
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoporous silica–carbon composites fabricated by a universal strategy of hydrothermal carbonization: controllable synthesis and applications†
Xitong Sun‡
a,
Wei Yu‡a,
Jingyu Yana,
Jiaqi Liab,
Gaowa Jina,
Jiatao Fenga,
Zhimou Guo *a and
Xinmiao Liang*a
*a and
Xinmiao Liang*a
aKey Lab of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, Liaoning 116023, China. E-mail: guozhimou@dicp.ac.cn; liangxm@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100149, China
First published on 31st July 2018
Abstract
Mesoporous silica–carbon composite materials, with homogeneous and thickness-controllable carbon coating, were synthesized by using a universal strategy of hydrothermal carbonization, and the carbon layer could be coated on the surface of ordered and disordered mesoporous silica. The electrostatic interaction between amino-modified silica and hydrothermal carbon was regarded as the main driving force for the formation of homogeneous carbon coverage on the silica surface. The obtained composites showed high graphitization degree, and controlled morphology (shape and particle size) and pore size by adjusting the species of carriers and hydrothermal conditions. The application results demonstrated that a thin carbon layer possessed high adsorption capacities for dyes, and the composite could be rapidly recovered by sedimentation (10 min) after adsorption with 30 μm spherical silica gel as the carrier. Besides, baseline chromatographic separation of oligosaccharide isomers could be achieved on the silica–carbon column. These results indicated that the silica–carbon composites should be promising functional materials for the large-molecule-involving processes such as adsorption and chromatographic separation.
1. Introduction
Carbon-based materials, ranging from activated carbon and one-dimensional carbon nanotubes to two-dimensional graphene, are recognized as one of the most important functional materials, which have been widely used in many crucial fields such as catalysis, separation and energy storage.1 Up to now, various methods have been developed to prepare carbon-based materials, including pyrolysis of carbon precursors, chemical vapor deposition, template methods, and hydrothermal carbonization (HTC).2 Among them, HTC is considered as a “green” and effective method to produce carbon materials, with special features of cheap and abundant precursors such as carbohydrates and biomass, water as the medium, mild processing conditions (below 200 °C), rich oxygen-containing groups on the surface and combination with other components.3 However, one limitation of the HTC materials is that they possess limited surface area and porosity. A widely used method to enhance these properties is thermal treatment under an inert atmosphere, where small organic molecules are removed to generate pores.4 In this case, thermal treated HTC materials mainly introduce microporous domains, and these micropores are not suitable for large-molecule-involving processes. Besides, the present studies of hydrothermal carbon are mainly focused on carbon spheres with small size (below 10 μm),5 and carbon particles with different shape and large size are still difficult to be synthesized based on the HTC method. Accordingly, the hydrothermal carbon materials (HC) have poor controllability in morphology and pore channel properties.Mesoporous carbon materials (MCs), including ordered and disordered mesoporous carbon materials (OMCs), have aroused great interest among researchers because of their outstanding properties, including high surface areas, large and tunable pore channels, exceptional thermal and chemical stability, and shown great potentials in catalysis, separation, electrodes and in particular for the large-molecule-involved processes.6 The template methods involving soft and hard templates proved to be the most effective method for the synthesis of mesoporous carbon with well mesostructures and narrow pore size contribution. Zhao et al. and Dai et al. directly synthesized OMCs using soft template strategy, but the reaction conditions and material structures could not be well controlled by this method because of the limitation of few soft templates.7 In the other method of hard template strategy, silica gel with controlled pore structure was prepared first, carbon precursors were subsequently filled into the template pores through wet impregnation or chemical vapor deposition and then carbonized, and the MCs were finally obtained by the removal of silica template. It is obviously that the reaction conditions and MC structures are easily controlled using hard template strategy due to the mature synthetic technique of mesoporous silica materials. Ryoo et al. prepared a series (CMK series) of OMCs using SBA and MCM series mesoporous silica as the hard templates.6a However, the requirement of multiple synthetic steps brings inconvenience to the large scale production. Hyeon et al. synthesized OMC via the carbonization of mesostructured silica/surfactant nanocomposites using P123 triblock copolymer as both structure-directing agent and carbon precursor.8 The advantage of this method was no need of external carbon precursor adding, but the main problems were that the pore size was difficult to be controlled, and the SiO2 template was also needed to be etched. Therefore, it is still a challenge to prepare the mesoporous carbon-based materials with controlled morphology and pore size by a simple method.
Considering the utilization of carbon-based materials is the surface properties of the inner and outer surfaces, the homogeneous coating of carbon layer on the mesoporous silica carriers would be a good choice to prepare novel carbon-based materials. Recently, Qin et al. directly prepared mesoporous silica–carbon composite nanoparticles by employing MCM-41 as the carrier and cetyltrimethyl ammonium bromide (CTAB) as the structure-directing agent and carbon precursor,9 which showed excellent glycan enrichment ability from complex biological samples. However, the pore size (or the thickness of carbon layer) of the composite could not be controlled because of the specific amount of template, and the used method was limited to the ordered mesoporous silica, which was not applicable to disordered mesoporous silica because of no usage of organic template in the preparation process.10 In contrast, some disordered mesoporous silica such as spherical silica gel exhibited great mechanical strength, and was wildly used as the stationary phase for liquid chromatography.10 In this work, mesoporous silica–carbon composite materials, were prepared by using a universal HTC strategy, with the controllability of morphology and pore size, and the carbon layer could be coated on the surface of ordered and disordered mesoporous silica. To the best of our knowledge, there is no study on the preparation of mesoporous silica–carbon composites by using this strategy, and the carbon-coated disordered spherical silica gel was first reported. These materials were then successfully applied to the pollutant adsorption and the oligosaccharide isomer separation.
