
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn iRGD peptide conjugated heparin nanocarrier for gastric cancer therapy†
Shichao Ai a,
Shuang Zhenc,
Zhijian Liua,
Feng Suna,
Xingchen Heb,
Feng Chub,
Wenxian Guan*a and
Jianquan Wang*b
a,
Shuang Zhenc,
Zhijian Liua,
Feng Suna,
Xingchen Heb,
Feng Chub,
Wenxian Guan*a and
Jianquan Wang*b
aDepartment of General Surgery, Drum Tower Hospital, Medical School of Nanjing University, Nanjing, Jiangsu 210008, China. E-mail: medguanwx@163.com; Fax: +86-25-68182097
bDepartment of Biomedical Engineering, College of Engineering and Applied Sciences, Nanjing University, Nanjing, Jiangsu Province 210093, China. E-mail: wangjianquan006@163.com
cDepartment of General Surgery, Jinling Hospital, Medical School of Nanjing University, Nanjing, China
First published on 24th August 2018
Abstract
The cis-diamminedichloroplatinum(II) (DDP, cisplatin) is an important antitumor drug for the therapy of gastric cancer in clinics, but it is limited by its nonspecific tissue distribution and severe side effects. Here, an integrin targeted drug delivery system iRGD-heparin nanocarrier (iHP) was successfully synthesized. The iHP has several unique properties. First, this nanocarrier has excellent biodegradation due to its heparin biopolymer frame. Second, it is biocompatible because succinic anhydride-modified heparin has no anticoagulant activity and cell toxicity. We proved that from anticoagulant function evaluation and a cytotoxicity test. Third, iRGD was conjugated to the nanoparticles as an integrin-targeting ligand. Our results showed that iHP has precise targeting to integrin-overexpressed human gastric cancer cells MKN-45P in vitro and tumor tissues in vivo. Hence, we synthesized targeted nanoparticles iHP-DDP (iHDDP) and untargeted nanoparticles HP-DDP (HDDP). In our result, iHDDP showed higher antitumor efficacy than HDDP in vitro and in vivo. And in comparison with free DDP, the iHDDP nanoparticle delivery system showed satisfactory antitumor activity of DDP without weight loss or liver and kidney damage in nude mice bearing MKN-45P tumors.
Introduction
Gastric cancer (GC) is one of the most common malignancies and is considered as the second underlying cause of cancer-related mortality in the world.1 Each year, nearly 950![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new diagnoses occur, and more than 50% of these cases occur in Eastern Asian, especially in China. An estimated 679100 new GC cases were diagnosed, and 498000 patients in China died from GC in 2015.2 The standardized treatment for GC now consists of radical surgery, a combination of chemotherapy and/or radiotherapy, which is dependent on the postoperative pathological stage. For advanced GC patients, 5 year overall survival increases by 10–15% with the addition of chemotherapy.1 Yet the lack of intended targets and the poor penetration into tumor tissue causes extensive systemic toxicity and low drug bioavailability.3 These drawbacks largely restrict the application of conventional chemotherapy. Thus, how to make these chemotherapy drugs targeting remains an urgent problem to be resolved in chemotherapy.
000 new diagnoses occur, and more than 50% of these cases occur in Eastern Asian, especially in China. An estimated 679100 new GC cases were diagnosed, and 498000 patients in China died from GC in 2015.2 The standardized treatment for GC now consists of radical surgery, a combination of chemotherapy and/or radiotherapy, which is dependent on the postoperative pathological stage. For advanced GC patients, 5 year overall survival increases by 10–15% with the addition of chemotherapy.1 Yet the lack of intended targets and the poor penetration into tumor tissue causes extensive systemic toxicity and low drug bioavailability.3 These drawbacks largely restrict the application of conventional chemotherapy. Thus, how to make these chemotherapy drugs targeting remains an urgent problem to be resolved in chemotherapy.
Targeting ligands, such as peptides and antibodies that bind to tumor-associated markers, have advantages of low toxicity, low immunogenicity, high specificity and biocompatibility. It has been proved that tumor targeting drug delivery systems modified by them, such as RGD and EGF,4–7 can selectively increase drug concentration and drug penetration at the tumor sites or tumor cells, thereby significantly improving the theranostic efficacy and security. It has been reported that a N-end cysteine peptide tumor-homing peptide known as iRGD (CRGDK/EGPD/EC) could interact with both integrin and neuropilin-1 receptors (NPR-1) and shows more efficient tumor penetration than conventional RGD peptides.8,9 The iRGD peptide targets tumor sites as following consecutive steps: once the RGD motif bind to αv integrins on tumor endothelium, the CendR (R/KXXR/K) motif of iRGD is exposed through proteolytic cleavage by a tumor-associated protease(s) such as matriptase,10 and then the CendR motif detaches from integrin and binds to NRP-1, triggering NPR-1-mediated cellular internalization.9,11 These properties of iRGD have been exploited to enhance the tumor targeting and internalization of imaging agents and drugs.9,12–15
It is known that functional nanocarriers constructed by synthetic and natural materials are playing an important role in drug delivery. In most cases, nanocarriers based on synthetic materials, such as polyethylene glycol (PEG), are the most common approach used in drug delivery through achieving longer circulation time of carriers in the bloodstream. However, the biological toxicity and immunological response of synthetic materials limit their application.16 Superior to various synthetic materials, nanocarriers based on naturally occurring polysaccharides, such as chitosan, hyaluronic acid, dextran, and heparin, are more desirable for drug delivery due to their nonimmunogenic, nontoxic and well-designed properties.17
In the present work, a succinic anhydride-modified heparin drug delivery system which is biocompatible and biodegraded with no anticoagulant activity was constructed. To improve the selectivity and penetration ability to tumor cell, the targeting peptide iRGD was further conjugated on the surface of heparin nanoparticles in this study. The iRGD-heparin (iHP) was characterized for size distribution and in vitro cytotoxicity. Cells imaging and tumor imaging were captured to demonstrate the tumor targeting ability of iHP. The cytotoxicity in vitro of iRGD-Heparin-DDP (iHDDP) was measured using MKN-4P human gastric cancer cell and human gastric epithelial cell line GES-1. Finally, the antitumor efficacy and toxicity of iHDDP nanoparticles in nude mice bearing MKN-45P tumors were evaluated.
Materials and methods
Materials
Heparin (sodium salt form, average Mw: 12000–16000) was purchased from Meryer (Shanghai) Chemical Technology Co., Ltd. iRGD (CRGDKGPDC) was purchased from Bankpeptide (Hefei) Biological Technology Co., Ltd. Milli-Q deionized (DI) water (Millipore, 18.2 MΩ cm−1) was adopted throughout the experiments. Anhydrous methylsulfoxide (DMSO) and N,N-dimethylformamide (DMF) were obtained from Sigma-Aldrich (St Louis, MO). Dialysis membrane Spectra/Por 3 (MWCO: 6–8k) was purchased from Spectrum Labs. All other reagents were obtained from Sigma-Aldrich (St Louis, MO) at highest purity available.
