing a staple strand which contains the Apt sequence.23 However, up to now, little attention has been paid on Apt modified DNA origami nanostructures for targeting delivery and gene therapy.
In our present work, Apt mediated tumor targeted and gene therapy systems were developed. Apt was anchored onto DNA origami nanostructures with different densities and the anti-cancer drug DOX was loaded for chemotherapy. A human anaplastic large cell lymphoma cell line (K299) which overexpress CD30 receptors, was chosen for the targeting study. Additionally, the mechanism and bioactivity of Apt and Apt-RE were explored. Our results showed that the DNA nanocarriers constructed in this work exhibited chemotherapy of DOX and biotherapy of Apt concurrently. The bioactivity of Apt was significantly increased after conjugation to DNA nanostructures.
2. Materials and methods
2.1. Materials
M13mp18 was purchased from New England Biolabs. K299 cells (a human anaplastic large cell lymphoma cell line) were obtained from CoBioer (Nanjing, China). Single stranded DNA was synthesized by BIONEER (Daejeon, Korea). Agarose was supplied by Biowest (Nuaillé, France). Fetal bovine serum (FBS) was purchased from BI (Kibbutz, Israel). All other agents and chemicals were obtained from Solarbio (Beijing, China). All these commercial chemicals and agents were used without any modification.2.2. Preparation of DNA origami nanostructures
Rectangular DNA origami nanostructures (RE) were assembled following Rothmund's method. Briefly, scaffold strands (M13mp18) and stable strands were mixed in water at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 in a PCR tube. Then folding buffer (final concentration: Tris base 10 mM, EDTA 5 mM, magnesium acetate 12.5 mM) was introduced into the system. Folding was carried out by cooling the system down from 95 °C to 20 °C slowly (0.016 °C s−1) and maintaining at 20 °C for another 20 s in a PCR Thermocycler (SENSO, Germany). To prepare 4 or 16 Apt conjugated DNA origami nanostructures (RE-4Apt and RE-16Apt), the pre-targeting DNA origami nanostructures were firstly synthesized. Basically, stable strands in the designated sites were replaced with capture single stranded DNA (Fig. S1†). After follow the same procedure, the pre-targeting DNA origami nanostructures was obtained, followed by incubation with Apt overnight at room temperature. Biotin labelled Apt was used to obtain RE-4Apt@Biotin and RE-16Apt@Biotin for atomic force microscopy (AFM) analysis.
:10 in a PCR tube. Then folding buffer (final concentration: Tris base 10 mM, EDTA 5 mM, magnesium acetate 12.5 mM) was introduced into the system. Folding was carried out by cooling the system down from 95 °C to 20 °C slowly (0.016 °C s−1) and maintaining at 20 °C for another 20 s in a PCR Thermocycler (SENSO, Germany). To prepare 4 or 16 Apt conjugated DNA origami nanostructures (RE-4Apt and RE-16Apt), the pre-targeting DNA origami nanostructures were firstly synthesized. Basically, stable strands in the designated sites were replaced with capture single stranded DNA (Fig. S1†). After follow the same procedure, the pre-targeting DNA origami nanostructures was obtained, followed by incubation with Apt overnight at room temperature. Biotin labelled Apt was used to obtain RE-4Apt@Biotin and RE-16Apt@Biotin for atomic force microscopy (AFM) analysis.
2.3. Characterization of DNA nanostructures
RE, RE-4Apt, and RE-16Apt were characterized by agarose (1%, m V−1) gel electrophoresis at 100 V for 60 min in an ice bath. In the preparation of the agarose gel, 2 μL of ethidium bromide (10 μg mL−1) was added for gel imaging (BIORAD, USA). Further characterization was carried out by AFM (Bruck, Germany). RE-4Apt@Biotin and RE-16Apt@Biotin were first purified by Microcon® Centrifugal filters (Merck, Germany), and the purified RE-4Apt@Biotin and RE-16Apt@Biotin were introduced onto two micas. After 5 min, streptavidin was dropped and incubated with RE-4Apt@Biotin and RE-16Apt@Biotin for several minutes, respectively.21 The micas were washed with 1× TAE buffer several times, followed by observation with AFM under liquid model.2.4. Drug loading
Doxorubicin (DOX) was loaded into DNA origami nanostructures by incubating DOX·HCl solution with DNA origami nanostructures in the dark at room temperature.14 To obtain the maximum DOX-loading, the amount of DOX initially added and the incubation time were optimized, respectively. In brief, various amounts of DOX (20 μL; 2000, 1000, 500, 100, 50, 25, 10, 5 μM) were incubated with DNA origami nanostructures (20 μL; 3.3 nM) for 6 h. Then the mixtures were centrifuged down at 12000 rpm for 10 min, followed by quantification using a microplate reader (BioTek, USA; λex = 485 nm, λem = 591 nm). The incubation time was also varied from 1 h to 36 h to obtain the maximum DOX loading, and the loading capacity was measured by the same method.The drug-loading capacity of DOX@RE was calculated by the differential measurement of the supernatant concentration, and the equation is as follow:
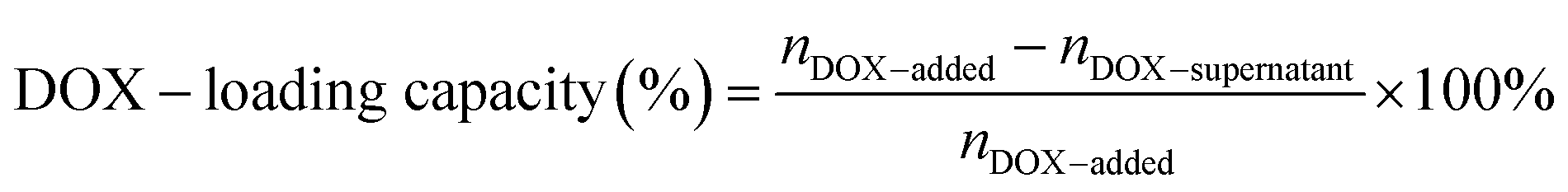
2.5. Stability evaluation
The stability of both RE and DOX were evaluated by incubating DOX@RE with cell culture medium in the dark at 37 °C. Samples were collected at different time points (1, 5, 8, 12, 24, 48, 72 h) and analyzed by agarose gel electrophoresis to study the stability of RE. The relative fluorescence intensity was determined using microplate reader at set time points (0, 0.5, 1, 3, 8, 15, 24, 48, 72 h).2.6. Drug release
Slide-A-Lyzer™ MINI Dialysis Devices (Thermo Fisher, Germany) were used to study the release of DOX from DOX@RE and DOX@RE-Apt in PBS (pH 5.0 and 7.4) at 37 °C. Firstly, DOX@RE and DOX@RE-Apt were separately suspended in folding buffer. Then suspensions (20 μL) of each were added to a dialysis device. At set time points (0.5, 1, 2, 4, 8, 12, 24, 48 h), three dialysis tubes of each sample were taken out and the DOX remaining inside the tube was quantified. The cumulative release was calculated by the following equation: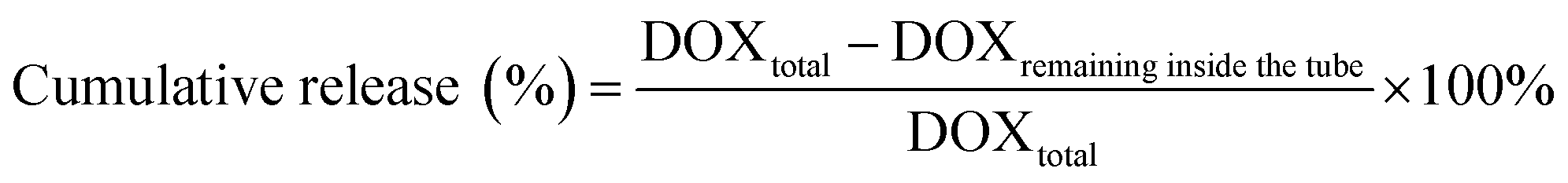
2.7. Cell culture
K299 cells were cultivated with RPMI Medium 1640 containing 5% FBS under standard conditions (37 °C, 5% CO2). Only cells in the exponential phase were used for the following experiments.2.8. In vitro anticancer activity
Cytotoxicity of free DOX, DOX@RE, DOX@RE-4Apt and DOX@RE-16Apt were tested against the K299 cell line by a standard CCK8 assay. Typically, K229 cells were seeded in 96 well plates (1×104 cells per well) and cultured under standard cell culture conditions. After 12 h, DOX, DOX@RE, DOX@RE-4Apt, or DOX@RE-16Apt (DOX final concentration: 10, 5, 1, 0.5, 0.1 μM; n = 5) was added and further cultivated for 24, 48, or 72 h, respectively. At the end of the experiment, CCK8 was added following the manufacturer's instructions for the cell counting kit. Cell culture medium was used as a blank control and cells cultivated with normal cell culture medium were applied as a positive group. The inhibition rate was calculated by the following equation: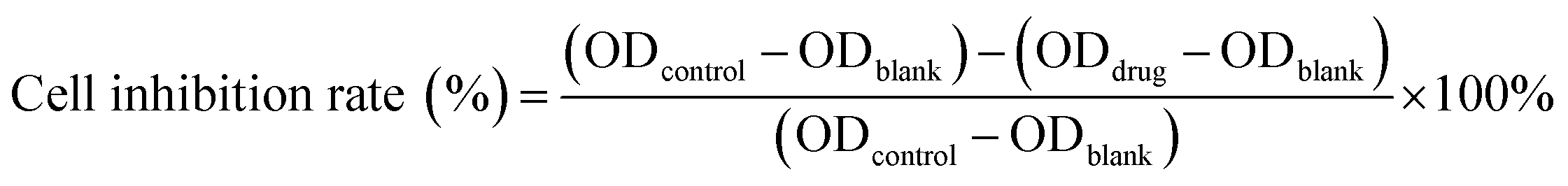
2.9. In vitro bioactivity of Apt
To evaluate the bioactivity of Apt, various amounts of free Apt, RE-4Apt, and RE-16Apt (Apt final concentration: 16, 32, 64, 128 nM) were incubated with K299 cells for 72 h. The inhibition rate was determined by the same method. With respect to chemotherapy and biotherapy of drug loaded systems, different concentrations of DOX@RE, DOX@RE-4Apt, and DOX@RE-16Apt were treated with K299 for 48 h, using free DOX as a positive control. Proliferation was analyzed by CCK8.2.10. Confocal laser scanning microscopy (CLSM)
5 × 105 of K299 cells per well were seeded in 24-well plates overnight before being treated with DOX, DOX@RE, DOX@RE-4Apt, and DOX@RE-16Apt (final concentration: RE: 4 nM, DOX: 2 μM). After incubation for 2 h, the uptake process was stopped by the addition of cold PBS to the plate. The cells were further washed 3 times with PBS to remove the redundant DOX or drug loaded systems. Subsequently, the cells were fixed with 4% paraformaldehyde. After a wash with PBS, the cells were suspended in PBS and observed by CLSM (Nikon A1, Japan).2.11. Cellular uptake
The cells were seeded in 24-well plates at a density of 5 × 105 cells per well and incubated overnight at standard cell culture conditions. DOX, DOX@RE, DOX@RE-4Apt, and DOX@RE-16Apt (final concentration: RE: 4 nM, DOX: 2 μM) were added and incubated for 2 h. Then, cellular uptake was stopped by introducing 1 mL of cold PBS to each well. The cells were washed with PBS for 3 times prior to flow cytometric analysis (BD FACSCanto II, USA).2.12. Cell apoptosis and cell cycle
Cell apoptosis was evaluated by a double staining technology with an annexin V-FITC and propidium iodide (PI) kit. Typically, cells (2 × 105 cells per mL, 2 mL per well) were seeded in 24-well plates and incubated at standard cell culture conditions overnight. 1 mL of upper cell culture medium was carefully removed and replaced by 1 mL of fresh cell culture medium containing DOX, DOX@RE, DOX@RE-4Apt, DOX@RE-16Apt, Apt, RE, RE-4Apt, or RE-16Apt (final concentration: RE: 4 nM, DOX: 2 μM). The cells were collected and washed thrice with PBS after incubation for 24 h. After that, the cells were suspended in 100 μL of binding buffer followed by treatment with FITC-Annexin V (5 μL, 5 min) and PI (5 μL, 10 min) in turn. Another 200 μL of binding buffer was added to dilute the cell mixtures before flow cytometric analysis.With respect to cell cycle analysis, cells were collected and fixed with 70% of ethanol at 4 °C overnight. Following the washing step (PBS 3×), the cells were treated with 100 μL of Rnase A at 37 °C for 30 min. Then, 200 μL of PI solution was introduced to stain the cells for another 30 min. The cells were analyzed by flow cytometry.