2. Experimental section
2.1 Chemicals
Poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) triblock copolymer Pluronic P123 (EO20PO70EO20, Mw = 5800) was purchased from Sigma-Aldich. Spherical silica gels (SiO2, 5 μm and 30 μm) were purchased from Fuji Silysia Chemical. Activated carbon (JH-767) was purchased from Shanghai Jinhu Activated Carbon Co., Ltd. 3-Aminopropyl trimethoxysilane (APTMS) was supplied by Gelest Inc. Glucose, tetraethyl orthosilicate (TEOS), hydrochloric acid (HCl) and toluene were the products of Sinopharm Chemical Reagent Co., Ltd and were used as obtained. Oligosaccharides including lacto-N-fucopentaose I (LNFP-I), lacto-N-fucopentaose II (LNFP-II), lacto-N-difucohexaose I (LNDFH-I) and lacto-N-neodifucohexaose II (LNnDFH-II) were obtained by purification from human milk in our laboratory.2.2 Preparation of amino-modified SBA-15 (SBA-15-NH2) and SiO2 (SiO2-NH2)
SBA-15 silica was synthesized according to the previous report.11 Briefly, 4 g of P123 was dissolved in 30 g of water and 120 g of 2 mol L−1 HCl solution with vigorously stirring. The mixture was then heated up to 40 °C and 8.5 g of TEOS was added, and the reaction system was kept stirring at 40 °C for 24 h. Subsequently, the resulting suspension was maintained at 100 °C for another 24 h under static conditions in a Teflon-lined autoclave. The solid product was filtered, and washed successively with water and methanol for several times, and dried at 80 °C for 12 h. To remove the surfactant, the as-synthesized product was finally calcined in air for 6 h at 550 °C and the SBA-15 silica was obtained. The modification of SBA-15 or SiO2 was carried out as follows: 10 g of SBA-15 or SiO2 were dried at 120 °C for 5 h, and then added into the 110 mL dry toluene containing 20 mL of APTMS. The mixture was kept stirring at 110 °C for 24 h under reflux and nitrogen atmosphere. The resulting product was washed successively with toluene, dichloromethane, methanol, water and methanol, and the SBA-15-NH2 and SiO2-NH2 were obtained after drying.2.3 Preparation of carbon-coated SBA-15 (SBA-15-C) and SiO2 (SiO2-C)
The silica carriers (SBA-15-NH2 or SiO2-NH2) and glucose with the proportions of 1 : 0.1–1 : 7 were dispersed into water (50 times volume of silica carriers), and then placed in a Teflon-lined autoclave and kept at a temperature of 160–220 °C between 2 and 8 h. The solid product was collected by filtration and washed with water and methanol, and dried in a drying oven at 80 °C. The as-prepared products were subsequently carbonized at the temperature of 600–800 °C for 3 h in argon atmosphere to obtain the final product.2.4 Characterization
The morphology of samples was characterized by SEM (JEOL JSM-2100, JSM-7800F, Japan), TEM (JEOL JEM-2100, Japan) and optical microscope (Keyence VHX-5000, Japan). The Raman spectra were recorded on a Bruker Senterra spectrometer (Germany). The SAXS patterns were recorded on an Antonparr Saxes M2 scattering system (Austria). The unit-cell parameters were calculated from the equation of a0 = 2d100/√3. The XPS measurement was conducted on a Thermo Fisher ESCALAB 250Xi spectrometer (USA) using Al Kα radiation (15 kV, 10.8 mA). The thermogravimetric analysis (TGA, Netzsch STA 449 F3) was performed from 40 to 900 °C with a heating rate of 10 °C min−1 under air atmosphere. The nitrogen adsorption/desorption isotherms were measured on a QuantaChrome Quadrasorb SI analyzer (USA) after vacuum degassing at 120 °C for 6 h. The specific surface areas and the pore volumes were calculated using the Brunauer–Emmett–Teller (BET) method. The pore size distributions were obtained from the adsorption branches of the isotherms using Barrett–Joyner–Halenda (BJH) model. The microporous specific surface areas and pore volumes were analyzed using t-plot method. The elemental analysis (C, N and H) of samples was carried out using an Elementar Vario EL III analyzer (Germany).2.5 Applications
Dye adsorption experiments were carried out by the batch method, an amount of 1 mg of adsorbent was transferred into 1 mL of a solution of 400 mg L−1 MC or RhB in 5 mL centrifuge tubes, and then the tubes were placed in a mixer at 1000 rpm and room temperature for 24 h. After adsorption, the solution was centrifuged at 5000 rpm for 5 min, and the dye concentration was determined using Thermo Fisher Scientific 1510 Multiskan spectrum (USA), and the wavenumbers of the detection were set as 665 and 554 nm for MC and RhB, respectively. The sedimentation experiments were performed by adding 1 g of adsorbent into 100 mL of water, the concentrations of adsorbent were analyzed using Persee TU-1810 Ultraviolet-visible (UV-vis) spectrophotometer (China), with the detection wavenumber of 650 nm. In order to prepare the HPLC column, 2.0 g of SiO2-C (5 μm) composite was slurry-packed into a stainless steel column (150 mm × 4.6 mm I.D.) under a pressure of 40 MPa, with 40 mL of methanol as slurry solvent and 80 mL of methanol as propulsion solvent. The separation of oligosaccharide isomers was evaluated on an Alliance HPLC system equipped with a Waters 2695 HPLC pump and a Waters 2489 UV-vis detector. The separation conditions were optimized as follows: flow rate, 1 mL min−1; mobile phase A, ACN; mobile phase B, H2O; gradient: 1–15 min, 5% A, 95% B → 30% A, 70% B; UV detection, 195 nm.3. Results and discussion
3.1 Synthetic strategy of silica–carbon composite materials
Mesoporous silica–carbon composite materials, were designed and synthesized by using a universal strategy of hydrothermal carbonization, with the advantages of controlled morphology (shape or size) and pore size. The synthesis route is shown in Scheme 1. First, the ordered and disordered mesoporous silica carriers were modified with amino groups via a silylation reaction. Following the hydrothermal treatment process of aqueous glucose and mesoporous silica, the pH value of the medium changed to acid (pH 3–4) because of the deprotonation of carboxyl groups on the polysaccharide intermediates and the acidic by-products such as formic acid and acetic acid.12 The amino-modified carriers were positively charged due to the protonation of amino groups at this pH region. In these circumstances, there should be a strong electrostatic interaction between carriers and polysaccharide intermediates, resulting in the homogeneous surface coverage of hydrothermal carbon layer. In contrast, because of the low isoelectric point of mesoporous silica,12a the mesoporous silica was negatively charged and exhibited a strong repulsion to polysaccharide intermediates, which leaded to the formation of hydrothermal carbon particles besides silica. The SBA-15 severed as a model to estimate the role of amino modification in the formation of hydrothermal carbon layer during the hydrothermal treatment. As shown in Fig. S1,† the SBA-15-NH2 after hydrothermal treatment and calcination (with no special description, the preparation of samples was carried out as follows: hydrothermal treatment temperature, 190 °C; hydrothermal treatment time, 5 h; the mass ratio of carriers to glucose, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3; calcination temperature, 800 °C and calcination time, 3 h) showed decreasing trend of pore diameter (from 7.74 nm to 5.49 nm), while the pure silica carriers had no significant change, indicating that the amino modification played an important role in the coating of homogeneous carbon layer. Finally, through calcination at high temperature, the heteroatom content of materials was reduced and the graphitization degree of materials was enhanced.
:3; calcination temperature, 800 °C and calcination time, 3 h) showed decreasing trend of pore diameter (from 7.74 nm to 5.49 nm), while the pure silica carriers had no significant change, indicating that the amino modification played an important role in the coating of homogeneous carbon layer. Finally, through calcination at high temperature, the heteroatom content of materials was reduced and the graphitization degree of materials was enhanced.