Preparation of iRGD-heparin (iHP)
Succinic anhydride heparin (Heparin-Su) were synthesized in line with published procedures.18 Succinic anhydride heparin (1000 mg) was dissolved in anhydrous DMSO (10 mL) together with iRGD (20 mg, 0.021 mmol), EDAC (0.17 g, 0.233 mmol) and NHS (0.05 g, 0.437 mmol), and the reaction was allowed to proceed at 35 °C for 24 h to prepare iRGD-heparin (iHP) nanocarriers. The reaction mixture was dialyzed against DI water for 24 h to remove the solvent and catalysts (Spectra/Por 3, MWCO: 3500), the solution was obtained after lyophilization at −50 °C to give a white powder. The structure of iHP was confirmed by NMR (NMR spectra were obtain from a Bruker 1H proton NMR 400 DRX Spectrometer). The morphology and size of iHP were measured using transmission electron microscopic (TEM) images (JEOL JEM-2010 (HR)) and dynamic light scattering (DLS) (ZetaSizer Nano-ZS90, Malvern Instrument).Preparation of iHP-cy5.5 and iHP-488
iHP-cy5.5/488 were prepared from reacting amine functionalized cy5.5/Oregon Green 488 and iHP in DMSO at ambient temperature overnight, followed by dialysis against water (Spectra/Por 6, MWCO: 8000). After lyophilization at −50 °C, the unreacted dye was removed by PD-10 column to give a blue/green powder.Preparation of HDDP and iHDDP
Succinic anhydride heparin (15 mg) and DDP (15 mg) were mixed in DI water (10 mL, pH ≈ 6), the mixture was allowed to stir at RT for 72 h in the dark to prepare HDDP nanoparticles. iHDDP nanoparticles were prepared through the same procedure, except iRGD-heparin (10 mg) was used other than succinic anhydride heparin. The free DDP was removed by dialyses against DI water (Spectroa/Por6, MWCO = 6–8k) for 24 h. The morphology and size of iHP were measured using transmission electron microscopic (TEM) images (JEOL JEM-2010 (HR)). The loading of DDP was determined using ICP-MS.Cells and cell culture
The human gastric cancer cell line MKN-45P and human gastric epithelial cell line GES-1 were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences (Shanghai, China). These cell lines were maintained in 5% CO2 gas at 37 °C in RPMI-1640 medium as supplemented with 10% (v/v) heat-inactivated FBS, 100 U mL−1 penicillin and 100 g mL−1 streptomycin.Confocal fluorescence microscopic imaging
The MKN-45P and GES-1 cells were implanted in glass bottom cell culture dishes and allowed to grow for 12 h. Then, cells were rinsed with PBS twice and incubated in medium containing iH-488 (3 mg mL−1) for 2 h. Images were then captured using confocal microscope.Fluorescence intensity measurement
The MKN-45P and GES-1 cells were seeded and incubated in 6-wells plate with and allowed to grow for 12 h. Then, cells were rinsed with PBS twice and incubated in medium containing iH-488 (3 mg mL−1) for 2 h. The cells were then rinsed with PBS twice and treated with trypsin. After centrifugation and suspended in PBS, samples of 1 × 104 cells were measured by FACSCalibur flow cytometer.In vitro cytotoxicity of iHP assay
The cytotoxicity of iHP to MKN-45P and GES-1 cells was measured by performing Cell Counting Kit-8 test (CCK-8), MKN-45P and GES-1 cells were inoculated on 96 well plates (104 cell per well) in 100 mL of RPMI-1640 medium supplemented with 10% FBS while the marginal wells were provided with sterile PBS. The inoculated cells were incubated in 5% CO2 atmosphere at 37 °C overnight. Next, the medium in each well was replaced with 100 mL of the fresh medium as supplemented with different concentrations of iHP (0, 0.01, 0.1, 1, 10, 100, 1000 μg mL−1, each of which was examined in six replicates), and the inoculated cells were incubated at 37 °C with 5% CO2 for 24 h. Then, 10 μL Cell Counting Kit-8 test (CCK-8) was added into each well followed by further incubation for 4 h. The absorbance value of each test well was measured at a wavelength of 450 nm using ELISA microplate reader following the description of the manufacturer.In vitro cytotoxicity of DDP, HDDP and iHDDP assay
