
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A H2O2/HBr system – several directions but one choice: oxidation–bromination of secondary alcohols into mono- or dibromo ketones†
Gennady I. Nikishin,
Nadezhda I. Kapustina,
Liubov L. Sokova,
Oleg V. Bityukov and
Alexander O. Terent'ev *
*
N. D. Zelinsky Institute of Organic Chemistry of the Russian Academy of Sciences, 47 Leninsky prosp., Moscow 119991, Russian Federation. E-mail: terentev@ioc.ac.ru
First published on 10th August 2018
Abstract
In this work we found that a H2O2–HBr(aq) system allows synthesis of α-monobromo ketones and α,α′-dibromo ketones from aliphatic and secondary benzylic alcohols with yields up to 91%. It is possible to selectively direct the process toward the formation of mono- or dibromo ketones by varying the amount of hydrogen peroxide and hydrobromic acid. The convenience of application, simple equipment, multifaceted reactivity, and compliance with green chemistry principles make the application of the H2O2–HBr(aq) system very attractive in laboratories and industry. The proposed oxidation–bromination process is selective in spite of known properties of ketones to be oxidized by the Baeyer–Villiger reaction or peroxidated with the formation of compounds with the O–O moiety in the presence of hydrogen peroxide and Bronsted acids.
Introduction
A wide variety of methods for the oxidation of secondary alcohols into ketones is regularly replenished with new oxidants and oxidizing systems. Hydrogen peroxide attracts increased attention as a cheap and environment-friendly oxidant. For the oxidation of secondary alcohols with H2O2, catalysts of different chemical nature were used: Br2,1 HBr,1–6 NaBr,7,8 BiBr3,9 FeBr3,10 MgBr2/[bmim] BF4,11 Mn(III) complex,12 Co(II) complex,13 Zn polyoxometalate,14 Na2WO4/PTC,15–18 [bmim]4[W10O23] in ionic liquids,19 W/phosphate,20 Keggin complexes,21,22 Al/W-polyoxoanions,23 W/Sc/P-salt.24The H2O2–HBr(aq) system is known in organic chemistry for its application in the following processes: oxidation of secondary and primary alcohols to ketones1–4 and esters,1,25 respectively, bromination of olefins,26,27 acetylenes,27 ketones,4,28 