2.13. Reactive oxygen species (ROS)
ROS in tumor cells were analyzed by a reactive oxygen species assay kit. Briefly, cells were seeded in a 24-well plate and treated with DOX, DOX@RE, DOX@RE-4Apt, DOX@RE-16Apt, Apt, RE, RE-4Apt, or RE-16Apt (final concentration: RE: 4 nM, DOX: 2 μM). After incubation for 24 h, the cells were collected and washed 3 times with PBS. Subsequently, the cells were suspended in 500 μL of cell culture medium (without FBS) containing DCFH-DA at a concentration of 10 μM. For the positive control group, an additional 10 μL of Rosup was added. The cells were incubated under standard conditions for 25 min and washed several times with cell culture medium (without FBS) and PBS before flow cytometric analysis.2.14. Statistical analysis
All data was expressed as the mean ± SD. The results of flow cytometric analysis were performed by Flow Jo software. All experiments in this study were conducted at least in triplicate.3. Results and discussion
3.1. Construction and characterization of DNA origami nanostructures
Aptamer C2NP anchored DNA origami nanostructures were designed and constructed in this work (Scheme 1). The list of DNA sequences used in this paper are shown in Table S1.† With the help of staples, M13mp18 was folded into a rectangle shape by Rothmund's method.2 The edged staples were removed to avoid coupling between the DNA origami nanostructures which was caused by the Ω–Ω accumulation effect of the DNA bases.19 The different densities of Apt were precisely anchored onto the top of the DNA origami nanostructures by the hybridization method with V-shaped structures (RE-4Apt, RE-16Apt).20 On one hand, Apt anchored RE could help DOX loaded carriers get into cells effectively, enhancing the anticancer activity of DOX. On the other hand, because Apt could induce cell apoptosis, DOX loaded and Apt anchored RE can exhibit synergic biological effects with chemotherapy on anticancer therapy.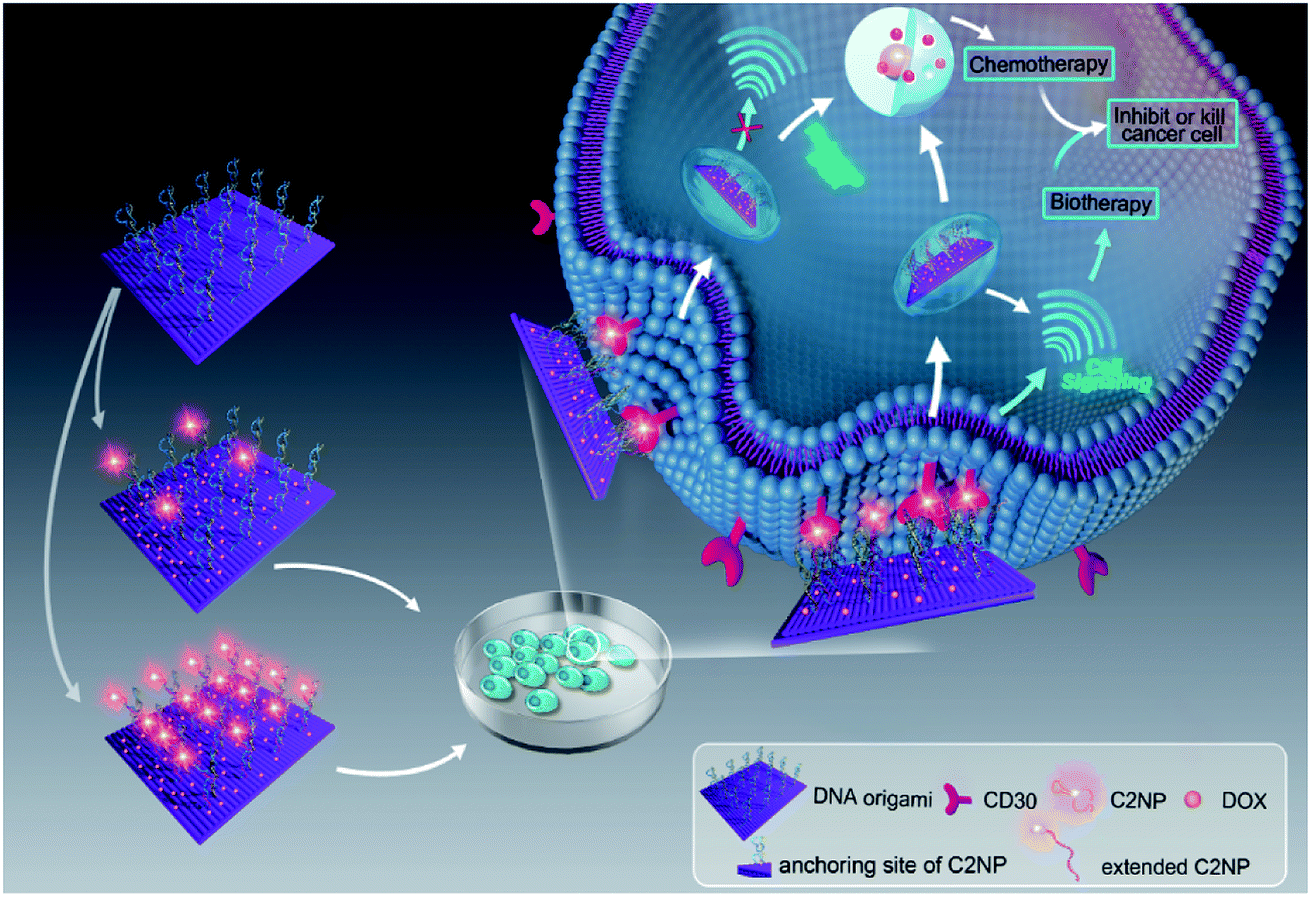 | ||
| Scheme 1 Schematic illustration of targeting DNA origami nanostructures for biotherapy and chemotherapy. Aptamer C2NP were anchored on the top of DNA nanostructures with different densities (RE-4Apt, RE-16Apt). On one hand, aptamer C2NP can help DOX loaded DNA origami nanostructures (DOX@RE-4Apt, DOX@RE-16Apt) get into cells effectively, enhancing the anticancer activity of DOX. On the other hand, C2NP can induce cell apoptosis, exhibiting synergic biological effect with chemotherapy on cancer therapy. | ||
The correct folding of DNA origami nanostructures was characterized by agarose gel electrophoresis and AFM. As shown in Fig. 1A, lane 3 shows a bright band between 6000 bp and 8000 bp which was fitted to the designated size of RE. Electrophoretic mobility of RE-4Apt was similar with RE, but much faster than RE-16Apt, indicating the successful conjugation of Apt.