The cytotoxicity of DDP, HDDP, iHDDP to MKN-45P and GES-1 cells was measured by performing Cell Counting Kit-8 test (CCK-8). MKN-45P and GES-1 cells were inoculated on 96 well plates (104 cells per well) in 100 mL of RPMI-1640 medium supplemented with 10% FBS while the marginal wells were provided with sterile PBS. The inoculated cells were incubated in 5% CO2 at 37 °C overnight. Next, the medium in each well was replaced with 100 mL of the fresh medium as supplemented with different concentrations of DDP, HDDP and iHDDP (0, 1, 2, 4, 8, 16, 32 μg mL−1, DDP equivalence, each of which was examined in six replicates), and the inoculated cells were incubated at 37 °C with 5% CO2 for 24 h, 48 h and 72 h. Then, 10 μL Cell Counting Kit-8 test (CCK-8) was added into each well followed by further incubation for 4 h. The absorbance value of each test well was measured at a wavelength of 450 nm using ELISA microplate reader, following the description of the manufacturer.Establishment of gastric cancer xenografts model
Five-week-old male mice (nu/nu) with severe combined immunodeficiency (SCID) were purchased from the jiesijie lab-animal company in Shanghai (Animal License no. SCXK (HU) 2013-0006) and maintained under specific pathogen-free conditions at Nanjing University. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Nanjing University and experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Nanjing University. The gastric cancer cell line, MKN-45P, were grown until logarithmic phase, harvested, the cell density measured, and a total of 1 × 107 cells were dissolved in 200 μL PBS. The nude mice were anesthetized with 1.5% pentobarbital sodium. The MKN-45P suspension containing 107 cells were injected subcutaneously into the left flank area of anesthetized nude mice.In vivo NIR fluorescence imaging
The nude mice with subcutaneous MKN-45P cells randomly fell into two groups. The iHP-cy5.5 and HP-cy5.5 were injected into tumor bearing mice, respectively (cy5.5: 2 mg kg−1, tail vein injection) and imaged by in vivo fluorescence imaging system (Cri Inc., Woburn, MA) at 1 h, 4 h, 8 h, 12 h, 24 h, 48 h, 72 h and 96 h postinjection. Then, the mice were sacrificed, and the tumor and organs were imaging by in vivo fluorescence imaging system (Cri Inc., Woburn, MA).In vivo antitumor efficacy study
The nude mice with subcutaneous MKN-45P cells randomly fell into four groups when tumor sizes were nearly 80 mm3, received free DDP, HDDP and iHDDP injected intravenously via the tail vein seven times at 3 day intervals at a dose of Pt: 2.5 mg kg−1 or DDP-loaded nanoparticles at the same dose, while the control group was administered saline. Each group consisted six mice. The body weight, tumor volume of each mouse were observed and recorded. After 21 days, the mice were sacrificed, and the tumor and organs were collected.Peripheral blood analysis
After treated with free DDP, HDDP and iHDDP nanoparticles (7 injections, Pt: 2.5 mg kg−1, 21 days), the mice were anesthetized with 1.5% pentobarbital sodium, and 1 mL blood was immediately drawn in a pro-coagulation tube, centrifuged at 3000 rpm for 10 min, and the serum was separated. By using alkaline phosphatase (ALT), alanine aminotransferase (AST), the serological liver function was studied, and renal function was detected using blood urea creatinine (CRE) and nitrogen (BUN).Histopathology evaluation
The heart, liver, spleen and kidney were dissected from the mice (21 days after treatment) for histopathological analysis. The organs collected from the mice were embedded in paraffin after immobilization in 4% paraformaldehyde at 4 °C for 4 h. Then, the sections were stained with hematoxylin and eosin (H&E), and observed using a light microscope. Besides, the representative images were captured.Results
Synthesis and characterization of iRGD-heparin (iHP)
In this work, iRGD-heparin (iHP) nanocarrier was prepared by making iRGD conjugated onto Heparin-Su through amide linkers (Scheme 1), and it was confirmed by NMR (Fig. S1†). iHP nanocarrier hydrodynamic size was about 30 ± 5 nm (Fig. 1B), consistent with TEM characterization of NPs, which was 20 ± 5 nm (Fig. 1A), as DLS found. To evaluate its biosafety, its anticoagulant function was tested. As the result suggests, iHP decrease thrombin time (TT) and activated partial thromboplastin time (APTT) significantly in comparison with the unfractionated heparin (Table S1†).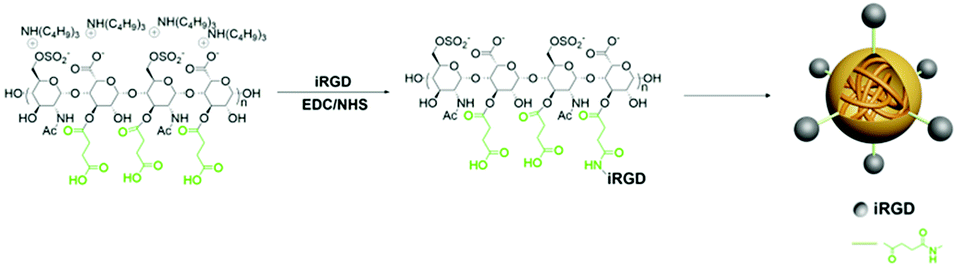 | ||
| Scheme 1 Synthetic scheme of iRGD-heparin (iHP). | ||
 | ||
| Fig. 1 (A) TEM image of iHP (diameter = 20 ± 5 nm), (B) DLS measure of iHP (hydrodynamic diameter = 35 ± 10 nm). (C and D) The cytotoxicity of iHP in GES-1 and MKN-45P, respectively. | ||