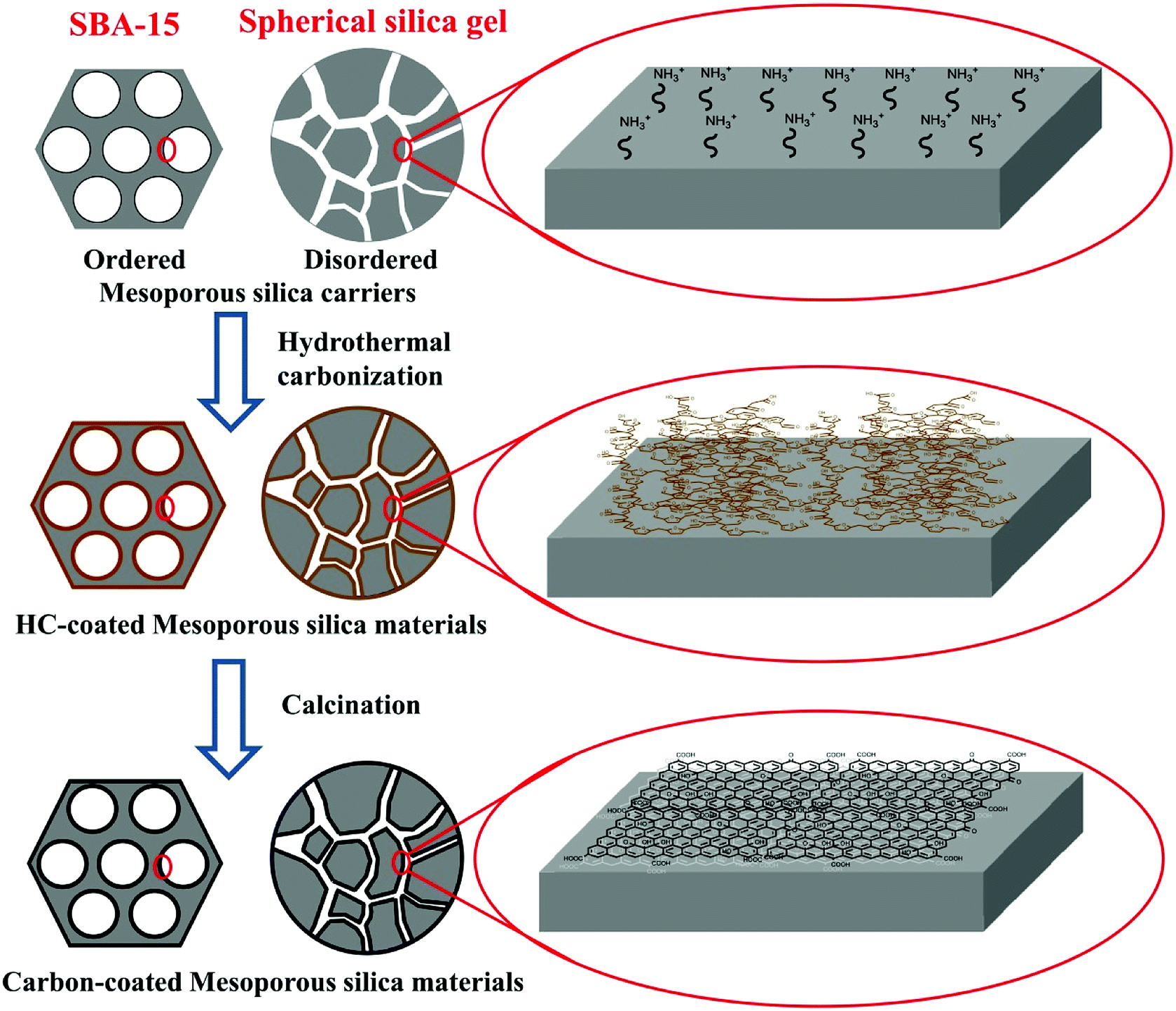 | ||
| Scheme 1 Schematic representation of the synthesis of the mesoporous silica–carbon composite materials. | ||
3.2 The properties of silica–carbon composite materials
The Raman spectra of SBA-15 and SBA-15-C and C1s core level spectrum of SBA-15-C are shown in Fig. 1. The Raman signals ranging from 1000 cm−1 to 1900 cm−1 is typical of carbonized materials.13 It was clear that the SBA-15 had no signals in this range. In contrast, there were two distinguishable peaks at 1330 cm−1 and 1600 cm−1 in the spectrum of SBA-15-C, which were corresponding to the D and G bands of carbonized materials, respectively.13 This result indicated that the carbon layer was successfully coated on the surface of SBA-15 by the hydrothermal method. In addition, the D band was related to the edges, defects and structural disorders in amorphous and quasi-crystalline forms of carbon materials, and the G band was associated with the first-order scattering of E2g mode in the basal plane of the crystalline graphite.14 The intensity ratio of two bands (ID/IG) provided a useful index for evaluating the graphitization degree of carbon materials. The ID/IG of SBA-15-C was estimated to be 0.90, implying that the SBA-15 had a relatively high graphitization degree according to the values in the previous literature.14 The X-ray photoelectron spectroscopy (XPS) was also used to characterize the graphitization degree and heteroatom content of SBA-15-C. As shown in Fig. 1(B), it could be seen that there were four peaks appeared at 284.6, 285.7, 287.2 and 289.0 eV, which were assigned to the sp2 bonded carbon in the SBA-15-C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and COOR, respectively.15 The sp2 bonded carbon could reach a value of 71.7%, which were in agreement with the result of the Raman characterization. The O/C atomic ratio was calculated to be 0.12, which was much lower than that of the hydrothermal carbon spheres under similar preparation conditions (0.387, as reported in the ref. 16). This result demonstrated that the SBA-15-C had a low heteroatom content and high graphitization degree due to the sufficient calcining treatment.
O and COOR, respectively.15 The sp2 bonded carbon could reach a value of 71.7%, which were in agreement with the result of the Raman characterization. The O/C atomic ratio was calculated to be 0.12, which was much lower than that of the hydrothermal carbon spheres under similar preparation conditions (0.387, as reported in the ref. 16). This result demonstrated that the SBA-15-C had a low heteroatom content and high graphitization degree due to the sufficient calcining treatment.
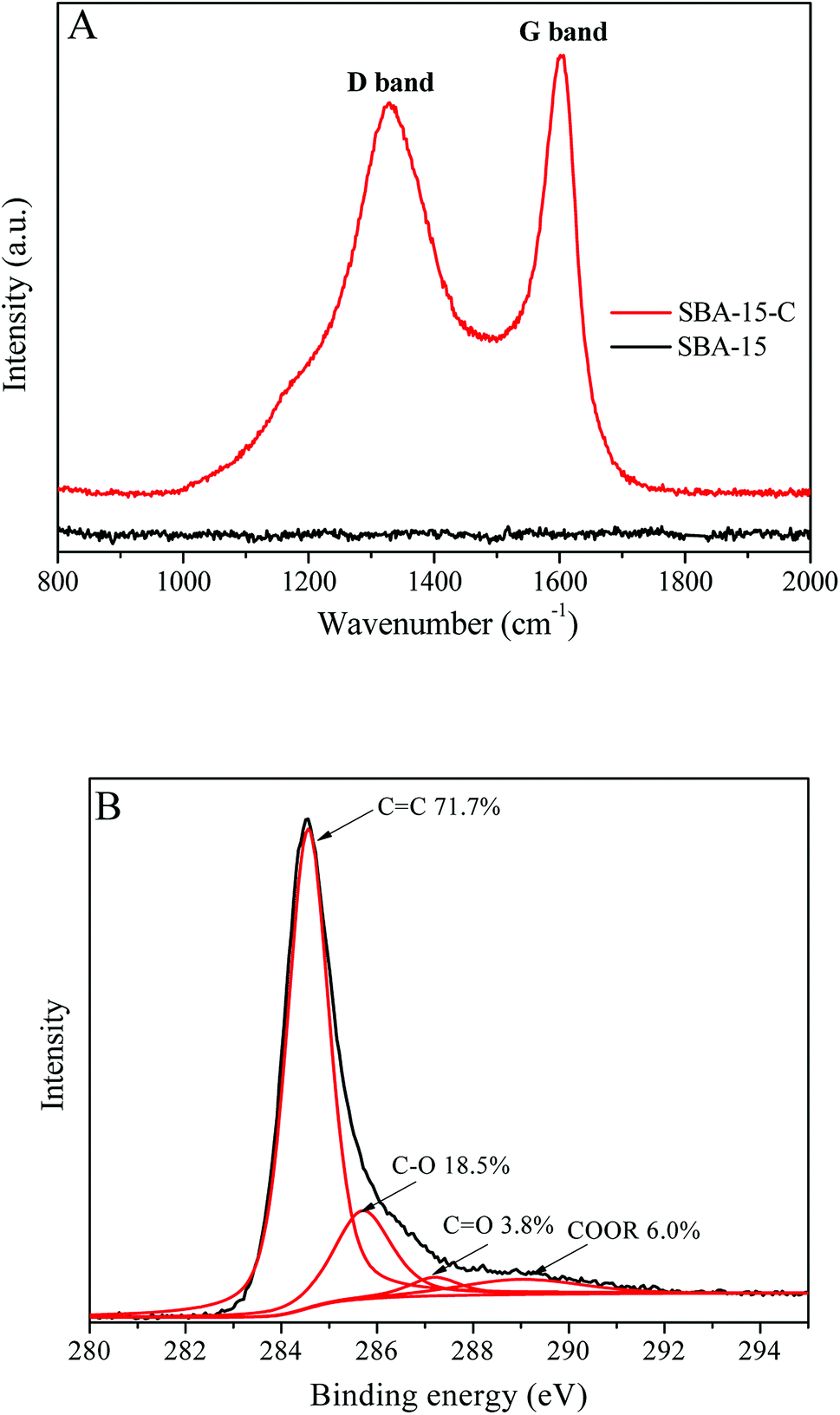 | ||
| Fig. 1 (A) Raman spectra of SBA-15 and SBA-15-C; (B) C1s core level spectrum of SBA-15-C. | ||