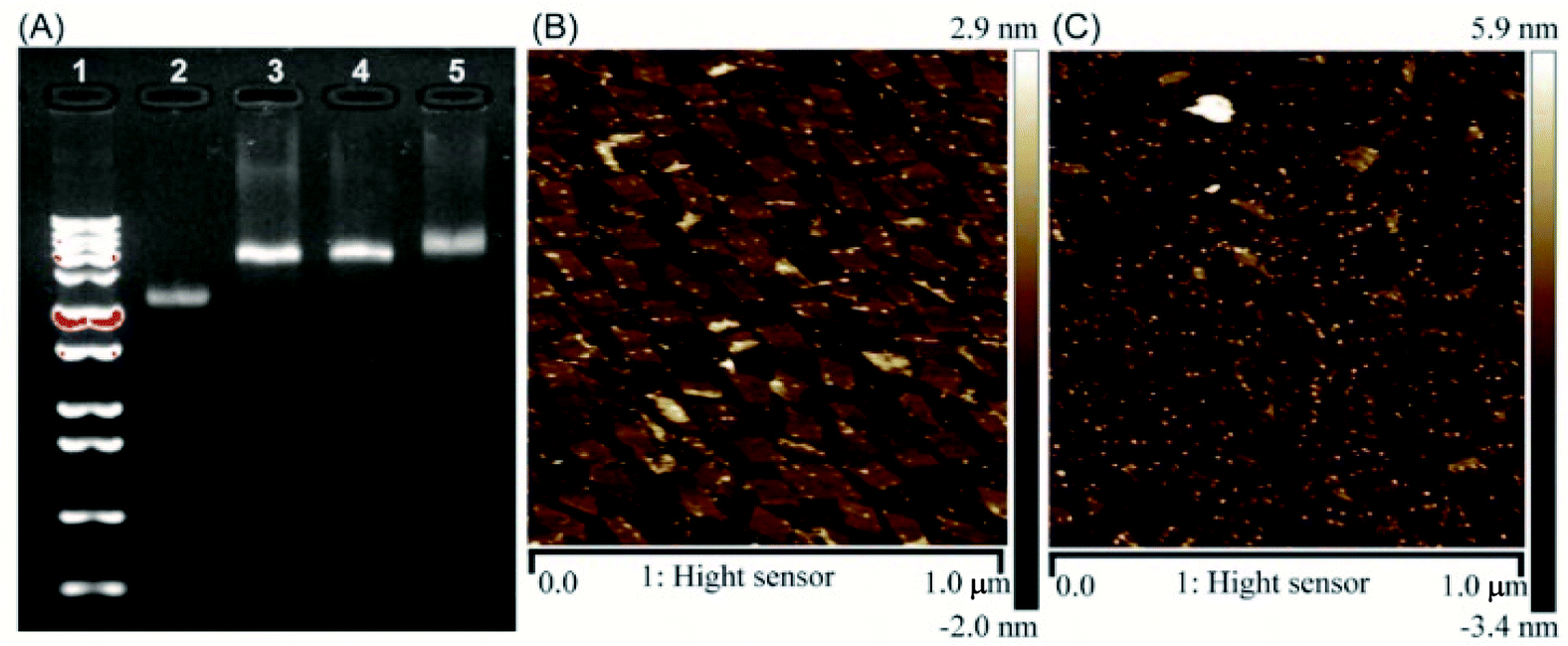 | ||
| Fig. 1 Characterization of DNA origami nanostructures. (A) Gel electrophoretic analysis of DNA origami nanostructures ((1) 1 kbp plus marker; (2) single M13mp18; (3) RE; (4) RE-4Apt; 5.RE-16Apt). (B) AFM image of RE-4Apt. (C) AFM image of RE-16Apt. | ||
The high affinity of biotin–streptavidin coupling was used to make the aptamer conjugated DNA origami nanostructures be easily observed by AFM. Fig. 1B and C exhibit details about the morphology of the DNA origami nanostructures (size: 90 nm × 60 nm). Because of their flexible properties, the aptamer could be wrapped into RE or pushed onto the other side of RE, making a less conjugated aptamer visible. Insufficient interactions between biotin and streptavidin was another reason for the fewer numbers.
3.2. Drug loading, stability and release
DOX could be intercalated into the DNA duplex with the help of hydrogen bonds between the amino group of DOX and the cytosine of DNA.13 Based on this feature, DNA nanostructures could be used as carriers to deliver DOX to living cells in vitro or in vivo. With respect to RE, DOX was loaded into RE by incubating them in the dark at room temperature. Maximum drug loading was obtained when 50 μM of DOX was incubated with 3.3 nM of RE (one DOX molecule per base pair).With respect to drug efficacy, the ability to release the drugs from their carriers is an important property. Drug burst release, usually caused by the structural damage of carriers, would shorten the half-life of drugs and show local or systemic toxicity.21,24 To avoid DOX burst release caused by damage to the DNA origami nanostructure, the stability of DOX@RE was evaluated in cell culture medium before further study. As shown in Fig. 2A, DOX@RE maintained its structure at least 72 h in a cell culture medium containing 5% FBS. At the same time, the relative stability of DOX was significantly increased (Fig. 2B). DOX could easily diffuse into the surrounding environment which exhibited a lesser DOX concentration.17 Compared with free DOX, DOX release from DOX@RE or DOX@RE-Apt showed a controlled and slower release property at different pH values. The release properties of DOX from RE or RE-Apt showed no differences at pH 7.0, while much more DOX defused into the surroundings at pH 5.5 than at pH 7.4 at the same time point (Fig. 2C), exhibiting that the acid conditions slightly accelerate the release of DOX. This is probably because acid might accelerate the degradation of the DNA origami nanostructures or weaken the hydrogen bonds between DOX and the DNA strands. These results showed that DNA origami based DOX delivery system was valuable for further anticancer study.
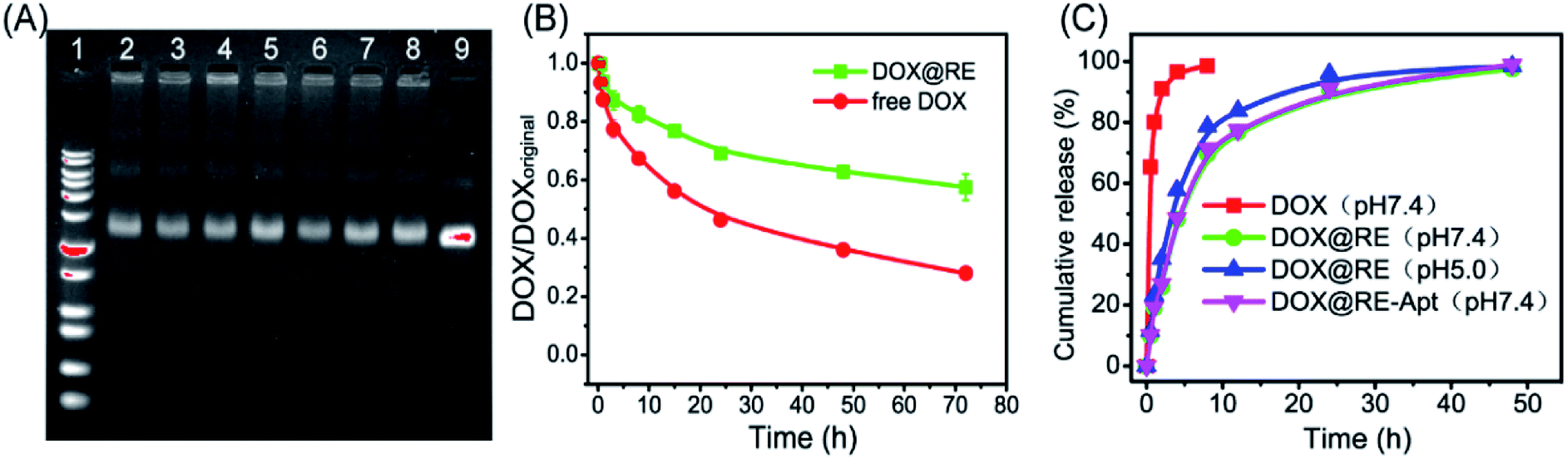 | ||
| Fig. 2 (A) Gel electrophoretic analysis of DOX-loaded DNA origami Nanostructures after incubation with cell culture medium at different time ((1) 1 kb plus marker; (2) 1 h; (3) 5 h; (4) 8 h; (5) 12 h; (6) 24 h; (7) 48 h; (8) 72 h; (9) contrast). (B) The relative stability of DOX in cell culture medium. (C) The release properties of DOX from DOX@RE or DOX@RE-Apt at pH 5.0 or 7.4. | ||
3.3. In vitro anticancer activity
In vitro anticancer activity of DOX@RE, DOX@RE-4Apt, and DOX@RE-16Apt were evaluated on K299 cells. To determine the cytotoxicity of the buffer and DNA materials, the folding buffer and RE were incubated with K299 cells, respectively. According to Table S2,† both folding buffer and RE had no significant effects on the cell inhibition rate at the concentration used in the following studies.Fig. 3 shows the cell inhibition rates of DOX, DOX@RE, DOX@RE-4Apt, and DOX@RE-16Apt. Each group showed a greater inhibition rate with a greater DOX concentration and longer incubation time. As shown in Fig. 2B, the relative stability of DOX was increased after being loaded into DNA origami nanostructure. DOX loaded into RE, RE-4Apt, and RE-16Apt revealed much greater cytotoxicity than free DOX at the same concentration and at the same incubation time. Compared with DOX@RE, DOX@RE-4Apt and DOX@RE-16Apt exhibited much stronger anticancer effects in vitro. Furthermore, the inhibition rate of DOX@RE-16Apt was much greater than DOX@RE-4Apt.