Next, the cytotoxicity of iHP was determined. The effect of different concentrations of iHP solution on cell viability was evaluated using the Cell Counting Kit-8 test. The viability of the cells remained unaffected by increasing concentration of iHP in the cell culture medium, as shown in Fig. 1C and D. Normal gastric epithelial GES-1 cells and gastric cancer MKN-45P cells retained cell viability above 80% even when the iHP concentration increased to 1 mg mL−1. As this result suggested, the iHP was non-toxic to the tested tissues and cells and showed good biocompatible, thus supporting its utilization in clinics.
Selective uptake of iHP by gastric cancer cell
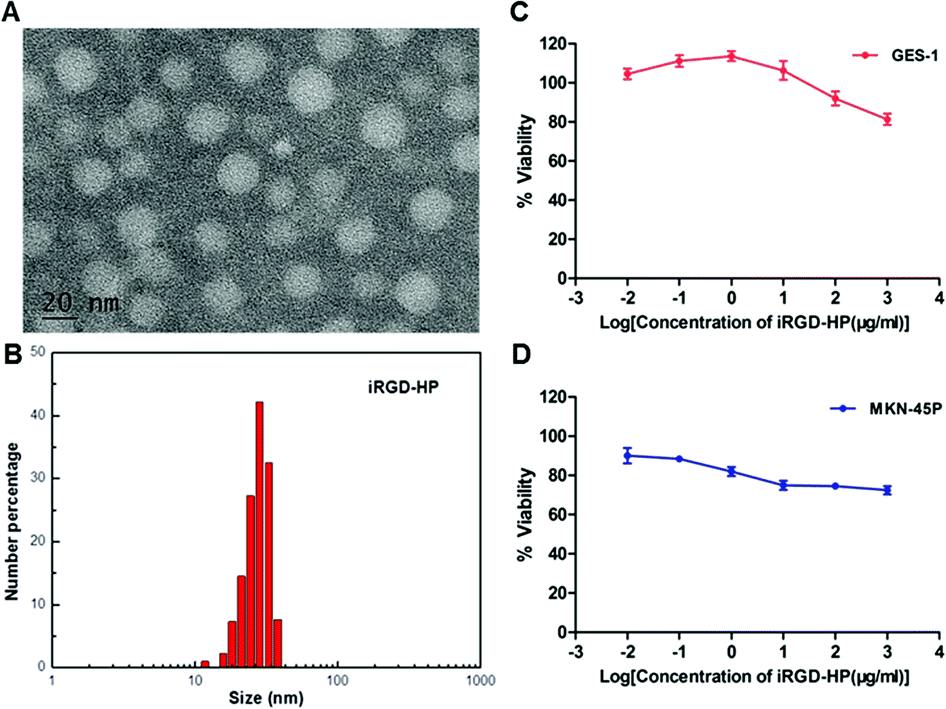
To validate the ability of the iHP to target gastric cancer cells, an iHP-488 fluorescent probe was synthetized (Oregon Green 488: 1.33%), while the gastric cancer cell line MKN-45P with high expression of integrin αv was used as the experimental group. The normal gastric epithelial cell line GES-1, with low expression of integrin αv, served as the control group. The MKN-45P and GES-1 cell line were treated with iHP-488 (0.3 mg mL−1) for 2 h, followed by detection of the fluorescence imaging using the confocal fluorescence microscope. Green fluorescent spots represent iHP-488 fluorescent probe, as shown in Fig. 2A. In comparison with GES-1 cells, it could be observed that there was great amount of green fluorescent spots inside MKN-45P cells which suggested much more uptake of iHP-488 in MKN-45P cells. Flow cytometry results further confirmed the result of confocal fluorescence microscope imaging. The green fluorescence signal intensity of MKN-45P cells was significantly higher than GES-1 cells after incubation with iHP-488 for 2 h and the mean fluorescence intensity of MKN-45P was nearly 2.27 folds higher than GES-1 cells (Fig. 2B and C). It is concluded that iHP-488 shows the capability of targeted delivery toward to MKN-45P cancer cells. Such targeted internalization of iHP-488 to MKN-45P cancer cells can be attributed to the iRGD mediated cellular internalization.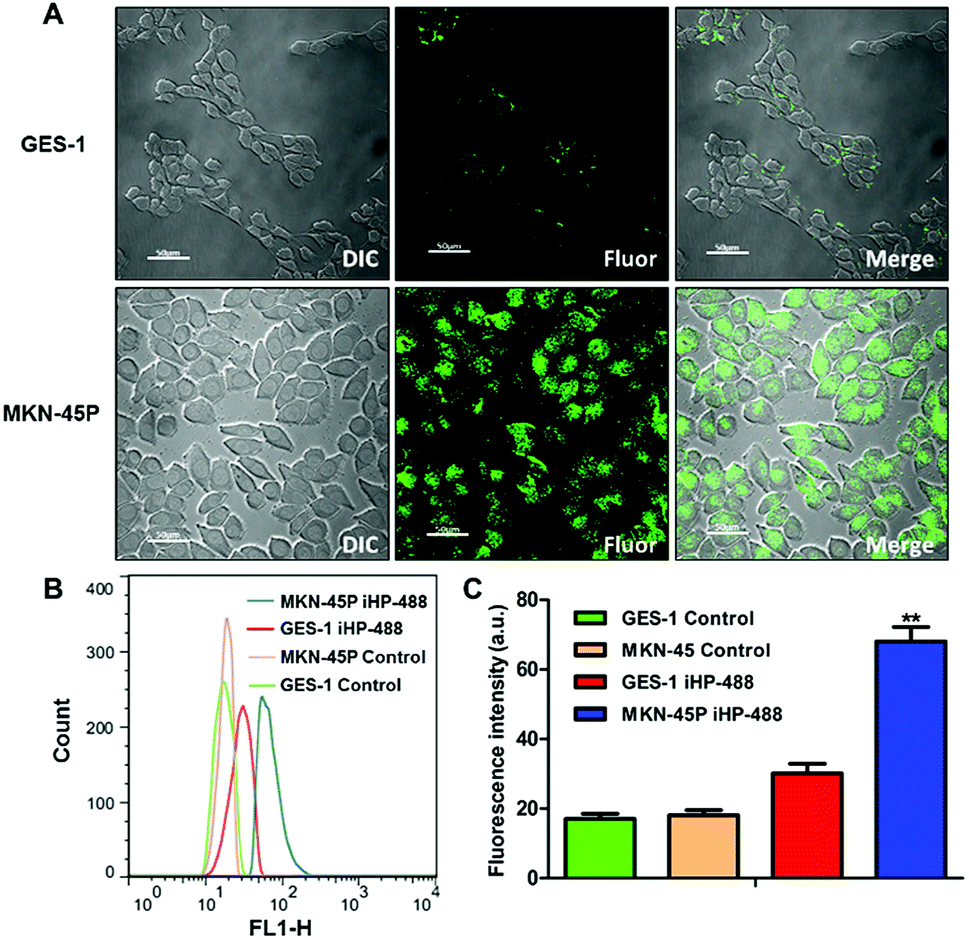 | ||
| Fig. 2 (A) fluorescence microscope imaging for uptake of iHP-488 in GES-1 (Upper), fluorescence microscope imaging for uptake of iHP-488 in MKN-45P (Lower). (B) Flow cytometry results of GES-1 and MKN-45P after incubation with iHP-488 for 2 h. (C) Mean fluorescence intensity of GES-1 and MKN-45P after incubation with iHP-488 for 2 h as displayed. | ||
Selective uptake of iHP by gastric cancer xenografts model
To investigate the targeting ability of the iHP to tumor tissues in vivo, an MKN-45P gastric cancer xenografts model was established. iHP and HP were labeled with cy5.5 (cy5.5: 5.53% and 4.50%, respectively) and injected into nude mice (cy5.5: 2 mg kg−1), and the fluorescence imaging results at different time points and same scale of fluorescence signal were compared and analyzed. Fig. 3A suggested that the fluorescence signal from iHP-cy5.5 began to accumulate in liver and kidney 1 h after injection. The signal then started to be observed in tumor tissue nearly 4 h after injection. The signal in other part of the body declined by 96 h, while the iHP-cy5.5 accumulating in tumor tissues remained a strong fluorescence signal intensity. No specific fluorescence was observed at the tumor sites at any time after injection with HP-cy5.5. To further determine the distribution of iHP-cy5.5 in the organs of mice, the nude mice were sacrificed 96 h after iHP-cy5.5 injected to obtain the organs for fluorescence signal detection (Fig. 3B). Ex vivo fluorescence imaging showed higher signal intensity in tumor tissue, liver and kidney. In comparison, no specific fluorescence was detected in tumor tissue from mice treated with HP-cy5.5, and the kidney was the major organ accumulated. As the results of in vivo imaging validated, iHP showed precise targeting to tumor tissues and complied with its ability to tumor cell in vitro. | ||
| Fig. 3 (A) Fluorescence imaging in vivo of the MKN-45P bearing nude mice postinjection of the HP-cy5.5 and iHP-cy5.5. (B) Ex vivo fluorescence imaging of tumors and organs at 96 h. From up to down, the 1st and 3rd rows are in bright light and the other two are fluorescent images (Lv: liver, Sp: spleen, Kd: kidney, Ht: heart, Lg: lung, Tu: tumor). | ||