arenes26,27,29,30 and heterocyclic compounds.31 This oxidation–bromination system is characterized by the use of inexpensive reagents, low environmental impact and the absence of organic wastes, which makes it a good alternative to existing oxidation and bromination methods.32 The H2O2–HBr(aq) system is also of interest for more extensive practical application.33 In our previous studies, this system was applied for the homo- and cross-condensation of alcohols into esters,34 the bromination of alkyl phenyl ketones into the aromatic ring and side-chain position.35
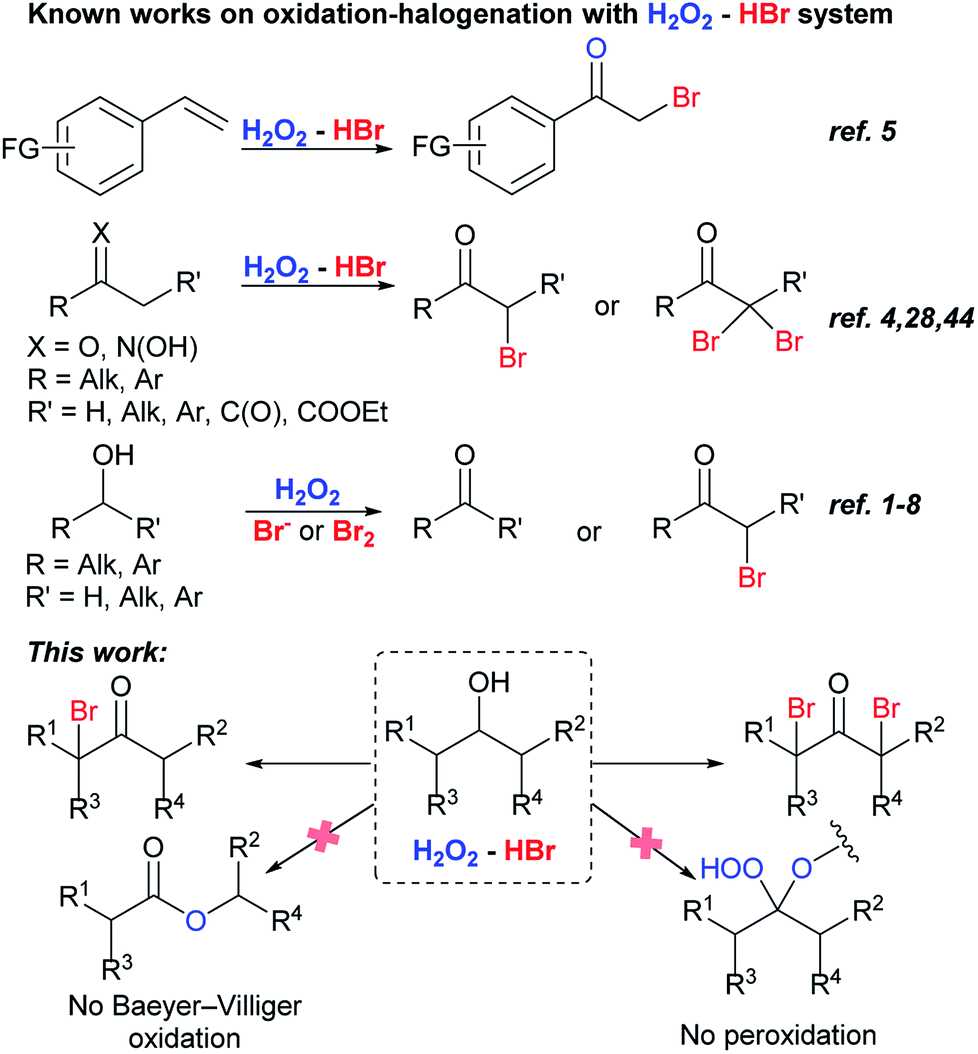
Bromo ketones are involved in organic chemistry as multipurpose reagents.36,37 They can be easily transformed into unsaturated ketones and ketones containing functional groups.38,39 Favorskii rearrangement of α-bromo ketones leads to the formation of esters.40 α,α′-Dibromo ketones are the precursors of 1,2-dialkyl cyclopropenones,41,42 and divinyl ketones,43 the latter ones are starting reagents for the synthesis of cyclopentenones by Nazarov reaction.40 H2O2–HBr(aq) system was used for the oxidative bromination of styrenes,5 ketones4,28 and oximes44 into bromo ketones. It is especially noteworthy that bromo ketones were not detected in the reactions with various substrates including alcohols under the action bromine containing systems: H2O2/HBr or hydrobromic acid sodium salt1–8 and H2O2/Br2.1 There is only one report of the oxidation of secondary alcohols into α-bromo ketones, but α,α′-dibromo ketones were not obtained (Scheme 1).4
 | ||
| Scheme 1 Oxidation-halogenation with H2O2–HBr(aq) system. | ||
This paper describes dibromo- and monobromo ketones synthesis by one-pot oxidation–bromination of secondary alcohols with H2O2–HBr(aq) (Scheme 1). The process is more experimentally simple, low-cost, and complies with green chemistry standards unlike the sole method proposed for the similar transformation, which is based on the oxidation–bromination of secondary alcohols with Ce(IV)/LiBr system.45 Change of hydrogen peroxide and hydrobromic acid amount can selectively direct the reaction routes towards the formation of mono- or dibromo ketones.