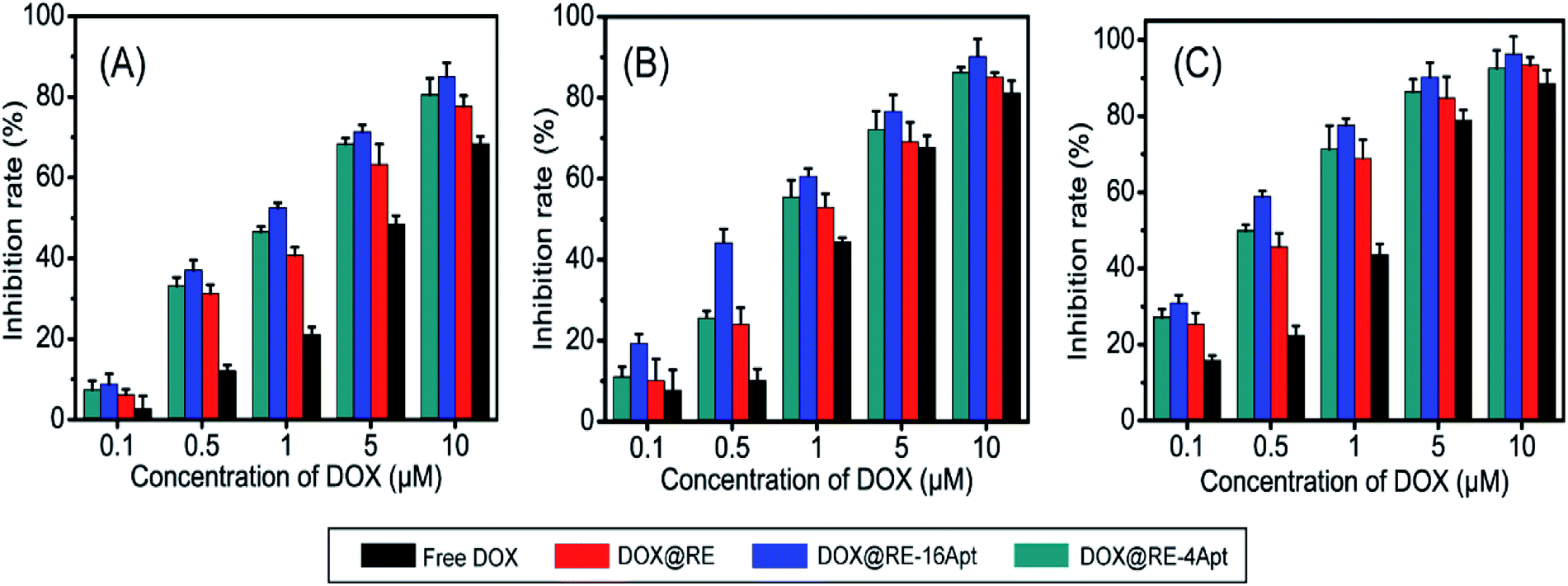 | ||
| Fig. 3 In vitro K299 cell inhibition analysis of free DOX, DOX@RE, DOX@RE-4Apt and DOX@RE-16Apt against at (A) 24 h, (B) 48 h, (C) 72 h (n = 5). | ||
The results demonstrated that aptamer conjugated RE enhanced the anticancer activity of DOX. The anticancer activity was further increased when additional Apt was modified onto DNA origami nanostructure. This is probably because DOX@RE-16Apt recognized cells easily, and was taken up effectively by activating more receptors simultaneously.25
Apt was selected by SELEX as a ligand to CD30. The activated CD30 would regulate the downstream signal pathway, resulting in cancer cell apoptosis. It was also reported that after incubation with serum at 37 °C for 24 h, Apt still maintained its ability to bind CD30.19 However, biotherapy of aptamer usually occurred after 72 h of treatment with cells at a high concentration.26 To evaluate if DNA origami carriers effected the bioactivity of Apt, K299 cells were treated with different concentrations of Apt, RE-4Apt, RE-16Apt. The results in Fig. 4A show that the bioactivities of Apt were enhanced with increasing concentration. At the same concentration, RE-4Apt and RE-16Apt resulted in an increase of inhibition rate, indicating that RE-4Apt and RE-16Apt enhanced the anticancer activity of Apt. The possible reason was that the stability of aptamer was increased in these two carriers.27 At the same concentration, more Apt maintain their activity in RE-4Apt and RE-16Apt groups. Compared with RE-4Apt and RE-16Apt, DOX@RE-4Apt and DOX@RE-16Apt further increased the inhibition rate, indicating that these carriers achieved chemotherapy and biotherapy simultaneously (Fig. 4B–D).