In vitro cytotoxicity of DDP, HDDP and iHDDP
To achieve targeted drug-delivery, heparin based nanoparticles for the delivery of cis-diamminedichloroplatinum(II) (DDP, cisplatin) through the coordination between the carboxyl groups and Pt2+ with folate modified heparin were reported in our previous study.19 In the present work, non-targeted nanoparticles heparin-DDP (HDDP) and targeted nanoparticles iHP-DDP (iHDDP) were synthetized for cancer treatment (Scheme 2). The content of platinum was around 32.90% and 29.45%, respectively, as detected by ICP-MS. TEM was employed investigate the morphology of HDDP and iHDDP. As displayed in Scheme 2A, HDDP showed a dispersed spherical morphology with a 40 nm mean diameter. Similar to HDDP, iHDDP also exhibited 40 nm mean diameters (Scheme 2B).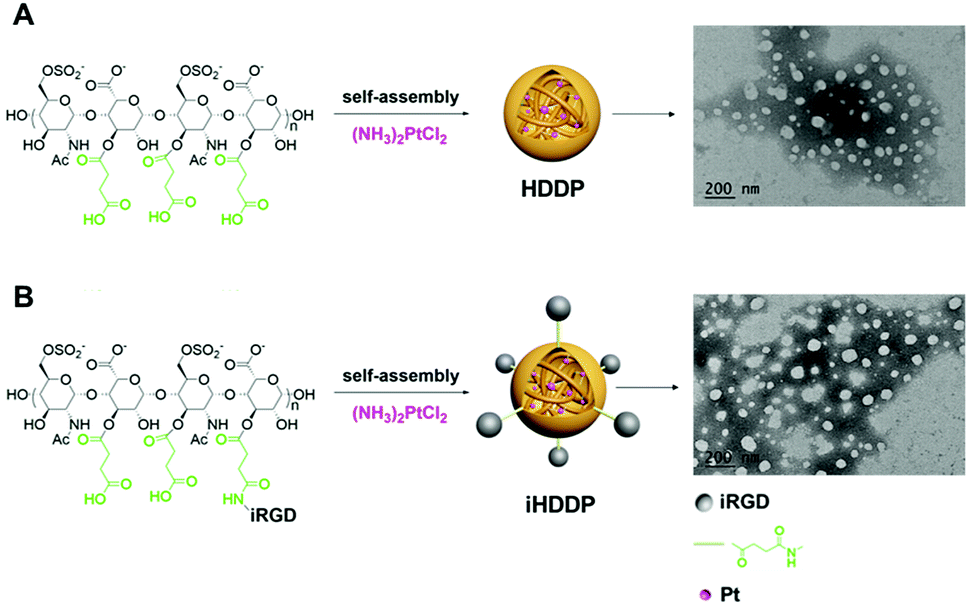 | ||
| Scheme 2 (A) Preparation of heparin-DDP (HDDP) and TEM image of HDDP, (B) preparation of iRGD-heparin-DDP (iHDDP) and TEM image of iHDDP. | ||
The Cell Counting Kit-8 test was performed to assess and compare the in vitro cytotoxicity induced by DDP among the three administration routes with different drug concentrations and treatment times. MKN-45P and GES-1 were cultured with free DDP, HDDP and iHDDP at a concentration of 1 μg mL−1 to 32 μg mL−1 (24 h, 48 h, 72 h, DDP equivalence). Compared to using nanocarriers, free DPP showed the greatest inhibition of cell proliferation to MKN-45P and GES-1 which indicated the side effects of free DDP to normal cell is as significant as its therapeutic effect to cancer cell, especially among high concentration administration (Fig. 4). The inhibition to MKN-45P by iHDDP was significant greater than that of nontargeted HDDP nanoparticles either cultured for 24 h, 48 h or 72 h, as shown in Fig. 4A–C. The IC50 of iHDDP (Pt concentration = 4.459 μg mL−1) was significantly lower than that of HDDP (Pt concentration = 6.654 μg mL−1) when cultured with them for 24 h. Comparatively, no significant difference in the inhibition of cell proliferation was found between iHDDP and HDDP to GES-1, which was low expression of integrin (Fig. 4D–F). Overall, these results clearly suggested that the iHDDP maintained targeted delivery function and had higher inhibitory effect against MKN-45P.
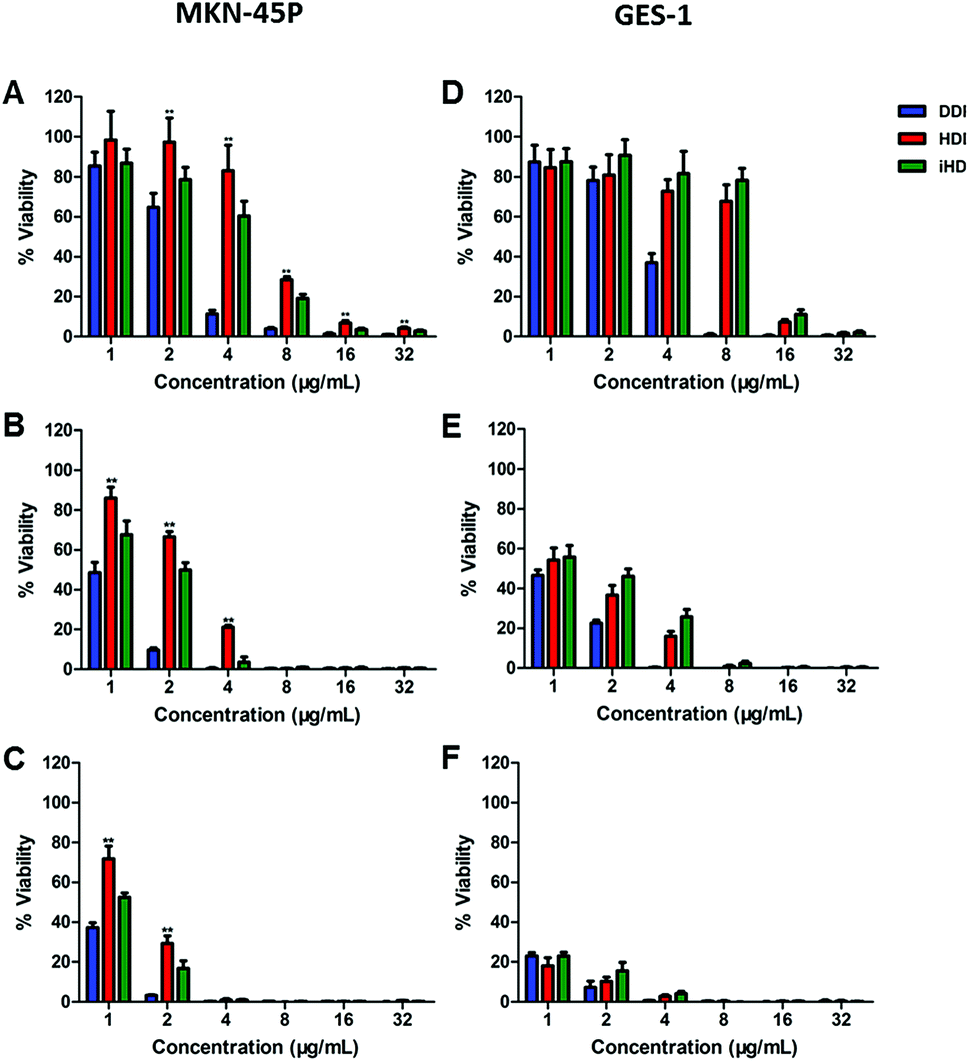 | ||
| Fig. 4 (A–C) In vitro cytotoxicity of DDP, HDDP and iHDDP in MKN-45P cells at different concentrations after 24 h, 48 h and 72 h, respectively. (D–F) in vitro cytotoxicity of DDP, HDDP and iHDDP in GES-1 cells at different concentrations after 24 h, 48 h and 72 h, respectively. The date are shown as mean ± SD, *P < 0.05, **P < 0.01. | ||
In vivo antitumor efficacy and safety evaluation
In this work, nude mice bearing MKN-45P tumors served as animal models to evaluate the efficacy of iHDDP in antitumor treatment. Mouse samples fell into four groups with six mice per group, each of which had a cell growth rate of 80 mm3 in average. A-saline-treated group served as the control group, yet other three groups were intravenously injected with free DDP, HDDP and iHDDP (Pt: 2.5 mg kg−1, 7 iv doses, 3 day intervals). The saline group showed rapid tumor growth, and tumor volume increased 29.8-fold on day 21 in comparison with day 0, as suggested in Fig. 5A. The free DDP and iHDDP showed similar levels of tumor inhibition (tumor volume: 770.03 ± 215.88 mm3 and 781.1 ± 156.99 mm3, respectively), yet the nontargeted HDDP showed much less efficacy (tumor volume: 1677.83 ± 1022.8 mm3). As a result, by the end of this test, tumor volumes increased approximately 20.49-fold, 9.79- and 10.89-fold for the HDDP, DDP and iHDDP groups, respectively, as compared with initial volumes at day 0. This suggested that iHDDP showed excellent therapeutic effects to the solid tumors (Fig. 5A and B). | ||
| Fig. 5 The effects of DDP, HDDP and iHDDP treatment on tumor growth (A and B) and body weight (C) of MKN-45P bearing nude mice treated with them (Pt: 2.5 mg kg−1, 7 iv doses, 3 day intervals). The date are shown as mean ± SD, *P < 0.05, **P < 0.01. | ||
The group receiving the free DDP showed a remarkable loss of body weight, with nearly 9.7%weight loss by the 12th day (p = 0.047) and 22.7% by the 21 day (p < 0.01) after treatment. Yet the mice treated with HDDP and iHDDP did not show any obvious weight loss compared with the saline group (Fig. 5C).