The morphology of mesoporous silica before and after the coating of carbon layer was investigated. Fig. 2(A and B) shows the scanning electron microscope (SEM) images of SBA-15 and SBA-15-C. Both of the materials exhibited typical rod-like structure, and there was no obviously change in the context of particle size. The spherical silica gels with particle size of 5 and 30 μm were also employed to estimate the morphology change of SiO2-C. As shown in Fig. 2(C–F), the shape and particle size of SiO2-C still maintained good. The color of carbon-coated SBA-15 and SiO2 changed from gray to black under optical microscope (Fig. S2†), indicating the successful coating of carbon layer. These results demonstrated that the carbon layer could be coated on the surface of ordered and disordered mesoporous silica by hydrothermal carbonization, no matter what the shape and particle size of the mesoporous silica were. In other words, the morphology of carbon layer was conveniently controlled by the regulation of carrier species, and showed better controllability compared with other carbon materials such as activated carbon and hydrothermal carbon spheres. The controlled morphology of carbon-based materials is of great value to practical applications, for example, microspheres can be used as adsorbents, chromatographic packings and catalyst carriers. The transmission electron microscope (TEM) images of SBA-15, HC-coated SBA-15 (SBA-15-HC) and SBA-15-C were also provided in Fig. 3(A–C). The SBA-15-HC and SBA-15-C possessed well-defined ordered mesoporous structure and uniform shape like SBA-15. Next, the SBA-15-C was washed with 1 M NaOH solution (50 vol% ethanol-50 vol% H2O) twice at 100 °C to remove the silica wall.17 The resulting carbon layer materials still exhibited good ordered mesoporous structure (Fig. 3(D)), and had high specific surface area and pore volume (Table S1†), suggesting that the carbon layer should be homogeneously coated on the mesoporous surfaces.
 | ||
| Fig. 2 SEM images of SBA-15 (A), SBA-15-C (B), SiO2 (5 μm, C), SiO2-C (5 μm, D), SiO2 (30 μm, E), and SiO2-C (30 μm, F), and the scale bars of (A and B) and (C–F) were 1 and 10 μm, respectively. | ||
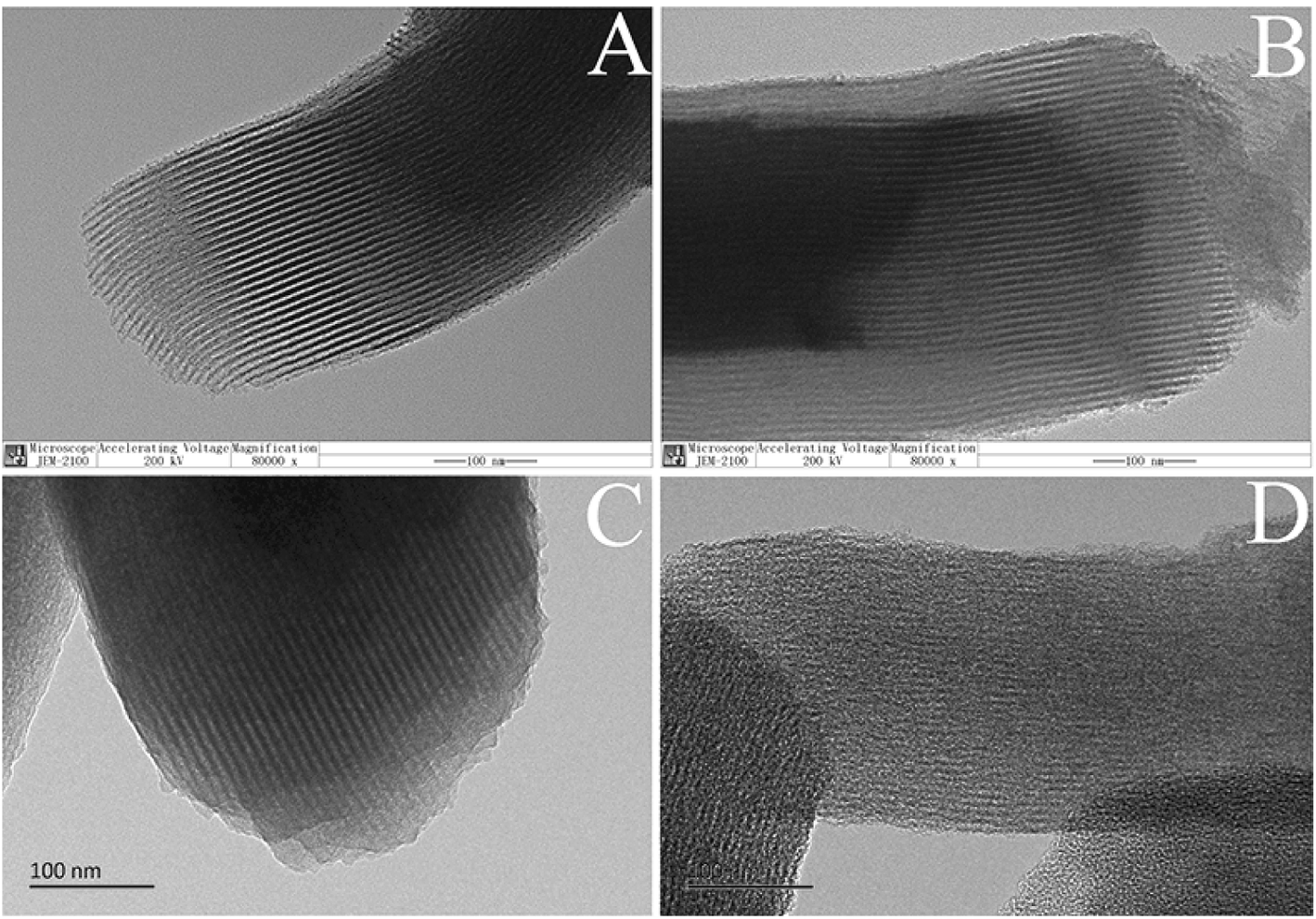 | ||
| Fig. 3 TEM images of SBA-15 (A), SBA-15-HC (B), SBA-15-C (C), and SBA-15-C etched by NaOH solution (D), and the scale bars were 100 nm. | ||
The preparation variables such as substrate proportion (the mass ratio of SBA-15 to glucose), hydrothermal treatment temperature, hydrothermal treatment time and calcination temperature were studied to analyze the regulative strategies of pore structures, and the SBA-15 still severed as the model. First, the SBA-15/glucose ratios of 1:0.5, 1:1, 1:3, 1:5 and 1:7 were used. Fig. 4 shows the nitrogen adsorption/desorption isotherms and pore size distributions of SBA-15, SBA-15-NH2 and SBA-15-C under different substrate proportions ranging from 1:0.5 to 1:7. All the materials exhibited typical curves of type-IV isotherms and H1-type hysteresis loops,18 which were the characteristics of mesoporous materials. The pore size distribution curves exhibited a narrow size range and gradually shifted to low value as the SBA-15/glucose ratio decreased from 1:0.5 to 1:5, indicating the uniform coverage of carbon layer and pore size controllability by the adjustment of the substrate proportion. However, the curve of 1:7 was in coincidence with that of 1:5, and this was might attributed to that the polysaccharide could not be filled into mesopores since the ratio of 1:5 during HTC process, which was proved by the similar dBJH values of the SBA-15-HC (1:5, 1:7) in Table S1.