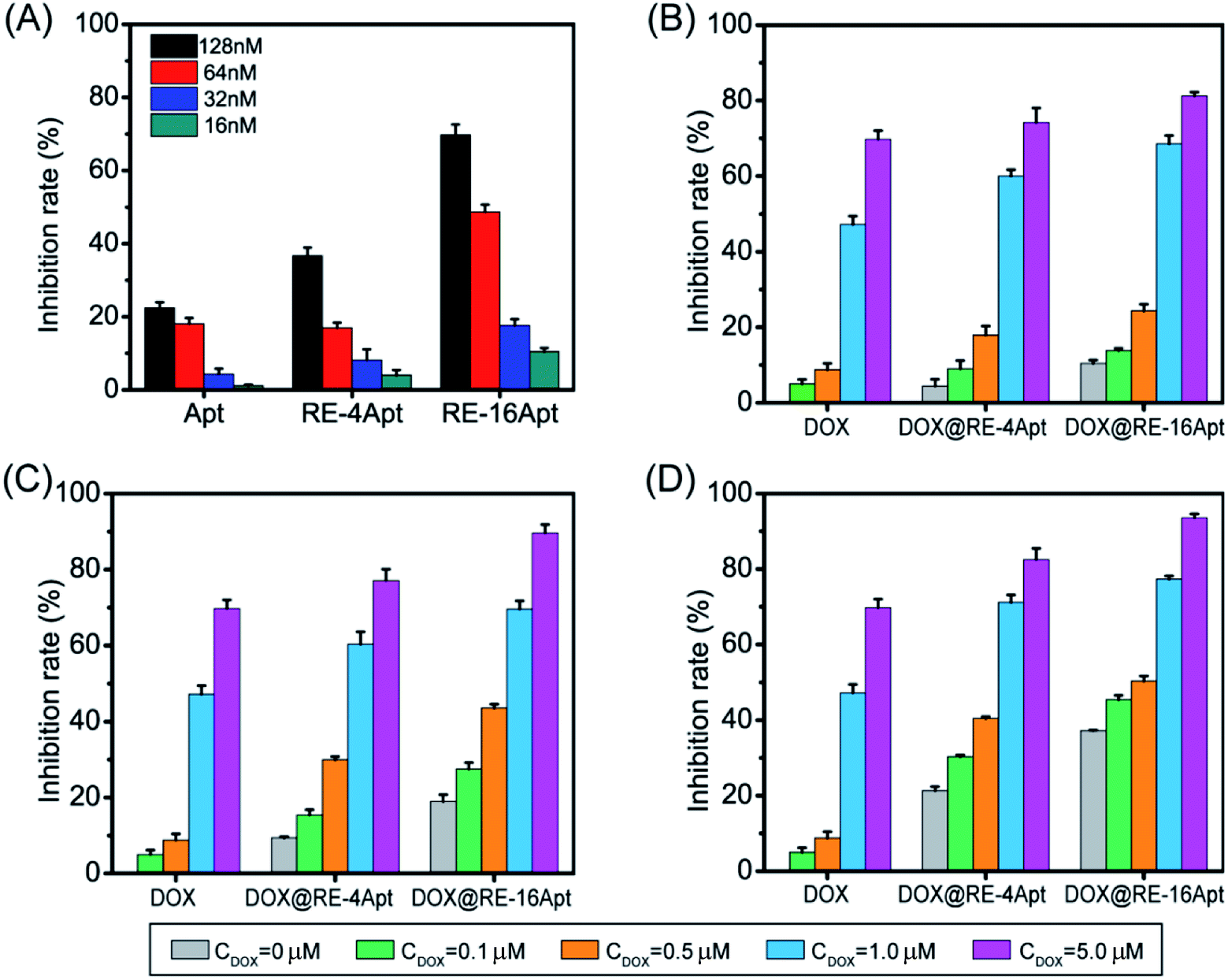 | ||
| Fig. 4 (A) Bioactivity of Apt, RE-4Apt, and RE-16Apt at different concentration on K299 cells. Synergetic bioactivity and chemotherapy of DOX@RE-4Apt and DOX@RE-16Apt which loaded with different amount of DOX on K299 cells with the final concentration of Apt 16 nM (B), 32 nM (C), 64 nM (D). (n = 5) (note: free DOX at different concentration was used as positive control). | ||
3.4. Cellular uptake
CLSM was performed to explore the cellular uptake of DOX@RE, DOX@RE-4Apt and DOX@RE-16Apt. As shown in Fig. 5A, the DOX signal in cells was detected after incubation with DOX@RE, DOX@RE-4Apt, and DOX@RE-16Apt for 4 h, indicating that DOX was successfully delivered into cells. Compared with the free DOX group, the DOX@RE group showed a brighter red fluorescence, probably due to the increased stability of DOX loaded into the RE carriers. In other words, at the same concentration of DOX, much more DOX could be effectively delivered into cells in the DOX@RE group. Besides, the signal of DOX was much stronger in the DOX@RE-4Apt group and the strongest in the DOX@RE-16Apt group. With the help of Apt, more DOX@RE-4Apt and DOX@RE-16Apt entered into cells by CD30 mediated endocytosis,28 especially in DOX@RE-16Apt which contained a high density of Apt. These results were further proved by flow cytometric analysis (Fig. 5B). The DOX level in the DOX@RE-16Apt group was the greatest (56.84%) while still much greater in the DOX@RE-4Apt (30.23%) and the DOX@RE (26.84%) groups, demonstrating that Apt promoted the cellular uptake of DOX, and that a high density of Apt exhibited an advantage in this process.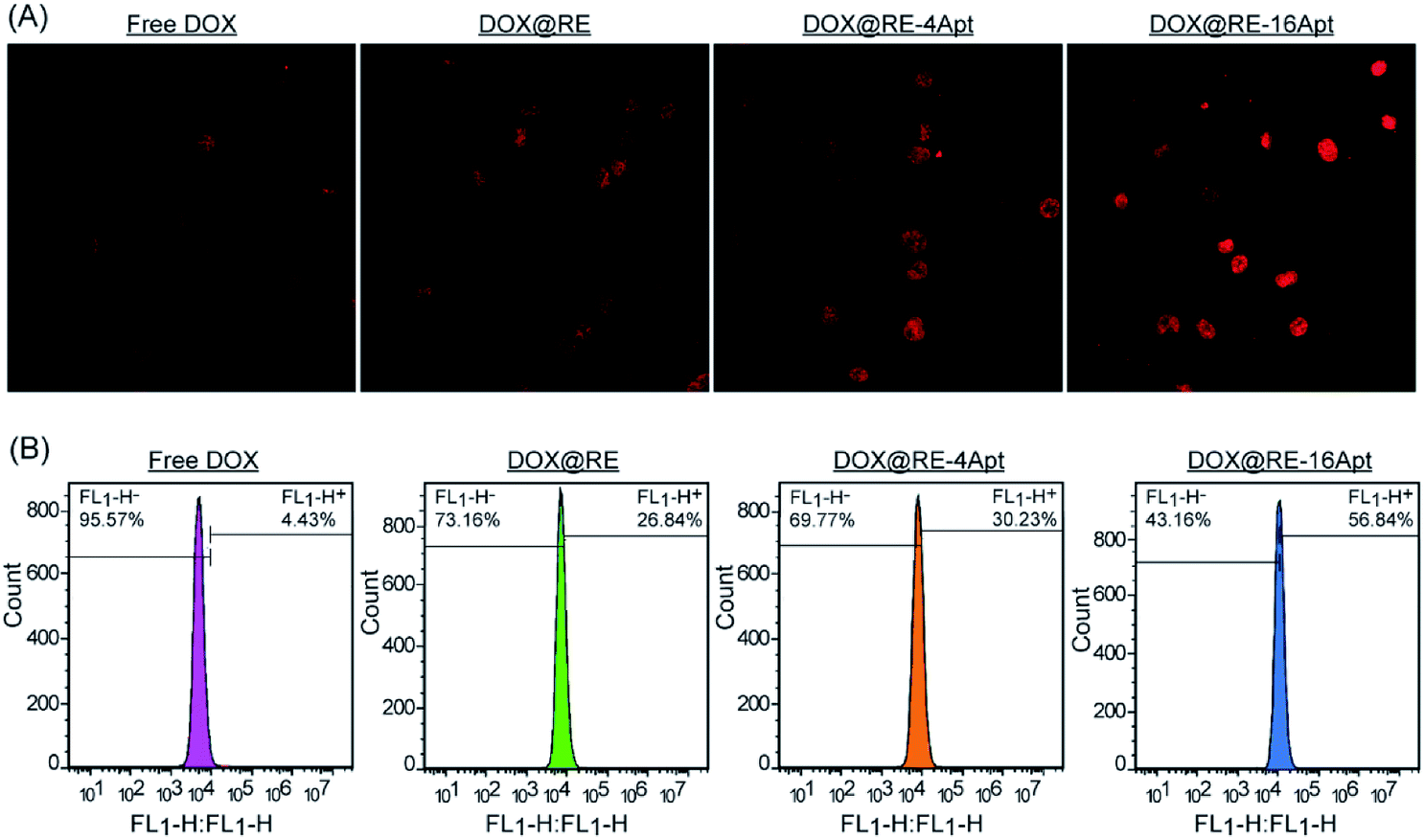 | ||
| Fig. 5 (A) CLSM images of K299 cells after incubation with free DOX, DOX@RE, DOX@RE-4Apt, and DOX@RE-16Apt for 2 h. (B) Flow cytometry histograms of intracellular DOX uptake after incubation with free DOX, DOX@RE, DOX@RE-4Apt and DOX@RE-16Apt for 2 h. | ||
3.5. Mechanism of cell inhibition