To evaluate the in vivo safety, heart, liver, spleen, lung, kidney and blood were collected after 21 days of treatment since the tumor volumes reached 2500 mm3 in the saline group.
Serum levels of alkaline phosphatase (ALT) and alanine aminotransferase (AST) were employed to assess liver function (Fig. 6A and B). The ALT levels did not show obvious difference in any of the treated groups in compassion with the control group. The AST levels in DDP-treated group (468.67 ± 198.62 U L−1) was notably different with the control group (255.33 ± 103.13 U L−1, P = 0.02), or HDDP-treated group (229.08 ± 41.52 U L−1, P = 0.008) and iHDDP-treated group (281.54 ± 65.62 U L−1, P = 0.03). Yet the AST levels of HDDP-treated group and iHDDP-treated group were slightly higher than the control group (P = 0.29 and 0.31, respectively). Though the level of AST in DDP-treated group was increased (Fig. 6E), the histopathological analysis showed no obvious changes in any treated group. We consider that free DDP is not toxic enough to cause severe liver impairment which can be observed in histopathology. But the higher level of AST in DDP-treated group indicated that hepatic cell damage actually occurred in these nude mice.
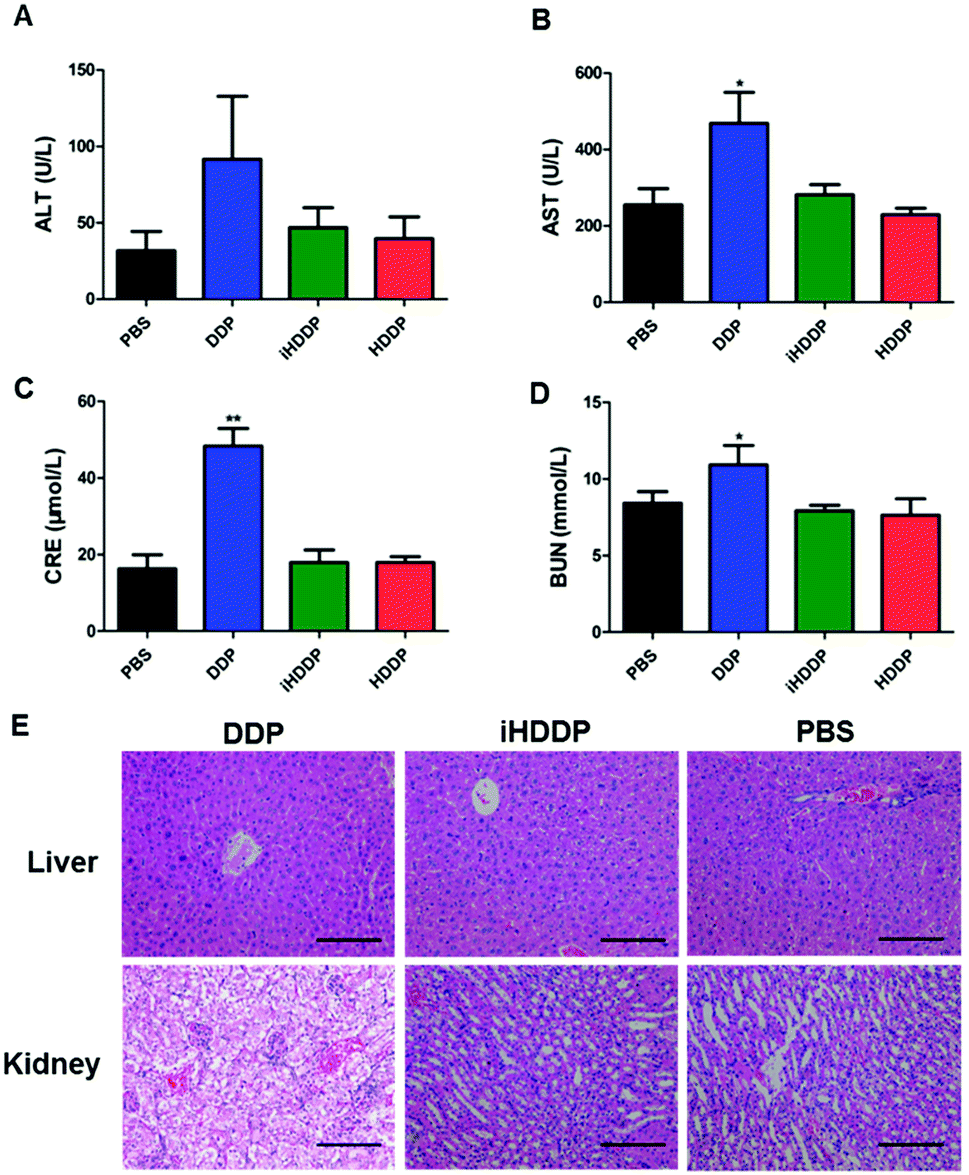 | ||
| Fig. 6 Side effects comparison of DDP, HDDP and iHDDP treatment to liver and kidney (Pt: 2.5 mg kg−1, 7 iv doses, 3 day intervals). (A) Serum levels of alkaline phosphatase (ALT); (B) serum levels of alanine aminotransferase (AST); (C) serum blood creatinine (CRE); (D) urea nitrogen (BUN); (E) histopathological results (×200). Scale bar, 100 μm. The date are shown as mean ± SD, *P < 0.05, **P < 0.01. | ||
Serum blood creatinine (CRE) and urea nitrogen (BUN) levels were employed to assess renal function (Fig. 6C and D), the DDP-treated mice showed an obviously higher concentration of CRE (48.33 ± 11.25 μmol L−1) than the control group (16.25 ± 9.05 μmol L−1 p < 0.001), while the CRE levels of HDDP (17.91 ± 3.68 μmol L−1) and iHDDP (17.91 ± 8.13 μmol L−1) slightly increased in comparison with the control group (P = 0.34 and 0.37, respectively). Serum BUN levels also showed increase in DDP-treated group (10.92 ± 3.12 mmol L−1) as compared with the control group (8.42 ± 1.84 mmol L−1), though the difference was insignificant (P = 0.06). Yet the BUN levels of HDDP-treated group (7.63 ± 2.64 mmol L−1) and iHDDP-treated group (7.92 ± 0.90 mmol L−1) showed obvious changes in comparison with the control group (P = 0.038 and 0.024, respectively). As the Histopathology study suggested, the changes of glomeruli structure was not noticeable in DDP-treated group. Yet the tubular epithelial cells became swollen and desquamate, even tubular necrosis. Conversely, the HDDP-treated group and iHDDP-treated group showed no significant renal toxicity in histopathology as compared with the control group (Fig. 6E).