† The mesoporous specific surface area (Smeso), mesopore volume (Vmeso) and average pore diameter (dBJH) showed a similar discipline (Table S1†), which decreased as the ratio decreased from 1:0.5 to 1:5, and kept constant at the ratio ranges of 1:5–1:7. The minimum dBJH of SBA-15-C reached approximately 4.8 nm. In contrast, the contents of carbon element and carbon layer of samples exhibited a reverse trend with the decreasing ratios, owing to the increasing carbon layer thickness. Therefore, it was concluded that the pore structure of mesoporous silica–carbon composite could be easily regulated by the changing of substrate proportion. However, for the ratio of 1:0.1, the nitrogen adsorption/desorption isotherm was in inconformity to type-IV isotherm compared with those of the above samples, and the pore size distribution curve covered a wide range (Fig. S3†). Besides, the specific surface area (SBET), pore volume (Vt) and dBJH reduced hugely (Table S1†). These results suggested that the structure of SBA-15-C partially collapsed. The same phenomenon was found in the characterization of SBA-15 and SBA-15-NH2 after hydrothermal treatment (Fig. S4†). It could be inferred that the pure ordered mesoporous molecular sieves had a poor hydrothermal stability, and the sufficient coating of carbon layer during the hydrothermal treatment process strongly enhanced the stability of materials.
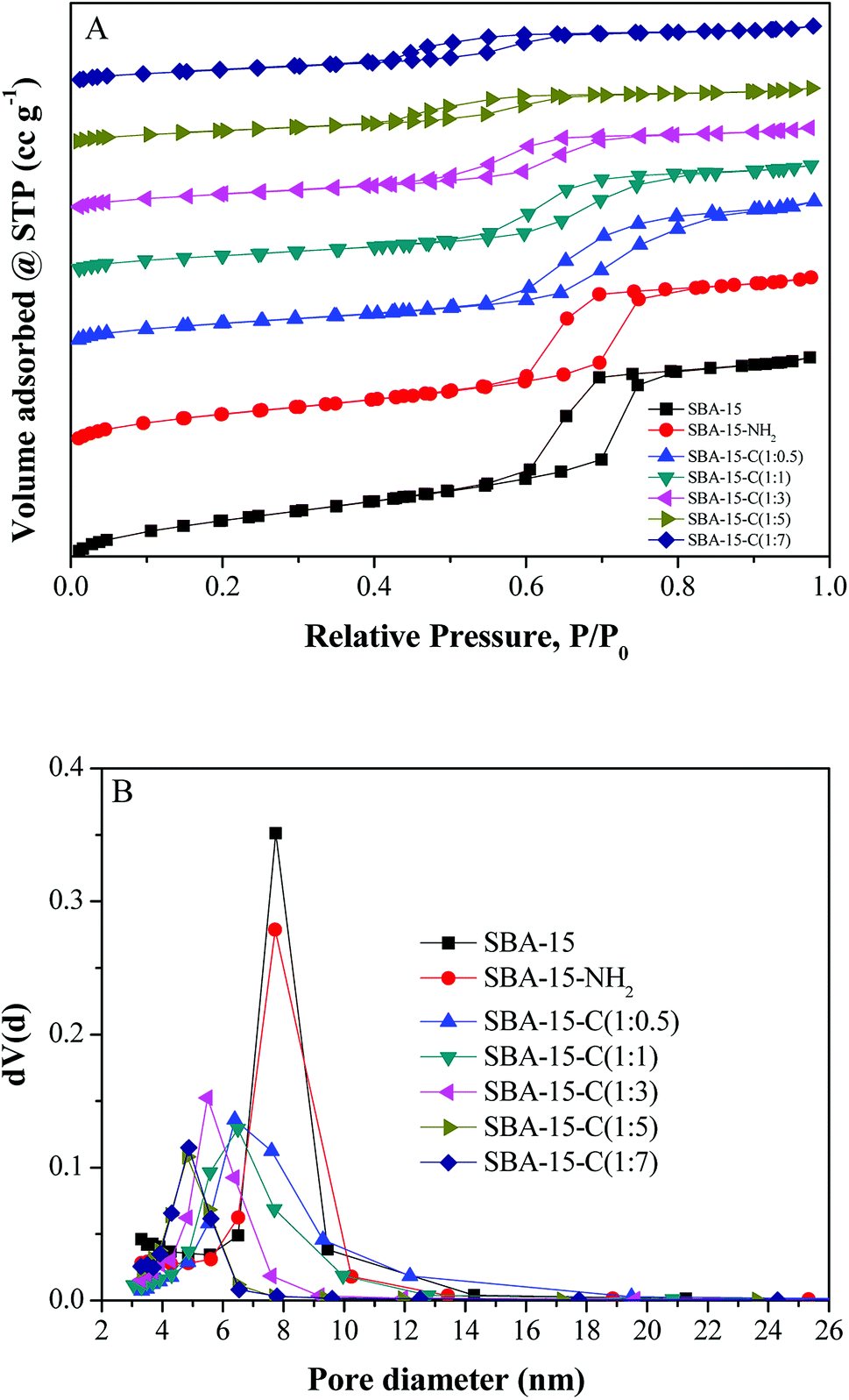 | ||
| Fig. 4 Nitrogen adsorption/desorption isotherms and pore size distributions of SBA-15, SBA-15-NH2 and SBA-15-C under different substrate proportions. | ||
In addition, the small-angle X-ray scattering (SAXS) patterns were recorded to determine the structural changes of SBA-15-C. As shown in Fig. 5, three well-resolved scattering peaks were observed in the pattern of mesoporous silica template SBA-15, which were assigned to 100, 110 and 200 reflections with unit cell parameter (a0) of 10.99 nm and pore wall thickness of 3.25 nm (Table S2†),19 implying a well-ordered two-dimensional (2-D) mesostructure (p6mm). However, only a wide 100 peak was found for SBA-15-C (1:0.1), suggesting a partial collapse of the mesostructural regularity after the hydrothermal and calcination treatment. This was consistent with the result of nitrogen sorption characterization, and was mainly attributed to the low hydrothermal stability of SBA-15 and insufficient coating of carbon layer. The samples of SBA-15-C (1:0.5–1:7) presented the distinct 100, 110 and 200 peaks, implying that the mesostructural regularity was well retained. Compared with that of the SBA-15, the scattering peaks of the SBA-15-C (1:1–1:7) slightly shifted to a high q value, and the calculated a0 value approximately in the range of 10.1–10.4, suggesting a structural slight shrinkage during the preparation processes. The pore wall thickness increased from 3.87 nm to 5.35 nm as the ratio ranging from 1:1 to 1:5, and then kept constant at the range of 1:5–1:7, further proving that the carbon layer was successfully coated on the inner surface of silica, and the thicker carbon layer was coated with the decreasing substrate proportions.