It is generally known that the anticancer activity of DOX is achieved by inhibiting topoisomerase II and resulting in DNA damage.29 Previous research has shown that DOX effected cell apoptosis, cell cycle and the intracellular ROS level. With respect to Apt, publications indicated that Apt activated CD30, resulting in cell apoptosis. However, more details are still unclear. In this paper, cell apoptosis, cell cycle, and ROS were analysed to understand the possible cell inhibition mechanism of Apt and whether the nanocarriers constructed in this study could enhance the activity of DOX or not. According to Fig. 6A, Apt, RE-4Apt and RE-16Apt enhanced early apoptosis. With the help of DOX, cell apoptosis was significantly increased in the DOX@RE, DOX@RE-4Apt and DOX@RE-16Apt group. Cell cycle results (Fig. 6B) showed that Apt increased cell arrest in the G2 phase slightly while DOX loaded RE systems enhanced the G2 phase arrest significantly.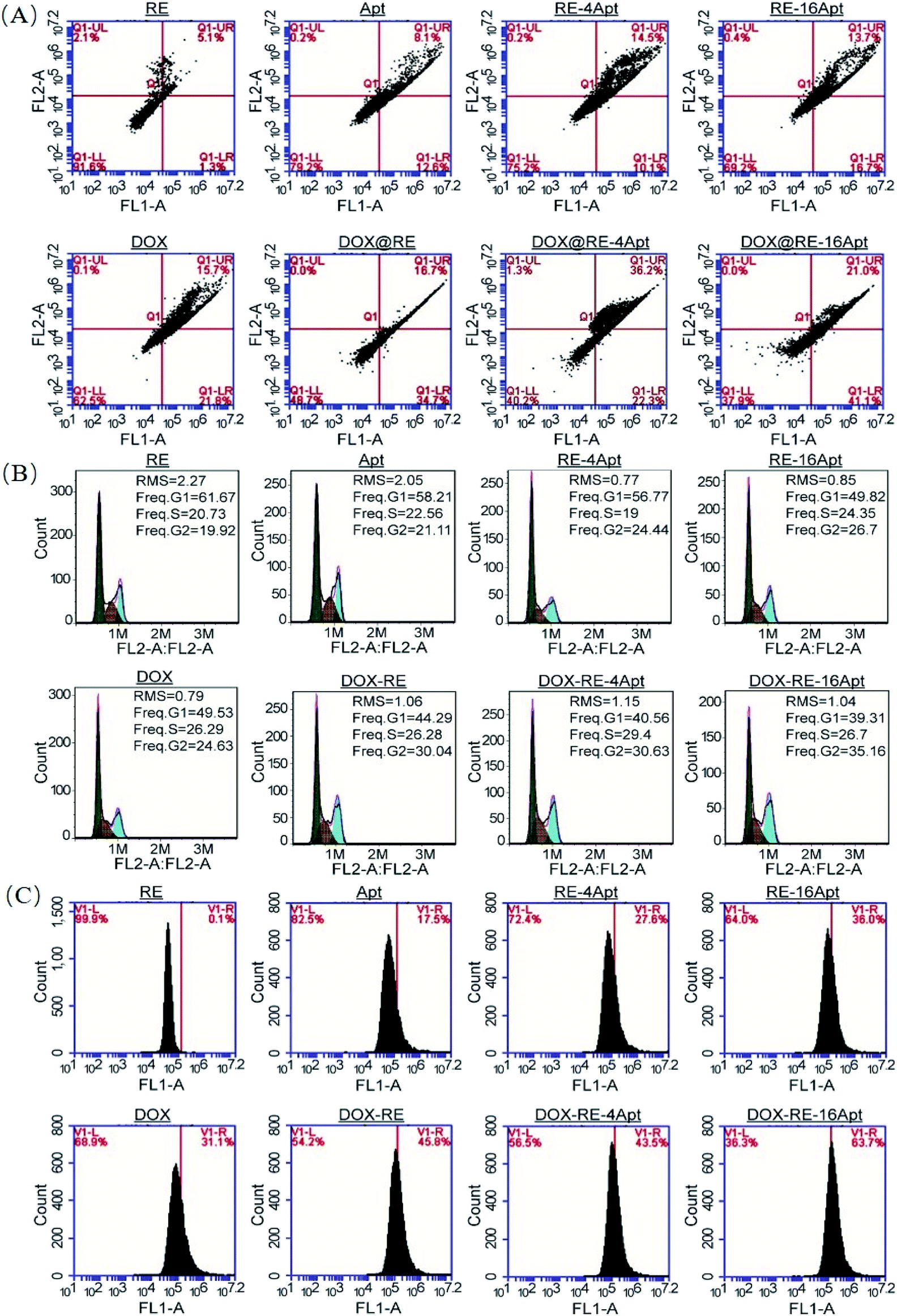 | ||
| Fig. 6 The impacts of free DOX and different DNA origami carriers on cell apoptosis (A) cell cycle (B) and RSO content (C) after incubation for 24 h. | ||
ROS play an important role in both normal bioactivities and abnormal pathological processes. On one hand, increased ROS stress could enhance cell proliferation. On the other hand, high levels of ROS would cause DNA damage and result in cell apoptosis.30 As shown in Fig. 6C, the intracellular level of ROS was increased after treatment with DOX, which agreed with the report that DOX could induce ROS generation in cancer cells.31 Interestingly, Apt also showed the ability to cause great levels of ROS, while RE alone had no significant influence on ROS. Compared with Apt, the RE-4Apt and RE-16Apt groups exhibited merits in increasing intracellular ROS. ROS was further upregulated in the DOX@RE, DOX@RE-4Apt and DOX@RE-16Apt groups. Fig. 6 indicates that Apt exhibited anticancer activity by enhancing early apoptosis and upregulating ROS levels. With the help of anticancer drug, DOX, cell apoptosis, cell cycle arrest and ROS were enhanced, revealing that DNA nanocarriers constructed in this study were more efficacious because of the synergistic effect of DOX and Apt.