Discussion
DDP is a potent chemotherapeutic drug having shown efficacy on GC therapy. As limited in ability to target to tumor tissues, the free drug causes severe systemic toxicity during the DDP treatment.20 Various nanocarriers have been developed to increase the drug delivery to tumor and decrease side-effects. Some nanocarriers classes, for example, lipid nanocarriers, inorganic nanocarriers and nanotubes, are limited by their intrinsic undesirable properties, though they have shown some success in drug delivery. There are many different liposomal formulations of platinum drugs have been explored currently, including cisplatin-liposome formulation.21 But the application of these liposomal formulations is limited by several aspects. For example, poor water solubility and low lipophilicity of cisplatin limits it efficiently encapsulated in a liposome. In addition, the liposomal formulations are easy to be removed from circulation by the macrophages of the reticuloendothelial system, thence the liposomes generally need to be modified with polyethylene glycol (PEG) to improve their stability.22 However, the non-biodegradability and immunological response of PEG need to be considered. Except for liposomal formulations, several carbon nanotubes (CNTs) are used in drug delivery of DDP, while the completely insoluble of pristine nanotubes, low biocompatibility and immunogenicity limitation also restrict their application.23,24 Despite many nanocarriers are developed to improve the biosafety and efficacy of DDP, few technologies have been clinically approved for platinum chemotherapeutics and the majority of them tend to accumulate within tumors due to the EPR effect which is considered as a way of passive tumor-targeting drug delivery. Therefore, the creation of a biocompatible and active tumor-targeting nanocarrier for drug delivery remains challenging.Heparin, as a commonly used drug in clinics approved by FDA, has the anti-inflammatory, anti-allergic, even anti-tumor properties in addition to the anticoagulant property. For the inherent biocompatibility and biodegradation of heparin, in previous study, we have developed a novel nanocarrier based on heparin biopolymers. Through modifying the polymers with folate targeting molecules, DDP targeting delivery was achieved to folate receptor over-expression cell lines.19 Many malignant cells merely show a mild increase expression of folate receptor, such as bladder cancer, gastric cancer,25 though folate receptor is a common surface marker on the tumor cell membrane. Except for the folate receptor, there are still many others type of targeted molecule overexpressed on the tumor cell membrane, such as EGFR and HER-2. If targeted, these molecules also could achieve drug target-delivery. Integrin are transmembrane receptors that mediate cell interactions with ECM glycoproteins, which have been identified highly expressed on many tumor cell membrane26 and considered a good targeted molecule for tumor-targeted drug delivery, inclusive of GC.27–29
iRGD peptide, as an armillary αv integrin ligand, is known to induce an excellent tumor-specific function, which not only completely binds to αv integrin highly expressed on tumor cells and tumor-associated vascular endothelium, but also improves vascular and tissue penetration of drugs.8 In this work, an integrin-targeted nanocarrier was synthesized using heparin conjugated with iRGD. Based on the good biocompatibility of heparin and iRGD, we propose that iRGD-heparin has no biotoxicity. To prove the biosafety properties of the nanocarriers, an in vitro study was conducted to determine their effect on tumor cells and normal cells. As the results suggested, iRGD-heparin nanocarriers at various concentrations did not inhibit the growth of any of these cell lines. And it is noteworthy that, to GES-1 cells, the cell vitality increased at concentration on 0.01 μg mL−1 to 10 μg mL−1 incubated with iHP, which is probably caused by the cell growth factor activation ability of heparin, such as VEGF, heparin-binding EGF (HB-EGF), bFGF.30–32 The size of drug delivery system is a key factor for designing drug carriers as well. Particles smaller than 10 nm are often cleaned from blood stream by renal filtration or via extravasation from tumors.33 However, particles larger than 200 nm are at risk of being filtered out by the liver or spleen or cleaned by the bone marrow.34 Nanoparticles with sizes of 10–100 nm are considered able to avoid renal clearance and accumulate at tumor sites after prolonged circulation.35 Our nanocarrier iHP and targeted nanoparticle iHDDP, exhibiting the mean sizes of 20 nm and 40 nm respectively, might therefore provide an appropriate biodistribution. After the iHP nanocarriers were synthesized, we evaluated the tumor targeting ability of iHP in vitro and in vivo. The mean fluorescence intensity of MKN-45P cells is significant higher than GES-1 cells which indicated iHP can be selectively uptaken by integrin αv-overexpressed gastric cancer cell. Interestingly, we observed weak fluorescence on the membrane of GES-1 cell, while strong fluorescence inside of MKN-45P cell. This might be due to the internalization function of iRGD as previous reported.13–15 In vivo, it was shown that iRGD-heparin nanocarriers also significantly improved the delivery of imaging agents into solid tumor.
Similar to the HFDDP that deliver more DDP to folate overexpressed tumor cells than HDDP, we considered that iHDDP have better antitumor effects through the targeted ability of iRGD to integrin overexpressed on GC cells. To validate our supposition, we assessed their effect in vitro first. As the results suggested, the iHDDP significantly inhibit more tumor cells in different treating times and doses in comparison with HDDP. Unexpectedly, compared with HDDP and iHDDP, the free DDP showed the greatest cytotoxicity to GC cells and normal cells. This is probably attributed to the slow release of DDP from the nanocarriers, while the free DDP rapidly act once it diffuses into intracellular. Similar results have been reported in other in vitro studies of nanoparticle drug-delivery systems.36 To confirm the antitumor effects of iHDDP to solid tumor, we further assessed it in vivo, the iHDDP-treated group showed more obvious tumor inhibition than HDDP-treated group. The possible reason might be that the iHDDP both homed to and penetrated through solid tumor rely on the target and internalization properties of iRGD,13 yet the untargeted HDDP accumulated merely in the tumor periphery through EPR effects. As the DDP release of HDDP is slower than free drug diffusion into cell membrane, HDDP cannot show efficiently tumor inhibition like free DDP. Although iHDDP also has a slow drug release, the antitumor effects of it is still similar to free DPP due to iRGD-mediated target and internalization properties. Our study suggested that target-delivery of DDP by iRGD-heparin significantly reduce the relevant toxicity in liver and kidney. It is doubted that as the DDP slowly released from the nanocarriers, the drug concentration is not high enough to cause organ damage when they are metabolized by liver and kidney. This suggests that iRGD-heparin nanocarriers may be employed to decrease side effects while maintaining drug efficacy.
Conclusions
In conclusion, we have presented that conjugated heparin with iRGD, a biocompatible and biodegraded drug-delivery system is highly efficient in targeting chemotherapeutic drug delivery. Surface modification of heparin with iRGD increased the intracellular uptake of the Oregon Green 488 and cy5.5 in GC cells and solid tumor, respectively. Furthermore, iRGD-heparin-DDP showed higher antitumor efficacy than untargeted nanoparticles in vitro and in vivo, while significantly reducing the toxicity of DDP in liver and kidney. Since the iRGD and heparin hold great promise for clinical applications with a low toxicity profile, iRGD-heparin nanocarrier has great future potential of application in gastric cancer therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from State Commission of Science & Technology of China (2016YFC0104100), Jiangsu Province Science & Technology Department (SBE2016750057), the Fundamental Research Funds for the Central Universities (021314380124), Key Research Plan and Social Development Project of Jiangsu Province, China (BE2016603) and the National Natural Science Foundation of China (No. 81372364).References
- E. Van Cutsem, X. Sagaert, B. Topal, K. Haustermans and H. Prenen, Lancet, 2016, 388, 2654–2664 CrossRef.
- W. Q. Chen, R. S. Zheng, P. D. Baade, S. W. Zhang, H. M. Zeng, F. Bray, A. Jemal, X. Q. Yu and J. He, Ca-Cancer J. Clin., 2016, 66, 115–132 CrossRef PubMed.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef PubMed.
- A. A. Bhirde, V. Patel, J. Gavard, G. F. Zhang, A. A. Sousa, A. Masedunskas, R. D. Leapman, R. Weigert, J. S. Gutkind and J. F. Rusling, ACS Nano, 2009, 3, 307–316 CrossRef PubMed.
- S. L. Luo, E. L. Zhang, Y. P. Su, T. M. Cheng and C. M. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef PubMed.
- S. Lee, J. Xie and X. Y. Chen, Chem. Rev., 2010, 110, 3087–3111 CrossRef PubMed.
- N. Graf, D. R. Bielenberg, N. Kolishetti, C. Muus, J. Banyard, O. C. Farokhzad and S. J. Lippard, ACS Nano, 2012, 6, 4530–4539 CrossRef PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, D. R. Greenwald and E. Ruoslahti, Science, 2010, 328, 1031–1035 CrossRef PubMed.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, O. M. Girard, D. Hanahan, R. F. Mattrey and E. Ruoslahti, Cancer Cell, 2009, 16, 510–520 CrossRef PubMed.
- K. Uhland, Cell. Mol. Life Sci., 2006, 63, 2968–2978 CrossRef PubMed.
- T. Teesalu, K. N. Sugahara, V. R. Kotamraju and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16157–16162 CrossRef PubMed.
- Y. P. Ye, L. Zhu, Y. Ma, G. Niu and X. Y. Chen, Bioorg. Med. Chem. Lett., 2011, 21, 1146–1150 CrossRef PubMed.
- C. Puig-Saus, L. A. Rojas, E. Laborda, A. Figueras, R. Alba, C. Fillat and R. Alemany, Gene Ther., 2014, 21, 767–774 CrossRef PubMed.
- L. Simon-Gracia, H. Hunt, P. Scodeller, J. Gaitzsch, V. R. Kotamraju, K. N. Sugahara, O. Tammik, E. Ruoslahti, G. Battaglia and T. Teesalu, Biomaterials, 2016, 104, 247–257 CrossRef PubMed.
- L. Wang, X. Xie, D. Liu, X. B. Fang, P. Li, J. B. Wan, C. W. He and M. W. Chen, RSC Adv., 2016, 6, 28331–28342 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef PubMed.
- X. Y. Yang, H. L. Du, J. Y. Liu and G. X. Zhai, Biomacromolecules, 2015, 16, 423–436 CrossRef PubMed.
- J. Q. Wang, D. S. Ma, Q. Lu, S. X. Wu, G. Y. Lee, L. A. Lane, B. Li, L. Quan, Y. Q. Wang and S. M. Nie, Nanoscale, 2015, 7, 15185–15190 RSC.
- J. Q. Wang, G. Y. Lee, Q. Lu, X. H. Peng, J. X. Wu, S. Y. Wu, B. A. Kairdolf, S. M. Nie, Y. Q. Wang and L. A. Lane, Bioconjugate Chem., 2017, 28, 1351–1355 CrossRef PubMed.
- T. Lazarevic, A. Rilak and Z. D. Bugarcic, Eur. J. Med. Chem., 2017, 142, 8–31 CrossRef PubMed.
- G. P. Stathopoulos, T. Boulikas, M. Vougiouka, G. Deliconstantinos, S. Rigatos, E. Darli, V. Viliotou and J. G. Stathopoulos, Oncol. Rep., 2005, 13, 589–595 Search PubMed.
- S. C. White, P. Lorigan, G. P. Margison, J. M. Margison, F. Martin, N. Thatcher, H. Anderson and M. Ranson, Br. J. Cancer, 2006, 95, 822–828 CrossRef PubMed.
- A. Guven, I. A. Rusakova, M. T. Lewis and L. J. Wilson, Biomaterials, 2012, 33, 1455–1461 CrossRef PubMed.
- G. Pastorin, Pharm. Res., 2009, 26, 746–769 CrossRef PubMed.
- Y. G. Assaraf, C. P. Leamon and J. A. Reddy, Drug Resist. Updates, 2014, 17, 89–95 CrossRef PubMed.
- J. S. Desgrosellier and D. A. Cheresh, Nat. Rev. Cancer, 2010, 10, 9–22 CrossRef PubMed.
- F. Danhier, A. Le Breton and V. Preat, Mol. Pharm., 2012, 9, 2961–2973 CrossRef PubMed.
- D. Arosio and C. Casagrande, Adv. Drug Delivery Rev., 2016, 97, 111–143 CrossRef PubMed.
- A. Lasorsa, M. Losacco, R. M. Iacobazzi, L. Porcelli, A. Azzariti, G. Natile and F. Arnesano, RSC Adv., 2016, 6, 29229–29236 RSC.
- N. Gunawardhana, S. Jang, Y. H. Choi, Y. A. Hong, Y.-E. Jeon, A. Kim, H. Su, J.-H. Kim, Y.-J. Yoo, D. S. Merrell, J. Kim and J.-H. Cha, Front. Cell. Infect. Microbiol., 2017, 7, 541 CrossRef PubMed.
- M. Forster and B. Mulloy, Biochem. Soc. Trans., 2006, 34, 431–434 CrossRef PubMed.
- Y. Inoue, M. Shimazawa, S. Nakamura, S. Takata, Y. Hashimoto, H. Izawa, T. Masuda, K. Tsuruma, T. Sakaue, H. Nakayama, S. Higashiyama and H. Hara, Arterioscler., Thromb., Vasc. Biol., 2018, 38, 174–185 CrossRef PubMed.
- Y. Liu, J. Tan, A. Thomas, D. Ou-Yang and V. R. Muzykantov, Ther. Delivery, 2012, 3, 181–194 CrossRef.
- Y. Li, Y. P. Lian, L. T. Zhang, S. M. Aldousari, H. S. Hedia, S. A. Asiri and W. K. Liu, Interface Focus, 2016, 6, 15 Search PubMed.
- S. M. Nie, Nanomedicine, 2010, 5, 523–528 CrossRef PubMed.
- J. K. Lu, X. X. Chuan, H. Zhang, W. B. Dai, X. L. Wang, X. Q. Wang and Q. Zhang, Int. J. Pharm., 2014, 471, 525–535 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05071f |
| This journal is © The Royal Society of Chemistry 2018 |