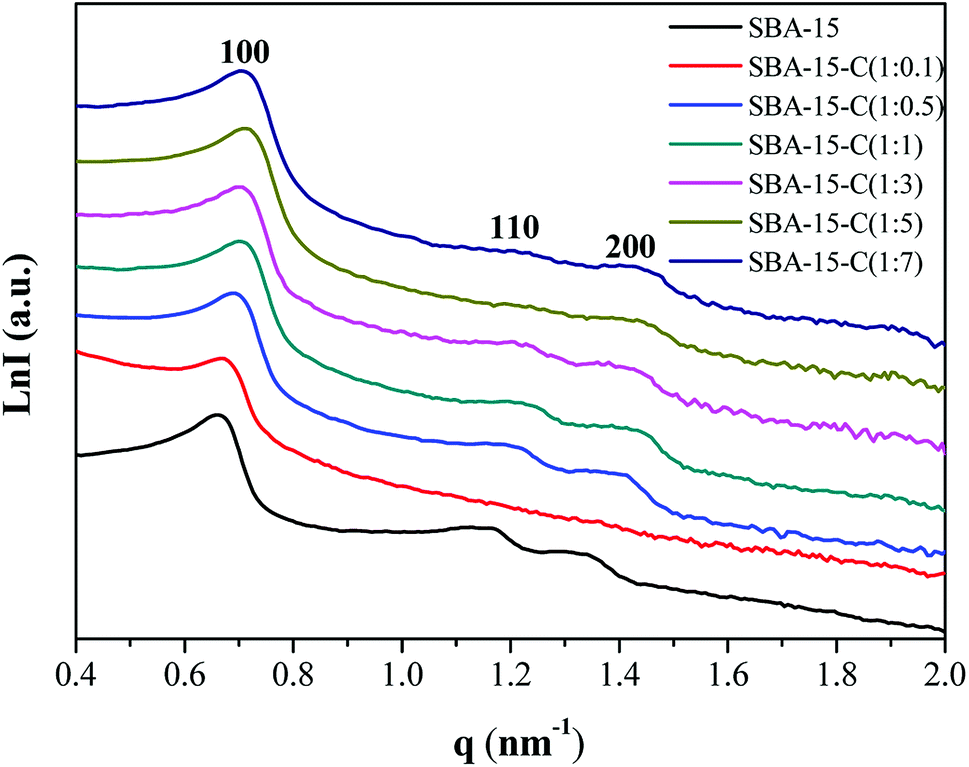 | ||
| Fig. 5 SAXS patterns of SBA-15 and SBA-15-C under different substrate proportions. | ||
Except the substrate proportion, the preparation variables of hydrothermal treatment temperature (160, 190 and 220 °C), hydrothermal treatment time (2, 5 and 8 h) and calcination temperature (400, 600 and 800 °C) were also investigated. The Smeso, Vmeso and dBJH of SBA-15-C decreased as the hydrothermal treatment temperature increased from 160 to 220 °C and the hydrothermal treatment time increased from 2 to 8 h (Table S1†). The nitrogen adsorption/desorption isotherms and pore size distributions are also given in Fig. S5 and S6.† Meanwhile, the contents of carbon element and carbon layer increased with the increasing hydrothermal temperature and time. These results indicated that the thickness of carbon layer could also be conveniently controlled by the hydrothermal treatment temperature and time. However, there was no significant difference of dBJH at the calcination temperature range of 400–800 °C, implying that the pore size could not be controlled by the calcination temperature. The carbon content of SBA-15-C at temperature of 600 °C was lower than that of SBA-15 at the temperature of 400 °C, and was similar to that of SBA-15 at the temperature of 800 °C, indicating that the materials could be effectively carbonized at the temperature of above 600 °C. Therefore, it was concluded that the pore structure of SBA-15-C could be regulated by the preparation variables in the hydrothermal carbonization process, including the substrate proportion, hydrothermal treatment temperature and time.
3.3 Applications of the silica–carbon composite materials
The practical application is the most important consideration of evaluating the value of materials. The silica–carbon composite materials had the advantages of homogeneous coating of carbon layer, relatively high graphitization degree and controlled morphology (shape and particle size) and pore size. Based on the properties of the silica–carbon composites, the materials were applied to the adsorption of bulky dyes and the chromatographic separation of oligosaccharide isomers in the present work.The bulky dyes of methylthionine chloride (MC, 1.26 × 0.77 × 0.65 nm) and rhodamine B (RhB, 1.59 × 1.18 × 0.56 nm) were used to estimate the adsorption capacities of the silica–carbon composites.20 In order to identify the effect of pore structure on the adsorption capacity, the SBA-15-C with different substrate ratio of 1:0.5–1:7 were first employed to adsorb the dyes from aqueous solution. As shown in Fig. 6, it was clear that the maximum equilibrium adsorption capacity was found at the ratio of 1:0.5. The adsorption capacity decreased as the ratio below 1:0.5 because of the decrease of Smeso. The adsorption capacity of SBA-15-C was not straightforward to uncover the super-performance of the carbon layer hereby obtained. The carbon layer content of SBA-15-C (1:0.5) was subsequently determined by TGA analysis and calculated as 7.58% (Fig. S7†). By simple math, the adsorption capacities of pure carbon layer were calculated to be 1172.8 and 3457.8 mg g−1 for MC and RhB, respectively, which was much higher than those of activated carbon and other ordered mesoporous carbons reported in the literatures.20,21 Therefore, it was obviously that the coating of carbon layer extremely improved the utilization efficiency of carbon materials. Besides, the SiO2-C with particle size of 30 μm was also used for dye adsorption, and the adsorption capacities were evaluated as 80.1 and 180.9 mg g−1 for MC and RhB, respectively. The sole carbon layer of SiO2-C was also estimated as 449.5 and 1015.2 mg g−1 for MC and RhB, respectively. Moreover, because of large particle size, the SiO2-C exhibited fast recovery by sedimentation after adsorption. The complete sedimentation of SiO2-C was achieved within 10 min, while it was unable to reach complete sedimentation for activated carbon even after 72 h, with the particle concentration of 0.10 g L−1 (Fig. S8 and S9†). As discussed above, the carbon layer of the silica–carbon composites showed good adsorption capacities for pollutants, and the silica carriers provided rapid recovery efficiency after adsorption.