4. Conclusion
For the first time aptamer C2NP was modified onto DNA origami nanostructures for a targeting drug delivery system. Apt enhanced the anticancer activity of DOX loaded DNA origami delivery systems, especially in the high density and closely positioned system (DOX@RE-16Apt). Besides, high concentrations of Apt induced additional cell apoptosis, cycle arrest in the G2 phase and high levels of ROS. After conjugation with DNA nanostructures, the bioactivity of Apt was significantly increased. What's more, DNA nanocarriers constructed in this work exhibited chemotherapy of DOX and biotherapy of Apt simultaneously. This work confirms that the DNA nanostructure is a promising carrier, not only for drugs, but also for aptamers. The controllable site immobilization of ligands and high drug loading capacity make DNA origami nanostructures a powerful carrier for drug delivery and a novel approach for the precise treatment against tumors. Apt anchored DNA nanostructures also supply a strategy to enhance the bioactivity of Apt itself.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC No. 81573011 and 81611130076).References
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef PubMed.
- N. V. Voigt, T. Torring, A. Rotaru, M. F. Jacobsen, J. B. Ravnsbaek, R. Subramani, W. Mamdouh, J. Kjems, A. Mokhir, F. Besenbacher and K. V. Gothelf, Nat. Nanotechnol., 2010, 5, 200–203 CrossRef PubMed.
- R. Chhabra, J. Sharma, Y. Ke, Y. Liu, S. Rinker, S. Lindsay and H. Yan, J. Am. Chem. Soc., 2007, 129, 10304–10305 CrossRef PubMed.
- B. A. Williams, K. Lund, Y. Liu, H. Yan and J. C. Chaput, Angew. Chem., Int. Ed. Engl., 2007, 46, 3051–3054 CrossRef PubMed.
- B. Ding, Z. Deng, H. Yan, S. Cabrini, R. N. Zuckermann and J. Bokor, J. Am. Chem. Soc., 2010, 132, 3248–3249 CrossRef PubMed.
- Y. Zhang, J. Chao, H. Liu, F. Wang, S. Su, B. Liu, L. Zhang, J. Shi, L. Wang, W. Huang, L. Wang and C. Fan, Angew. Chem., Int. Ed. Engl., 2016, 55, 8036–8040 CrossRef PubMed.
- J. Chao, H. Liu, S. Su, L. Wang, W. Huang and C. Fan, Small, 2014, 10, 4626–4635 CrossRef PubMed.
- V. J. Schuller, S. Heidegger, N. Sandholzer, P. C. Nickels, N. A. Suhartha, S. Endres, C. Bourquin and T. Liedl, ACS Nano, 2011, 5, 9696–9702 CrossRef PubMed.
- J. Mikkila, A. P. Eskelinen, E. H. Niemela, V. Linko, M. J. Frilander, P. Torma and M. A. Kostiainen, Nano Lett., 2014, 14, 2196–2200 CrossRef PubMed.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef PubMed.
- V. Bagalkot, O. C. Farokhzad, R. Langer and S. Jon, Angew. Chem., Int. Ed., 2006, 45, 8149–8152 CrossRef PubMed.
- D. Agudelo, P. Bourassa, G. Berube and H. A. Tajmir-Riahi, Int. J. Biol. Macromol., 2014, 66, 144–150 CrossRef PubMed.
- P. C. Sun, N. Zhang, Y. F. Tang, Y. N. Yang, X. Chu and Y. X. Zhao, Int. J. Nanomed., 2017, 12, 2657–2672 CrossRef PubMed.
- Q. Jiang, C. Song, J. Nangreave, X. W. Liu, L. Lin, D. L. Qiu, Z. G. Wang, G. Z. Zou, X. J. Liang, H. Yan and B. Q. Ding, J. Am. Chem. Soc., 2012, 134, 13396–13403 CrossRef PubMed.
- Q. Zhang, Q. Jiang, N. Li, L. R. Dai, Q. Liu, L. L. Song, J. Y. Wang, Y. Q. Li, J. Tian, B. Q. Ding and Y. Du, ACS Nano, 2014, 8, 6633–6643 CrossRef PubMed.
- Y. X. Zhao, A. Shaw, X. H. Zeng, E. Benson, A. M. Nystrom and B. Hogberg, ACS Nano, 2012, 6, 8684–8691 CrossRef PubMed.
- J. Li, H. Pei, B. Zhu, L. Liang, M. Wei, Y. He, N. Chen, D. Li, Q. Huang and C. Fan, ACS Nano, 2011, 5, 8783–8789 CrossRef PubMed.
- P. Parekh, S. Kamble, N. X. Zhao, B. P. Portier and Y. L. Zu, Biomaterials, 2013, 34, 8909–8917 CrossRef PubMed.
- Z. H. Zeng, P. Parekh, Z. Li, Z. Z. Shi, C. H. Tung and Y. L. Zu, Theranostics, 2014, 4, 945–952 CrossRef PubMed.
- V. J. O'Sullivan, I. Barrette-Ng, E. Hommema, G. T. Hermanson, M. Schofield, S. C. Wu, C. Honetschlaeger, K. K. S. Ng and S. L. Wong, PLoS One, 2012, 7, e35203 CrossRef PubMed.
- J. Yang, Z. C. Song, S. Liu, Q. Zhang and C. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 22451–22456 CrossRef PubMed.
- Y. Ke, S. Lindsay, Y. Chang, Y. Liu and H. Yan, Science, 2008, 319, 180–183 CrossRef PubMed.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef PubMed.
- A. Angelin, S. Weigel, R. Garrecht, R. Meyer, J. Bauer, R. K. Kumar, M. Hirtz and C. M. Niemeyer, Angew. Chem., Int. Ed., 2015, 54, 15813–15817 CrossRef PubMed.
- H. Kaur, J. J. Li, B. H. Bay and L. Y. L. Yung, PLoS One, 2013, 8, 11 Search PubMed.
- P. Charoenphol and H. Bermudez, Mol. Pharm., 2014, 11, 1721–1725 CrossRef PubMed.
- M. K. Kennedy, C. R. Willis and R. J. Armitage, Immunology, 2006, 118, 143–152 CrossRef PubMed.
- G. Capranico, F. Zunino, K. W. Kohn and Y. Pommier, Biochemistry, 1990, 29, 562–569 CrossRef PubMed.
- H. Pelicano, D. Carney and P. Huang, Drug Resist. Updates, 2004, 7, 97–110 CrossRef PubMed.
- S. W. Wang, E. A. Konorev, S. Kotamraju, J. Joseph, S. Kalivendi and B. Kalyanaraman, J. Biol. Chem., 2004, 279, 25535–25543 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04589e |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2018 |