It is important to note that in the reaction conditions we didn't detect the formation of traditional Baeyer–Villiger oxidation products46–48 or expected peroxides,49–61 which are products of a reaction between C-atom of carbonyl group and hydrogen peroxide. Possible explanation of these reactions failure can be based on the higher rate of ketone bromination in competition with peroxidation and (or) following transformations. Steric hindrance in mono- and dibrominated ketones prevents the attack of hydrogen peroxide on the electrophilic carbon atom of the carbonyl group. To the best of our knowledge the only one example of α-brominated ketone peroxidation exists: 3-bromo-2,2-dihydroperoxy-1,7,7-trimethyl-bicyclo[2.2.1]heptane was obtained with H2O2–AlCl3 × 6H2O system.62
Results and discussion
The starting substrates for the oxidation–bromination were symmetrical 1a–e, 1h and unsymmetrical aliphatic alcohols 1f-g and benzylic alcohols 4a–e (Schemes 2 and 3). | ||
| Scheme 2 Oxidation–bromination of secondary alcohols 1a–h with the formation of α-bromoketones 2a–h or α,α′-dibromo ketones 3a–g. | ||
 | ||
| Scheme 3 Oxidation–bromination of secondary alcohols 4a–e bearing only one α-CH2 group near the carbonyl group with the formation of α-bromoketones 5a–e or α,α-dibromo ketones 6a,b,d,e. | ||
Depending on the experimental conditions and the structure of the alcohol 1a–h, it is possible to change the direction of the reaction, towards the formation of either α-bromo ketones 2a–h (molar ratio of 1a–h![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HBr:H2O2 = 1:1.2:10) or α,α′-dibromo ketones 3a–g (molar ratio of 1a–h:HBr:H2O2 = 1:6:15) (Scheme 2).
:HBr:H2O2 = 1:1.2:10) or α,α′-dibromo ketones 3a–g (molar ratio of 1a–h:HBr:H2O2 = 1:6:15) (Scheme 2).
α-Bromo ketones 5a–e (molar ratio of 4a–e:HBr:H2O2 = 1:1.2:10) and α,α-dibromo ketones 6a,b,d,e (molar ratio of 4a–e:HBr:H2O2 = 1:6:15) can be selectively prepared in the oxidation–bromination reactions with starting alcohols 4a–e bearing only one α-CH2 group near the carbonyl group (Scheme 3).
The influence of the experimental conditions on the selectivity of oxidation–bromination and on the yield of α-bromo ketone 2a, α,α′-dibromo ketone 3a, and ketone 7a was determined using pentanol-3 1a as an example. Generally, the reaction was carried out by the addition of 35% aqueous solution of H2O2 in acetonitrile for 6–10 hours to the solution of pentanol-3 1a and 48% HBr(aq) in acetonitrile or water (Table 1).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Molar ratio: mole HBr and H2O2/mole 1a | Solvent | τ, h | Yield, % | |||
| HBr | H2O2 | 2a | 3a | 7a | |||
| a General procedure: to a solution of alcohol 1a (1 mmol, 88.2 mg) and HBr (48% aq., 1.2–6 mmol, 0.136–0.679 ml) in 1 ml of a solvent at 65–70 °C (for entry 9 and 10, 60 °C) and vigorous stirring a solution of H2O2 (35% aq., 10–15 mmol, 0.860–1.290 ml) was added portionwise (0.2–0.3 ml) during 0.6–10 h. Yields were determined by GC analysis. | |||||||
| 1 | 1.2 | 5 | CH3CN | 6 | 38 | Trace | 55 |
| 2 | 1.2 | 10 | CH3CN | 6 | 80 | Trace | Trace |
| 3 | 1.4 | 10 | CH3CN | 6 | 76 | 13 | — |
| 4 | 2 | 10 | CH3CN | 6 | 60 | 35 | Trace |
| 5 | 2.5 | 10 | CH3CN (50% aq.) | 6 | 14 | 44 | 33 |
| 6 | 2.5 | 10 | H2O | 6 | 38 | 43 | Trace |
| 7 | 2.5 | 15 | CH3CN | 6 | Trace | 80 | — |
| 8 | 6 | 15 | CH3CN | 10 | Trace | 90 | — |
| 9 | 1.2 | 10 | MeOH | 6 | 40 | — | 52 |
| 10 | 1.2 | 10 | THF | 6 | 31 | — | 47 |
| 11 | 1.2 | 10 | DCE | 6 | 71 | — | 20 |

According to the obtained data, the highest yield of mono bromo ketone 2a was achieved with a 10-fold molar excess of H2O2 and 1.2 equivalents of HBr (entry 2). 5-Fold molar excess of H2O2 is insufficient for selective preparation of 2a. An increase of the HBr amount from 1.2 to 2.5 equivalents (entries 3–5) resulted in a decrease in the selectivity of products 2a, 3a, and 7a formation. When water was used as the solvent (entry 5), a mixture of monobromo ketones 2a and dibromo ketones 3a was also formed. The best yield of dibromo ketone 3a was achieved with a 15-fold molar excess of H2O2 (entries 7, 8). An increase of the HBr amount up to 6 equivalents and the reaction time up to 10 hours (entry 8) afforded the product 3a with the maximum yield (90%). In the best conditions for 2a preparation (entry 2), using of MeOH, THF, and DCE as a reaction media leads to the synthesis of 2a and 7a mixtures (entries 9–11).