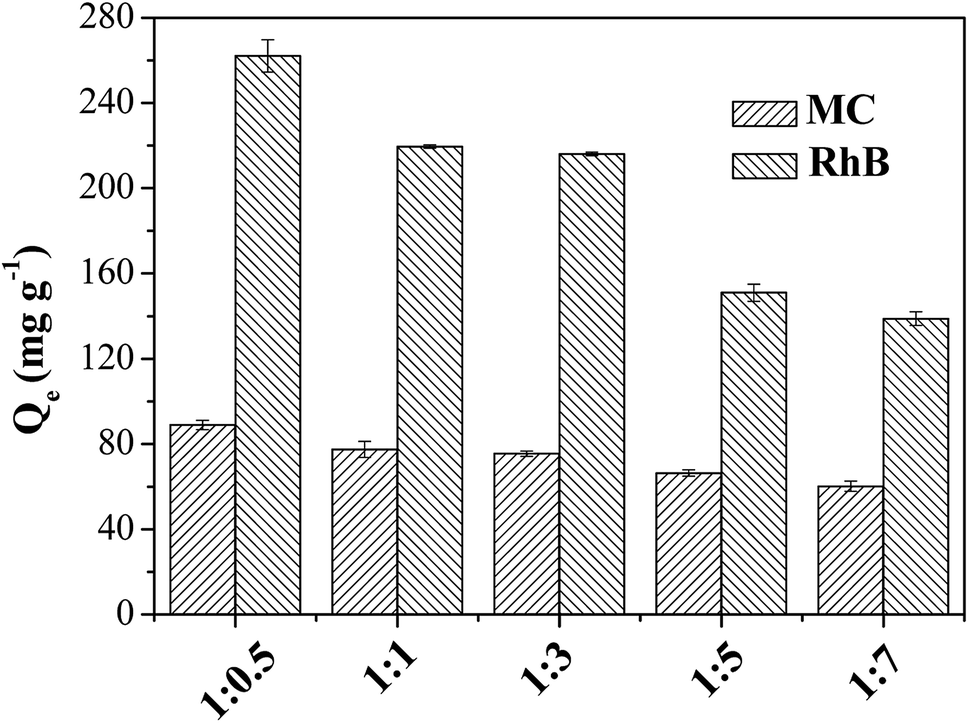 | ||
| Fig. 6 Adsorption capacities for MC and RhB on SBA-15-C. | ||
Glycans existed widely in organisms, which attached to cell surface and extracellular proteins and lipids, and were involved in many biological processes such as interaction, recognition and defense.22 The separation of glycan before detailed characterization of the glycan moieties is crucial for a molecular understanding of various biological processes. Porous graphitic carbon (PGC) is available commercially as chromatographic stationary phase under the trade name of Hypercarb, and is very suitable for the separation of glycan isomers.22 But the synthesis of PGC required multiple steps, including polymer impregnation in silica gel, carbonization at 1000 °C, dissolution of silica and graphitization at 2500 °C, resulting in long manufacturing time and high price of this material. In contrast, the silica-based material is the most commonly used matrix in the high performance liquid chromatography (HPLC), with the advantages of excellent mechanical strength and facile modification of surface properties. The silica–carbon composite in this report showed high graphitization degree, and was easy prepared with simple method, which might provide an alternative to PGC for the separation of oligosaccharide isomers. In this work, four oligosaccharides with two pairs of isomers, including LNFP-I, LNFP-II, LNDFH-I and LNnDFH-II, were used to evaluate the separation ability of the silica–carbon composite. The corresponding information of these four oligosaccharide isomers can be found at Table S3† and the result is shown in Fig. 7. The oligosaccharides had good retention on the SiO2-C (5 μm) column under reverse phase liquid chromatography (RPLC) mode, which was consistent with the properties of graphitized carbon stationary phase for oligosaccharides separation. And the baseline separation of the two pairs of oligosaccharide isomers was achieved on the column, which showed great potential in the separation of glycans. Moreover, owing to the incomplete graphitization, the retention and α/β anomeric splitting of oligosaccharide on the SiO2-C (5 μm) column were lower than those on the hypercarbon column. The α/β anomeric splitting of oligosaccharide was always undesired and usually recognized as a drawback of the hypercarbon column. Because of the strong anomeric splitting ability, the isomers of LNDFH-I and LNFP-II could not be separated on the hypercarbon column. These results implied that the SiO2-C with special selectivity could be considered as a great potential HPLC stationary phase to separate oligosaccharide isomers.
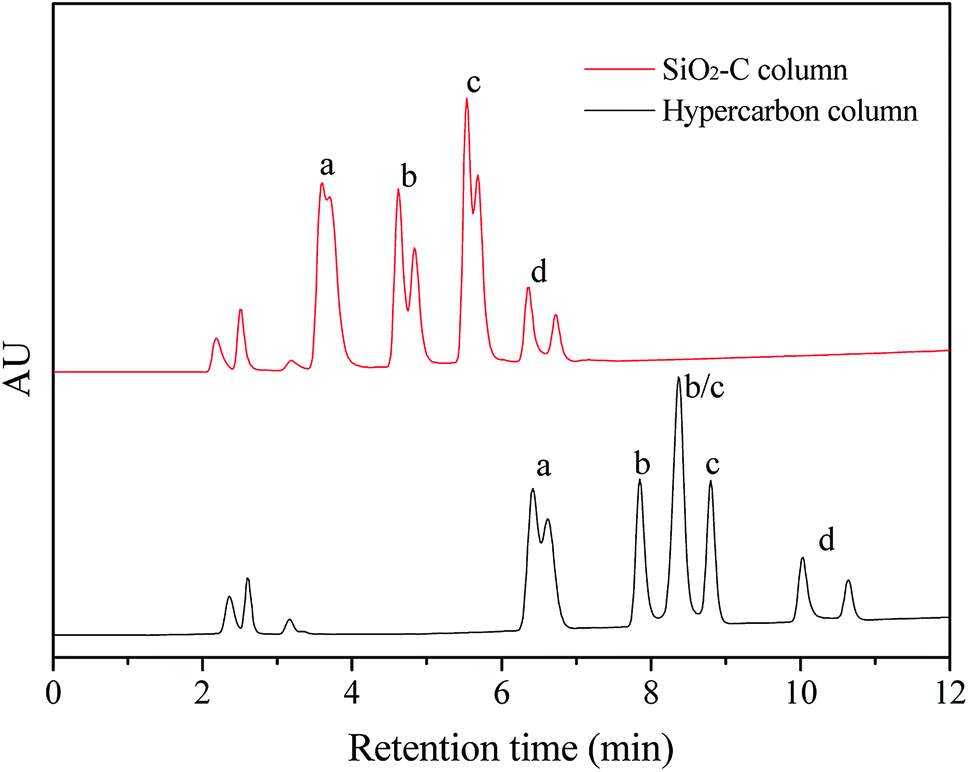 | ||
| Fig. 7 The separation of oligosaccharide isomers on the SiO2-C (5 μm) and hypercarbon columns (150 mm × 4.6 mm): (a) LNnDFH-II, (b) LNDFH-I, (c) LNFP-II, (d) LNFP-I; (a and b) and (c and d) were two pairs of isomers, and each compound corresponded to double peaks due to the effect of α/β anomeric splitting of saccharide. | ||
4. Conclusions
Mesoporous silica–carbon composite materials, with homogeneous and thickness-controllable carbon coating, were fabricated by a universal synthetic route of hydrothermal carbonization. The electrostatic interaction between amino-modified carriers and hydrothermal carbon played a crucial role in the formation of homogeneous carbon coverage, and the carbon layer could be coated on the surface of ordered and disordered mesoporous silica. The obtained composites showed high graphitization degree and controlled morphology (shape and particle size) and pore size. The composites were subsequently applied to the adsorption of bulky dyes and chromatographic separation of oligosaccharide isomers. The application results demonstrated that the composites possessed high adsorption capacity for dyes, and exhibited rapid recovery by sedimentation (10 min) after adsorption when 30 μm spherical silica gel as the carrier. Besides, the baseline separation of oligosaccharide isomers could be obtained on the silica–carbon column, which showed typical properties of graphitized carbon. These results indicated that the silica–carbon composites should be promising functional materials for the pollutant removal and oligosaccharide isomer separation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported in part by the National Key Research and Development Program of China (2016YFB0301502), the Joint Funds of the National Natural Science Foundation of China (U1608255), and the National Key Research and Development Program of China (2017YFD0400600).References
- (a) H. J. Lee, W. Cho, E. Lim and M. Oh, Chem. Commun., 2014, 50, 5476–5479 RSC; (b) E. Lam and J. H. T. Luong, ACS Catal., 2014, 4, 3393–3410 CrossRef; (c) L. Chen, T. Ji, L. Mu, Y. Shi, L. Brisbin, Z. Guo, M. A. Khan, D. P. Young and J. Zhu, RSC Adv., 2016, 6, 2259–2269 RSC; (d) A. Bello, N. Manyala, F. Barzegar, A. A. Khaleed, D. Y. Momodu and J. K. Dangbegnon, RSC Adv., 2016, 6, 1800–1809 RSC.