Results of the optimization (Table 1) demonstrate that the reaction direction is mainly influenced by the molar ratio of the secondary alcohol, hydrogen peroxide, and hydrobromic acid. In the optimized reaction conditions (entry 2), a number of monobromo ketones 2a–h and 5a–e were prepared (Table 2). Optimized conditions (entry 7, Table 1) were also used for the synthesis of dibromo ketones 3a–g and 6a,b,d,e (Table 3).
| a General procedure: to a solution of alcohol 1a–h, 4a–e (1 mmol, 88.2–256.5 mg) and HBr (48% aq., 1.2 mmol, 0.136 ml) in CH3CN (1 ml) at 65–70 °C and vigorous stirring, a solution of H2O2 (35% aq.,10 mmol, 0.860 ml) in CH3CN (1 ml) was added portionwise (0.2–0.3 ml) for 6 hours.b The ratio of isomers 2f:2f′ ∼ 1:1 according to NMR data.c The ratio of isomers 2g:2g′ ∼ 2:1 according to NMR data. |
|---|
 |
| a General procedure: to a solution of alcohol 1a–h, 4a–c (1 mmol, 88.2–256.5 mg) and HBr (48% aq., 6 mmol, 0.679 ml) in CH3CN (1 ml) at 65–70 °C and vigorous stirring, a solution of H2O2 (35% aq., 15 mmol, 1.290 ml) in CH3CN (1 ml) was added portionwise (0.2–0.3 ml) for 10 hours.b The ratio of diastereoisomers meso:rac = 1:3 according to NMR data. |
|---|
 |
Alcohols of unbranched structure 1a–f are easily converted into α-bromo ketones (Table 2) and into α,α′-dibromo ketones (Table 3) with good yields. Diisopropyl carbinol 1h is converted only to the corresponding α-bromo ketone 2h (Table 2), the formation of α,α′-dibromo ketone from diisopropyl carbinol 1h is probably prevented by the steric hindrance. In the case of isopropyl amyl carbinol 1g, two isomeric mono bromides, 2-bromo-2-methyloctan-3-one 2g and 4-bromo-2-methyloctan-3-one 2g′ are formed in a ratio of 1:1 (Table 2). Oxidation–bromination of propyl amyl carbinol 1f also results in two monobromo ketones, 3-bromononan-4-one 2f and 5-bromononan-4-one 2f′ (∼1:1) (Table 2). Methyl tert-butyl carbinol 4a and methyl phenyl carbinol 4b are converted to α,α-dibromo ketone 6a and α,α-dibromo ketone 6b (Table 3) under optimized conditions for dibromo ketone synthesis (entry 7, Table 1). Amyl phenyl carbinol 4c undergoes only monobromination with the formation of 5c (Table 2). In the reaction medium symmetric α,α′-dibromo ketones are formed as a mixture of meso- and rac-isomers with a rac-form preference (ratio 1:3), as evidenced by 1H and 13C NMR spectra.
Conclusions
Thus, we have developed a convenient one-pot method for the synthesis of monobromo- and dibromo ketones by oxidation–bromination of secondary alcohols with the H2O2/HBr system in acetonitrile. Formation of peroxides and products of Baeyer–Villiger reaction was not detected. Amount and molar ratio of hydrogen peroxide and hydrobromic acid are the key factors for the achievement of a high degree of selectivity. Optimized conditions permits to synthesize α,α′-dibromo- or α,α-dibromo ketones with yields up to 91%. Conversion of unbranched secondary alcohols into α,α′-dibromo ketones is the most effective.Experimental
Experimental for the Table 1
To a solution of alcohol 1a (1 mmol, 88.2 mg) and HBr (48% aqueous) (1.2–6 mmol, 0.136–0.679 ml) in 1 ml of solvent (CH3CN, CH3CN (50% aq.), H2O, DCE) at 65–70 °C (for MeOH (entry 9) and THF (entry 10), 60 °C) and vigorous stirring, a solution of H2O2 (35% aqueous, 10–15 mmol, 0.860–1.290 ml) was added portionwise (0.2–0.3 ml) during 6–10 h. After the addition of the first portion, brown vapours and a bright orange colour were observed. The next portions of H2O2 were added after the decolorization of the reaction mixture (a pale-yellow colour), then the reaction mass was cooled, diethyl ether (15 mL) and Na2SO3 (1 g) were added. The organic layer was decanted and washed with water (5 ml), then dried over Na2SO4. The solvent was evaporated in a vacuum of a water jet pump (20 mmHg). Yields of 2a, 3a, 7a were determined by GC analysis with heptan-4-one and undecan-6-one as the internal standard.Experimental for the Table 2
To a solution of alcohol 1a–h, 4a–e (1 mmol, 88.2–256.5 mg) and HBr (48% aqueous, 1.2 mmol, 0.136 ml) in CH3CN (1 ml) at 65–70 °C and vigorous stirring, a solution of H2O2 (35% aqueous, 10 mmol, 0.860 ml) in CH3CN (1 ml) was added portionwise (0.2–0.3 ml) for 6 hours. After the addition of the first portion, brown vapors and a bright orange color were observed. The next portions of H2O2 were added after the decolorization of the reaction mixture (a pale-yellow color), then the reaction mass was cooled, diethyl ether (15 mL) and Na2SO3 (1 g) were added. The organic layer was decanted and washed with water (5 ml), then dried over Na2SO4. The solvent was evaporated in a vacuum of a water jet pump (20 mmHg). The products 2a–h and 5a–e were isolated by column chromatography on silica gel in a solvent system PE:EA (100:1).
Experimental for the Table 3
To a solution of alcohol 1a–h, 4a–e (1 mmol, 88.2–256.5 mg) and HBr (48% aqueous, 6 mmol, 0.679 ml) in CH3CN (1 ml) at 65–70 °C and vigorous stirring, a solution of H2O2 (35% aqueous, 15 mmol, 1.290 ml) in CH3CN (1 ml) was added portionwise (0.2–0.3 ml) for 6 hours. After the addition of the first portion, brown vapors and a bright orange color were observed. The next portion of hydrogen peroxide was added after the decolorization of the reaction mixture (a pale-yellow color), then the reaction mass was cooled, diethyl ether (15 mL) and Na2SO3 (1 g) were added. The organic layer was decanted and washed with water (5 ml) and then dried over Na2SO4. The solvent was evaporated in a vacuum of a water jet pump (20 mmHg). Products 3a–g and 6a,b,d,e were isolated by column chromatography on silica gel in a solvent system PE:EA (100:1).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Foundation for Basic Research (Grant no 18-03-00122 A).Notes and references
- A. Amati, G. Dosualdo, L. Zhao, A. Bravo, F. Fontana, F. Minisci and H.-R. Bjørsvik, Org. Process Res. Dev., 1998, 2, 261–269 CrossRef.
- V. B. Sharma, S. L. Jain and B. Sain, Synlett, 2005, 173–175 Search PubMed.
- S. L. Jain, V. B. Sharma and B. Sain, Tetrahedron, 2006, 62, 6841–6847 CrossRef.
- A. Podgorsek, S. Stavber, M. Zupan and J. Iskra, Green Chem., 2007, 9, 1212–1218 RSC.
- R. D. Patil, G. Joshi and S. Adimurthy, Ind. Eng. Chem. Res., 2010, 49, 8100–8105 CrossRef.
- M. Rasouli, M. A. Zolfigol, M. H. Moslemin and G. Chehardoli, Green Chem. Lett. Rev., 2017, 10, 117–120 CrossRef.
- X. Qi, J. Wang, L. Zheng and L. Qi, Synlett, 2011, 555–558 CrossRef.
- G. Joshi, R. D. Patil and S. Adimurthy, RSC Adv., 2012, 2, 2235–2239 RSC.
- M.-k. Han, S. Kim, S. T. Kim and J. C. Lee, Synlett, 2015, 26, 2434–2436 CrossRef.
- S. E. Martín and A. a. Garrone, Tetrahedron Lett., 2003, 44, 549–552 CrossRef.
- H. J. Park and J. C. Lee, Synlett, 2009, 79–80 Search PubMed.
- H. R. Mardani and H. Golchoubian, Tetrahedron Lett., 2006, 47, 2349–2352 CrossRef.
- S. Das and T. Punniyamurthy, Tetrahedron Lett., 2003, 44, 6033–6035 CrossRef.
- W. Kasprzyk, M. Galica, S. Bednarz and D. Bogdał, Synlett, 2014, 25, 2757–2760 CrossRef.
- K. Sato, M. Aoki, J. Takagi, K. Zimmermann and R. Noyori, Bull. Chem. Soc. Jpn., 1999, 72, 2287–2306 CrossRef.
- D. Bogda and M. Lukasiewicz, Synlett, 2000, 143–145 Search PubMed.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977–1986 RSC.
- B. S. Chhikara, R. Chandra and V. Tandon, Synlett, 2005, 872–874 Search PubMed.
- B. S. Chhikara, S. Tehlan and A. Kumar, Synlett, 2005, 63–66 Search PubMed.
- C. Venturello and M. Gambaro, J. Org. Chem., 1991, 56, 5924–5931 CrossRef.
- J. Wang, L. Yan, G. Li, X. Wang, Y. Ding and J. Suo, Tetrahedron Lett., 2005, 46, 7023–7027 CrossRef.
- S. Zhang, G. Zhao, S. Gao, Z. Xi and J. Xu, J. Mol. Catal. A: Chem., 2008, 289, 22–27 CrossRef.
- M. Carraro, B. S. Bassil, A. Soraru, S. Berardi, A. Suchopar, U. Kortz and M. Bonchio, Chem. Commun., 2013, 49, 7914–7916 RSC.
- L. Balbinot, U. Schuchardt, C. Vera and J. Sepúlveda, Catal. Commun., 2008, 9, 1878–1881 CrossRef.
- S. Samanta, V. Pappula, M. Dinda and S. Adimurthy, Org. Biomol. Chem., 2014, 12, 9453–9456 RSC.
- V. Arutyunyan, T. Kochikyan, N. Egiazaryan and A. Avetisyan, Russ. J. Org. Chem., 1995, 31, 100 Search PubMed.
- N. B. Barhate, A. S. Gajare, R. D. Wakharkar and A. V. Bedekar, Tetrahedron, 1999, 55, 11127–11142 CrossRef.
- V. H. Tillu, P. D. Shinde, A. V. Bedekar and R. D. Wakharkar, Synth. Commun., 2003, 33, 1399–1403 CrossRef.
- C. U. Dinesh, R. Kumar, B. Pandey and P. Kumar, J. Chem. Soc., Chem. Commun., 1995, 611–612 RSC.
- B. M. Choudary, Y. Sudha and P. N. Reddy, Synlett, 1994, 450 CrossRef.
- S. Yang, J. Liu, Z. Jin, W. Tian, H. Sun and M. Wang, Heterocycl. Commun., 2018 Search PubMed.
- G. I. Nikishin, N. I. Kapustina, L. L. Sokova, O. V. Bityukov and A. O. Terent'ev, Tetrahedron Lett., 2017, 58, 352–354 CrossRef.
- INTEROX CHEMICALS LTD (GB), EP19890302334, 1989.
- G. I. Nikishin, L. L. Sokova, A. O. Terent'ev and N. I. Kapustina, Russ. Chem. Bull., 2015, 64, 2845–2850 CrossRef.
- A. O. Terent'ev, S. V. Khodykin, I. B. Krylov, Y. N. Ogibin and G. I. Nikishin, Synthesis, 2006, 1087–1092 CrossRef.
- Á. M. Montaña, J. A. Barcia, P. M. Grima and G. Kociok-Köhn, Tetrahedron, 2016, 72, 6794–6806 CrossRef.
- A. S. Ionkin, W. J. Marshall and B. M. Fish, Org. Lett., 2008, 10, 2303–2305 CrossRef PubMed.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7042 CrossRef PubMed.
- A. O. Terent'ev, V. A. Vil, I. A. Yaremenko, O. V. Bityukov, D. O. Levitsky, V. V. Chernyshev, G. I. Nikishin and F. Fleury, New J. Chem., 2014, 38, 1493–1502 RSC.
- J. J. Li, Name Reactions, Springer-Verlag, Berlin Heidelberg, 2003 Search PubMed.
- R. Breslow, J. Posner and A. Krebs, J. Am. Chem. Soc., 1963, 85, 234 CrossRef.
- J. Ciabattoni and E. C. Nathan, J. Am. Chem. Soc., 1969, 91, 4766–4771 CrossRef.
- S. Lemaire-Audoire and P. Vogel, Tetrahedron: Asymmetry, 1999, 10, 1283–1293 CrossRef.
- A. O. Terent'ev, I. B. Krylov, V. A. Vil', Z. Y. Pastukhova, S. A. Fastov and G. I. Nikishin, Cent. Eur. J. Chem., 2012, 10, 360–367 Search PubMed.
- G. I. Nikishin, L. L. Sokova and N. I. Kapustina, Russ. Chem. Bull., 2010, 59, 391–395 CrossRef.
- G. R. Krow, in Organic Reactions, ed. L. A. Paquette, John Wiley and Sons, 1993, vol. 43, pp. 251–798 Search PubMed.
- R. Michael and M. Bernard, Eur. J. Org. Chem., 1999, 737–750 Search PubMed.
- I. A. Yaremenko, V. A. Vil', D. V. Demchuk and A. O. Terent'ev, Beilstein J. Org. Chem., 2016, 12, 1647–1748 CrossRef PubMed.
- H. Gandhi, K. O'Reilly, M. K. Gupta, C. Horgan, E. M. O'Leary and T. P. O'Sullivan, RSC Adv., 2017, 7, 19506–19556 RSC.
- A. O. Terent'ev, M. M. Platonov, Y. N. Ogibin and G. I. Nikishin, Synth. Commun., 2007, 37, 1281–1287 CrossRef.
- K. Žmitek, M. Zupan, S. Stavber and J. Iskra, Org. Lett., 2006, 8, 2491–2494 CrossRef PubMed.
- C. Schwartz and P. H. Dussault, in PATAI'S Chemistry of Functional Groups, John Wiley & Sons, Ltd., 2014, pp. 1–38 Search PubMed.
- D. Opsenica, G. Pocsfalvi, Z. Juranić, B. Tinant, J.-P. Declercq, D. E. Kyle, W. K. Milhous and B. A. Šolaja, J. Med. Chem., 2000, 43, 3274–3282 CrossRef PubMed.
- Y. Li, H.-D. Hao, Q. Zhang and Y. Wu, Org. Lett., 2009, 11, 1615–1618 CrossRef PubMed.
- A. Bunge, H.-J. Hamann and J. Liebscher, Tetrahedron Lett., 2009, 50, 524–526 CrossRef.
- P. Ghorai, P. H. Dussault and C. Hu, Org. Lett., 2008, 10, 2401–2404 CrossRef PubMed.
- B. Das, M. Krishnaiah, B. Veeranjaneyulu and B. Ravikanth, Tetrahedron Lett., 2007, 48, 6286–6289 CrossRef.
- A. O. Terent'ev, M. M. Platonov and A. V. Kutkin, Open Chem., 2006, 4, 207 Search PubMed.
- A. O. Terent'ev, M. M. Platonov, I. B. Krylov, V. V. Chernyshev and G. I. Nikishin, Org. Biomol. Chem., 2008, 6, 4435–4441 RSC.
- D. Azarifar, K. Khosravi and F. Soleimanei, Molecules, 2010, 15, 1433 CrossRef PubMed.
- K. Starkl Renar, S. Pecar and J. Iskra, Org. Biomol. Chem., 2015, 13, 9369–9372 RSC.
- D. Azarifar and K. Khosravi, J. Iran. Chem. Soc., 2011, 8, 1006–1013 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization of new compounds. See DOI: 10.1039/c8ra04885a |
| This journal is © The Royal Society of Chemistry 2018 |