- (a) C. D. Liang, Z. J. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef PubMed; (b) E. Raymundo-Pinero, M. Cadek and F. Beguin, Adv. Funct. Mater., 2009, 19, 1032–1039 CrossRef; (c) M. M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796–6822 RSC; (d) Y. Zhang, L. Y. Zhang and C. W. Zhou, Acc. Chem. Res., 2013, 46, 2329–2339 CrossRef PubMed.
- (a) M. M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC; (b) M. M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC; (c) X. Qi, N. Liu and Y. Lian, RSC Adv., 2015, 5, 17526–17531 RSC.
- (a) M. Sevilla, A. B. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 4, 1400–1410 RSC; (b) A. J. Romero-Anaya, M. Ouzzine, M. A. Lillo-Rodenas and A. Linares-Solano, Carbon, 2014, 68, 296–307 CrossRef.
- (a) Q. Wang, H. Li, L. Q. Chen and X. J. Huang, Carbon, 2001, 39, 2211–2214 CrossRef; (b) M. T. Zheng, Y. L. Liu, Y. Xiao, Y. Zhu, Q. Guan, D. S. Yuan and J. X. Zhang, J. Phys. Chem. C, 2009, 113, 8455–8459 CrossRef.
- (a) J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef; (b) T. Y. Ma, L. Liu and Z. Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC; (c) L. Sun, C. Tian, Y. Fu, Y. Yang, J. Yin, L. Wang and H. Fu, Chem.–Eur. J., 2014, 20, 564–574 CrossRef PubMed.
- (a) F. Q. Zhang, Y. Meng, D. Gu, Y. Yan, C. Z. Yu, B. Tu and D. Y. Zhao, J. Am. Chem. Soc., 2005, 127, 13508–13509 CrossRef PubMed; (b) C. D. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 CrossRef PubMed.
- J. Kim, J. Lee and T. Hyeon, Carbon, 2004, 42, 2711–2719 CrossRef.
- H. Q. Qin, Z. Y. Hu, F. J. Wang, Y. Zhang, L. Zhao, G. J. Xu, R. A. Wu and H. F. Zou, Chem. Commun., 2013, 49, 5162–5164 RSC.
- C. du Fresne von Hohenesche, V. Ehwald and K. K. Unger, J. Chromatogr. A, 2004, 1025, 177–187 CrossRef PubMed.
- Y. Duan, M. Zheng, D. Li, D. Deng, C. Wu and Y. Yang, RSC Adv., 2017, 7, 3343–3449 RSC.
- (a) S. Ikeda, K. Tachi, Y. H. Ng, Y. Ikoma, T. Sakata, H. Mori, T. Harada and M. Matsumura, Chem. Mater., 2007, 19, 4335–4340 CrossRef; (b) M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281–2289 CrossRef.
- H. Wang, L. Y. Shi, T. T. Yan, J. P. Zhang, Q. D. Zhong and D. S. Zhang, J. Mater. Chem. A, 2014, 2, 4739–4750 RSC.
- J. Y. Liang, S. L. Chen, M. J. Xie, Y. Z. Wang, X. K. Guo, X. F. Guo and W. P. Ding, J. Mater. Chem. A, 2014, 2, 16884–16891 RSC.
- Y. Chen, B. H. Song, M. Li, L. Lu and J. M. Xue, Adv. Funct. Mater., 2014, 24, 319–326 CrossRef.
- M. Sevilla and A. B. Fuertes, Chem.–Eur. J., 2009, 15, 4195–4203 CrossRef PubMed.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef.
- (a) C. W. Lee, S. B. Yoon, H. K. Kim, H. C. Youn, J. Han, K. C. Roh and K. B. Kim, J. Mater. Chem. A, 2015, 3, 2314–2322 RSC; (b) G. H. Wang, Z. W. Cao, D. Gu, N. Pfander, A. C. Swertz, B. Spliethoff, H. J. Bongard, C. Weidenthaler, W. Schmidt, R. Rinaldi and F. Schuth, Angew. Chem., Int. Ed., 2016, 55, 8850–8855 CrossRef PubMed.
- (a) J. W. Fan, X. Jiang, H. Y. Min, D. D. Li, X. Q. Ran, L. Y. Zou, Y. Sun, W. Li, J. P. Yang, W. Teng, G. M. Li and D. Y. Zhao, J. Mater. Chem. A, 2014, 2, 10654–10661 RSC; (b) X. M. Gao, Z. Chen, Y. Yao, M. Y. Zhou, Y. Liu, J. X. Wang, W. D. Wu, X. D. Chen, Z. X. Wu and D. Y. Zhao, Adv. Funct. Mater., 2016, 26, 6649–6661 CrossRef.
- X. Zhuang, Y. Wan, C. M. Feng, Y. Shen and D. Y. Zhao, Chem. Mater., 2009, 21, 706–716 CrossRef.
- (a) Z. X. Wu and D. Y. Zhao, Chem. Commun., 2011, 47, 3332–3338 RSC; (b) Y. Dong, H. M. Lin, Q. M. Jin, L. Li, D. Wang, D. Zhou and F. Y. Qu, J. Mater. Chem. A, 2013, 1, 7391–7398 RSC.
- (a) R. Balogh, P. Jankovics and S. Beni, J. Chromatogr. A, 2015, 1422, 140–146 CrossRef PubMed; (b) S. Y. Zhou, Y. F. Huang, X. Dong, W. J. Peng, L. Veillon, D. A. S. Kitagawa, A. J. A. Aquino and Y. Mechref, Anal. Chem., 2017, 89, 6590–6597 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra04641g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |