
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric ring-opening reaction of meso-epoxides with aromatic amines using homochiral metal–organic frameworks as recyclable heterogeneous catalysts†
Koichi Tanaka *a,
Maya Kinoshitaa,
Jun Kayaharaa,
Yutaro Uebayashia,
Kazusada Nakajia,
Maja Morawiakb and
Zofia Urbanczyk-Lipkowskab
*a,
Maya Kinoshitaa,
Jun Kayaharaa,
Yutaro Uebayashia,
Kazusada Nakajia,
Maja Morawiakb and
Zofia Urbanczyk-Lipkowskab
aDepartment of Chemistry and Materials Engineering, Faculty of Chemistry, Materials and Bioengineering, Kansai University, Suita, Osaka 564-8680, Japan. E-mail: ktanaka@kansai-u.ac.jp; Fax: +81-06-6368-0861; Tel: +81-06-6368-0861
bInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warszawa, Poland. E-mail: ocryst@icho.edu.pl; Fax: +48 22 6326681; Tel: +48 22 3432207
First published on 6th August 2018
Abstract
An efficient asymmetric ring-opening (ARO) reaction of meso-epoxides with aromatic amines catalysed by a series of homochiral metal–organic frameworks (MOFs) was carried out. Excellent results (up to 95% ee) for the ARO of cyclohexene oxide with several aromatic amines were achieved with a homochiral MOF derived from the ligand (R)-2,2′-dihydroxyl-1,1′-binaphthalene-5,5′-dicarboxylic acid. Furthermore, homochiral MOFs based on (R)-2,2′-dihydroxy-1,1′-binaphthyl-4,4′-di(4-benzoic acid) and (R)-2,2′-diethoxy-1,1′-binaphthyl-4,4′-di(5-isophthalic acid) catalysed ARO reactions of cis-stilbene oxide with 1-naphthylamine in high yield (up to 95%) and excellent enantioselectivity (up to 97%) of the β-amino alcohol. The MOF catalysts were recoverable and recyclable with retention of their performance.
Introduction
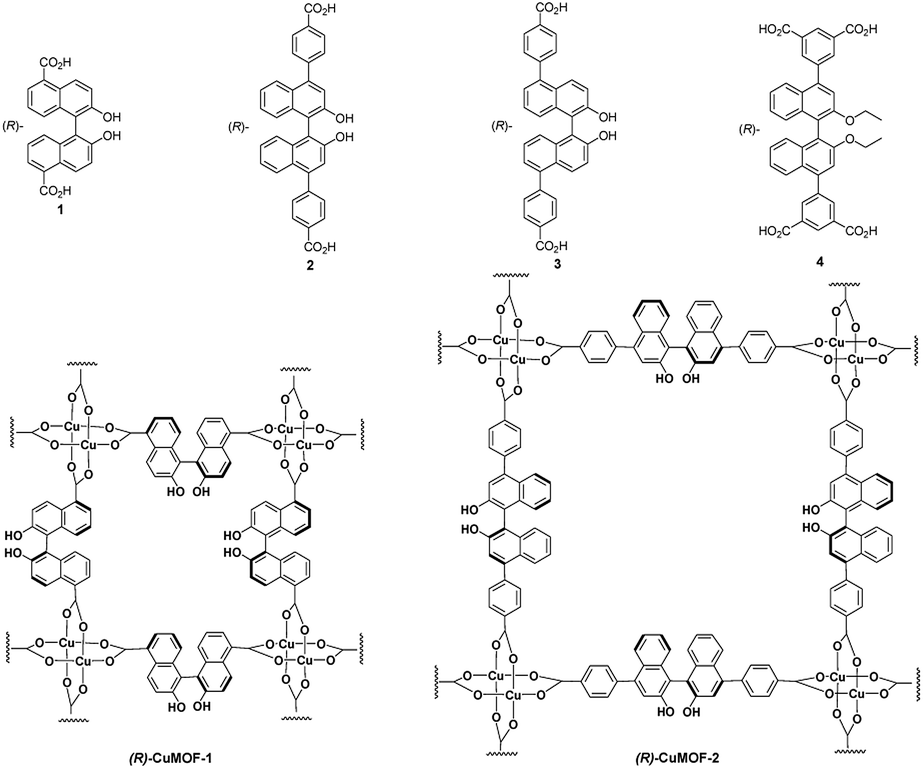
Chiral β-amino alcohols are useful building blocks for biologically active natural products such as therapeutic agents, β-blockers, insecticidal agents, antimalarial agents and oxazolines,1 as well as important auxiliaries and ligands in asymmetric synthesis.2 Several methods for the asymmetric synthesis of these compounds have been developed. Among them, the catalytic asymmetric ring opening (ARO) of meso-epoxides with amines is one of the most important and straightforward strategies to synthesize optically enriched β-amino alcohols.3 Several reports have appeared for the synthesis of β-amino alcohols through the ARO of meso-epoxides with anilines.4 However, homogeneous catalysts have been widely applied. Porous metal–organic frameworks (MOFs) with high surface areas and thermal stabilities have attracted much attention recently owing to their versatile applications in storage,5 separation,6 sensing,7 and catalysis.8 In particular, chiral MOFs, which are assembled using chiral organic ligands and metal ions, are of interest for applications in enantioselective separation and catalysis, which are important for the pharmaceutical industry. In 2008, we reported the synthesis of (R)-CuMOF-19a by treating (R)-2,2′-dihydroxyl-1,1′-binaphthalene-5,5′-dicarboxylic acid 1 with Cu(NO3)2, and investigated its catalytic activity toward ARO reactions of cyclohexene oxide with some aniline derivatives to furnish the corresponding β-amino alcohols in moderate enantioselectivities ranging from 43–51% ee.9a Recently, we synthesized (R)-CuMOF-2 from (R)-2,2′-dihydroxyl-1,1′-binaphthalene-4,4′-di(4-benzoic acid) 2 and its application to the efficient heterogeneous catalyst for the asymmetric Diels–Alder reaction of isoprene with N-ethyl maleimide has been reported.10 We also constracted (R)-CuMOF-3 from (R)-2,2′-dihydroxyl-1,1′-binaphthalene-5,5′-di(4-benzoic acid) 3, which exhibited high catalytic activity in the Diels–Alder reaction of acrolein and 1,3-cyclohexadiene.11In this study, we report the synthesis of a novel homochiral (R)-ZnMOF-4 from (R)-2,2′-dihydroxy-1,1′-binaphthyl-4,4′-di(5-isophthalic acid) 4 and the application of (R)-CuMOF-1, (R)-CuMOF-2, (R)-CuMOF-3 and (R)-ZnMOF-4 as recyclable heterogeneous catalysts for the ARO reaction of meso-epoxides with some aromatic amines to form optically active β-amino alcohols.
Results and discussion
Synthesis of (R)-CuMOF-1, (R)-CuMOF-2 and (R)-CuMOF-3
(R)-CuMOF-1,9 (R)-CuMOF-210 and (R)-CuMOF-311 were synthesized by the methods reported earlier.Synthesis and characterization of a new (R)-ZnMOF-4
Chiral organic ligand (R)-2,2′-dihydroxy-1,1′-binaphthyl-4,4′-di(5-isophthalic acid) 4 was synthesized by Suzuki cross-coupling of dimethyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isophthalate and (R)-4,4′-dibromo-2,2′-diethoxy-1,1′-binaphthyl (prepared from (R)-4,4′-dibromo-2,2′-dihydroxy-1,1′-binaphthyl),12 followed by hydrolysis and acidification. (R)-ZnMOF-4 was obtained as pale-yellow prisms after a solvothermal reaction of the chiral organic linker (R)-4 and Zn(NO3)2·6H2O in a mixed solvent (N,N-dimethylacetamide–EtOH) at 110 °C for 2 days. The structure was characterized by IR spectroscopy (Fig. S1†), thermogravimetric analysis (Fig. S2†), solid CD spectroscopy (Fig. S3†). The observed powder XRD patterns (Fig. S4†) are the same as the simulated ones (Fig. S5†), demonstrating phase purity of the bulk samples. N2 adsorption isotherm (Fig. S6†) and BET plot (Fig. S7†) of (R)-ZnMOF-4 were also determined.
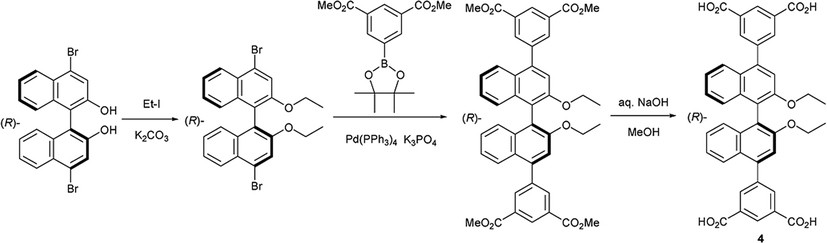
X-ray diffraction analysis revealed that a rigid metal–organic framework was constructed around two pentacoordinated Zn(II) cations with the nearest Zn1⋯Zn2 distance 3.384(2) Å, which were bridged by three carboxy groups from three different chiral ligands (Fig. 1). The Zn1–O distances were 1.977(4) Å–2.062(6) Å, and the Zn2–O lengths span was in the range of 1.940(5)–2.476(5) Å. A trigonal bipyramidal coordination sphere of Zn1 cation was completed by another monodentate carboxylate, one water and one C, whereas the coordination sphere of the Zn2 had as geometry of a very distorted trigonal bipyramid and was completed by one monodentate carboxy groups (Fig. S6†). Other structural features that may be essential for the catalytic process is the presence of three N,N-dimethylacetamide molecules in the structure – one coordinated to Zn1 and two trapped in the crystal lattice by a few C–H⋯O hydrogen bonds. The crystal contained two N,N-dimethyalcetamide, one water and one ethanol molecules, all with half occupancy. A low R-factor (0.0664), reasonable goodness-of-fit (1.08) and small residual density (0.83 e) suggested that no additional solvent molecules were present in crystals. A collection of separate voids present in the unit cell of (R)-ZnMOF-4 occupies only ca. 275 Å3 (8.6%) of the total unit cell volume (Fig. S7†).
 | ||
| Fig. 1 X-ray structure of (R)-ZnMOF-4, shown down a-axis. Color codes: Zn(II) – green, O – red, N – blue, C, H – grey. | ||
When all solvent molecules excluding one scaffold-firming and coordinated to Zn(II) N,N-dimethylacetamide molecule were removed the structure became more porous. In such case, the calculated volume of Connolly's surface13 that was accessible for substrates (solvent) adsorption–desorption process was enhanced to 1329 Å3, i.e. 41% of the unit cell volume. The analysis of a spatial distribution of possible transportation pathways in the chiral (R)-ZnMOF-4 revealed that there are two channels present in the crystals that are oriented along a-axis. Their estimated cross-sections are: for channel 1 – 4.7 × 7.7 Å and for channel 2 – 5.6 × 5.6 Å (Fig. 2 and S6†). It is obvious from these estimations that all solvated epoxides as well as aniline derivatives fit well to both channels. Moreover, both channels have large chiral side compartments where reaction might proceed and products might be temporarily stored. Molecular dimensions of the reaction product 8a calculated from X-ray data (CCDC 281038)14 suggest that such molecules might migrate down the channels, particularly when raising temperature facilitates higher motions of both, molecules constructing MOF scaffold and reaction products. Efficient catalyst turnover and lack of strong H-bond donor or acceptor in solid (R)-ZnMOF-4 suggests that probably N,N-dimethylacetamide molecule coordinated to Zn(II) does not contribute significantly to the stability of the MOF's scaffold and can be replaced by epoxide molecule in catalytic process.
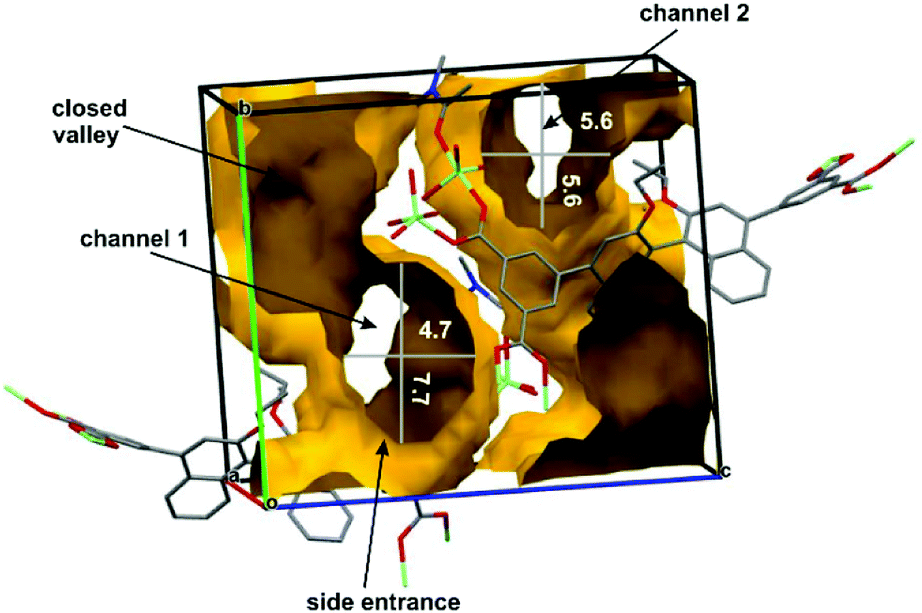 | ||
| Fig. 2 Connolly's surface calculated from X-ray structure of (R)-ZnMOF-4 showing diameters and topology of two channels. | ||
Asymmetric ring-opening reaction
First, we evaluated the catalytic asymmetric ring opening reaction of cyclohexene oxide 5b with N-methylaniline using evacuated (R)-CuMOF-1 as a heterogeneous chiral catalyst. When an equimolar mixture of cyclohexene oxide (0.5 mmol) and N-methylaniline (0.5 mmol) was stirred in various solvents (1 mL) in the presence of (R)-CuMOF-1 (10 mol%) at 50 °C for 48 h, optically active β-amino alcohol 6b was formed in the yields shown in Table 1. Among the various solvents tested, only CHCl3 furnished the desired product 6b in good yield (86%) with excellent enantioselectivity (85%) (Table 1, entry 2). MeOH, THF or toluene led to almost racemic 6b in lower yields with poor enantioselectivities (Table 1, entries 1, 3 and 4). The A-B-type stacking mode of (R)-CuMOF-19a makes it likely that the catalytic reaction takes place on the MOF surface. MeOH and THF solvents interfere the coordination between epoxide and the MOF metal center. On the other hand, toluene molecules might interact with naphthol group through CH–π and π–π interactions and prevent the access of the substrates to the copper-paddlewheel unit affording only the racemic product.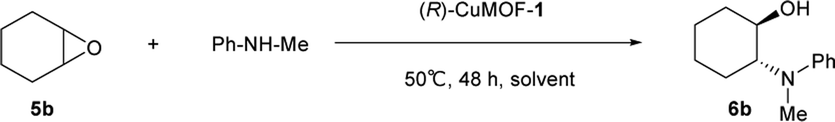
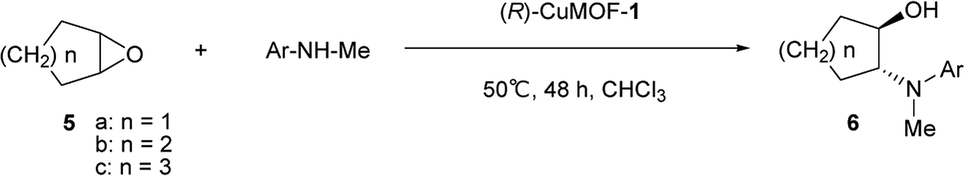
To show the scope of this reaction, we extended it to several cyclic epoxides and N-methylanilines using (R)-CuMOF-1 as a catalyst (Table 2). The reaction worked well on five-, six-, and seven-membered epoxides with N-methylaniline to form the corresponding β-amino alcohols 6a–6c with moderate to good enantioselectivities (Table 2, entries 1–3). Of the substituted N-methylanilines, N-methyl-p-toluidine, N-methyl-p-anisidine and N-methyl-p-chloroaniline furnished the desired β-amino alcohols 6f–6h in good yields (85–94%) with excellent enantioselectivities (92–95%) (Table 2, entries 6–8). In contrast, N-methyl-m-toluidine gave the product 6e in good yield (90%) but with lower enantioselectivity (78%). The N-methyl-o-toluidine afforded 6d in lower efficiency (19% yield) with moderate enantioselectivity (43%), maybe because of the steric constraint (Table 2, entry 4).
|
|||||
|---|---|---|---|---|---|
| Entry | Epoxide | Ar | Product | Yield (%) | Ee (%) |
| a HPLC column: Chiralcel OD-H, eluent: hexane/i-PrOH = 98/2, flow rate: 0.3 mL min−1, detection: UV 254 nm.b HPLC column: Chiralcel OB-H, eluent: hexane/i-PrOH = 99/1, flow rate: 0.5 mL min−1, detection: UV 254 nm.c HPLC column: Chiralcel OB-H, eluent: hexane/i-PrOH = 95/5, flow rate: 1.0 mL min−1, detection: UV 254 nm.d (1S,2S)-isomer.e (1R,2R)-isomer. | |||||
| 1 | 5a | Ph | 6ad | 93 | 68a |
| 2 | 5b | Ph | 6be | 86 | 85a |
| 3 | 5c | Ph | 6c | 20 | 89a |
| 4 | 5b | 2-MePh | 6d | 19 | 43a |
| 5 | 5b | 3-MePh | 6e | 90 | 78a |
| 6 | 5b | 4-MePh | 6f | 94 | 92b |
| 7 | 5b | 4-MeOPh | 6g | 87 | 95c |
| 8 | 5b | 4-ClPh | 6h | 85 | 92c |
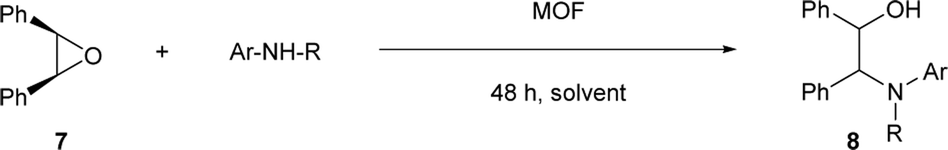
Next, we investigated the aminolysis of cis-stilbene oxide with aniline, N-methylaniline and 1-naphthylamine by using (R)-CuMOF-1, (R)-CuMOF-2, (R)-CuMOF-3 or (R)-ZnMOF-4 as heterogeneous catalysts (Table 3). When an equimolar mixture of cis-stilbene oxide and aniline were stirred in various solvents in the presence of evacuated chiral MOFs at several temperatures for 48 h, optically active β-amino alcohols 8a–c were formed in the yields shown in Table 3. In the presence of (R)-CuMOF-1 as a catalyst, cis-stilbene oxide 7 reacted with aniline and N-methylaniline at 70 °C for 48 h afforded 8a and 8b in 19% and 60% ee, respectively (Table 3, entries 1 and 2). The reaction of cis-stilbene oxide 7 with bulky 1-naphthylamine gave 8c in only 2% yield with 46% ee at 80 °C (Table 3, entry 3).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | MOF | Ar | R | Solv. | Product | T (°C) | Yield (%) | Eea (%) |
| a HPLC column: Chiralcel AD-H, eluent: hexane/i-PrOH = 95/5, flow rate: 1.0 mL min−1, detection: UV 254 nm.b (1S,2S)-isomer.c (1R,2R)-isomer. | ||||||||
| 1 | (R)-1 | Ph | H | MeOH | 8a | 70 | 20 | 19b |
| 2 | (R)-1 | Ph | Me | MeOH | 8b | 70 | 38 | 60b |
| 3 | (R)-1 | 1-Naph | H | 2-PrOH | 8c | 80 | 2 | 46c |
| 4 | (R)-2 | Ph | H | 2-PrOH | 8a | 80 | 8 | 20c |
| 5 | (R)-2 | Ph | Me | 2-PrOH | 8b | 80 | 11 | 25c |
| 6 | (R)-2 | 1-Naph | H | 2-PrOH | 8c | 80 | 58 | 97c |
| 7 | (R)-3 | Ph | H | 2-PrOH | 8a | 80 | 1 | 15c |
| 8 | (R)-3 | Ph | Me | 2-PrOH | 8b | 80 | 19 | 14b |
| 9 | (R)-3 | 1-Naph | H | 2-PrOH | 8c | 80 | 11 | 14b |
| 10 | (R)-4 | Ph | H | 2-PrOH | 8a | 80 | 80 | 57b |
| 11 | (R)-4 | Ph | Me | CHCl3 | 8b | 50 | 51 | 83b |
| 12 | (R)-4 | 1-Naph | H | 2-PrOH | 8c | 80 | 95 | 90b |
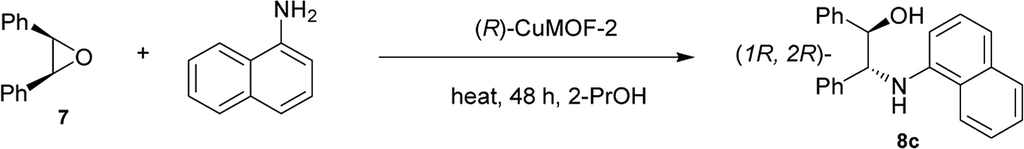
The reactions of cis-stilbene oxide with aniline and N-methyl aniline using (R)-CuMOF-2 as a catalyst showed poor reactivity and enantioselectivity (Table 3, entries 4–5), whereas the reaction with bulky 1-naphthylamine furnished the product in 58% yield and with excellent enantioselectivity (97% ee) at 80 °C (Table 3, entry 6). (R)-ZnMOF-4 also exhibited moderate to good enantioselectivities toward the reaction with aniline or N-methyl aniline (Table 3, entries 10–11). While (R)-ZnMOF-4 catalyzed reaction with sterically bulky 1-naphthylamine furnished in excellent enantioselectivity of 90% ee at 80 °C (Table 3, entry 12).
In contrast, the ARO reactions catalyzed by (R)-CuMOF-3 afforded the products in lower yields and enantioselectivities (Table 3, entries 7–9).
Then we examined the temperature effect on the ARO reaction of cis-stilbene oxide with 1-naphthylamine using (R)-CuMOF-2 as a catalyst (Table 4). As shown in Table 4, the efficient reaction did not occur at temperatures below 50 °C (Table 4, entries 1–4). When the temperature was higher than 60 °C, the ARO reaction occurred efficiently with excellent enantioselectivity up to 97% (Table 4, entries 5–7). A similar temperature effect on the ARO reaction of cis-stilbene oxide with anilines has been reported recently.15
We also performed control experiments to study the confinement effect of (R)-ZnMOF-4 on the catalyst. When the reaction was performed using Zn(OAc)2 as the catalyst, the product was obtained in 7% yield (Table 5, entry 1). When the reaction was performed using (R)-1,1′-bi-2-naphthol (BINOL), the (1R,2R)-8c was obtained in only 4% yield (Table 5, entry 2). When a combination of (R)-BINOL and Zn(OAc)2 were used as a catalyst, the reaction proceeded with a similar low efficiency, affording a racemic mixture in 4% yield (Table 5, entry 3). These results clearly showed that the reaction within the framework can develop the enantioselectivity, and this may be attributed to the restricted movement of the substrates in combination with a multiple chiral induction in the confined system. It is also interesting to note that the catalytic activity of (R)-ZnMOF-311 with higher solvent-accessible volume (66% of crystal volume) was lower than that of (R)-ZnMOF-210 (50% of crystal volume) (Table 5, entries 5 and 6).
| Entry | Cat. | Yield (%) | Eea (%) | Config. |
|---|---|---|---|---|
| a HPLC column: Chiralcel AD-H, eluent: hexane/i-PrOH = 95/5, flow rate: 1.0 mL min−1, detection: UV 254 nm. | ||||
| 1 | Zn(OAc)2 | 7 | 0 | — |
| 2 | (R)-BINOL | Trace | 3 | (1R,2R) |
| 3 | Zn(OAc)2 + (R)-BINOL | 4 | 0 | — |
| 4 | (R)-CuMOF-1 | 4 | 46 | (1R,2R) |
| 5 | (R)-CuMOF-2 | 58 | 97 | (1R,2R) |
| 6 | (R)-CuMOF-3 | 11 | 14 | (1S,2S) |
| 7 | (R)-ZnMOF-4 | 95 | 90 | (1S,2S) |
The reusability of the (R)-ZnMOF-4 catalyst was also investigated for the asymmetric ring-opening reactions of cis-stilbene oxide and 1-naphthylamine. As shown in Table 6, (R)-ZnMOF-4 was recovered from the catalytic reaction by filtration, and the recovered catalyst showed a similar high activity in conversion and enantioselectivity. The decreasing reactivity of recovered MOF in 3rd cycle may be due to the loss of the catalyst during the filtration process. To confirm the heterogeneous nature of the reaction, the asymmetric ring-opening reaction of cis-stilbene oxide and 1-naphthylamine was performed under the optimized reaction conditions. As shown in Fig. 3, the solid (R)-ZnMOF-4 catalyst was filtered from the reaction mixture after 24 h. The formation of product was interrupted by removing the catalyst from the reaction mixture.
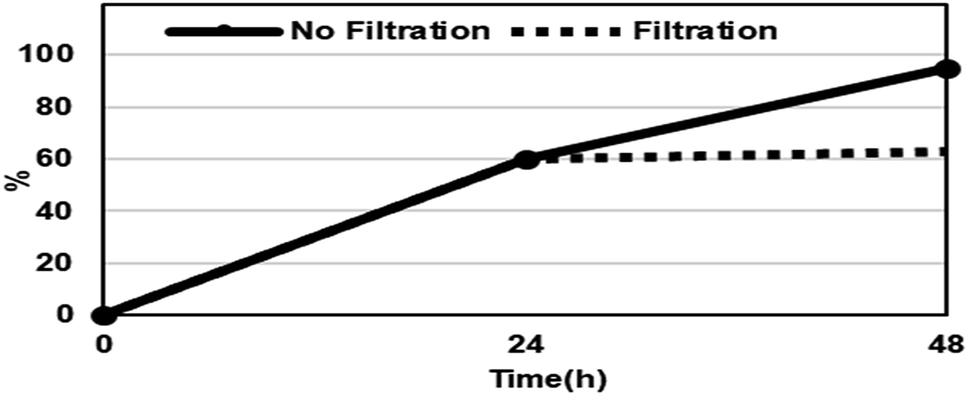 | ||
| Fig. 3 Heterogeneity test of asymmetric ring-opening reactions of cis-stilbene oxide and 1-naphthylamine in the presence of (R)-ZnMOF-4 (straight line) or after filtration of the catalyst at 24 h (dotted line). | ||
The reaction may proceed as follows: first, cis-stilbene oxide coordinates to the Lewis acidic Zn site of (R)-ZnMOF-4 to form an adduct: meanwhile, 1-naphthylamine is fixed by the NH–O hydrogen bond with the carboxylate oxygen and preferentially attacks the adjacent (R)-carbon atom of cis-stilbene oxide from the backside position to give (1S,2S)-β-amino alcohol with inversion of stereochemistry (Scheme 1).9b
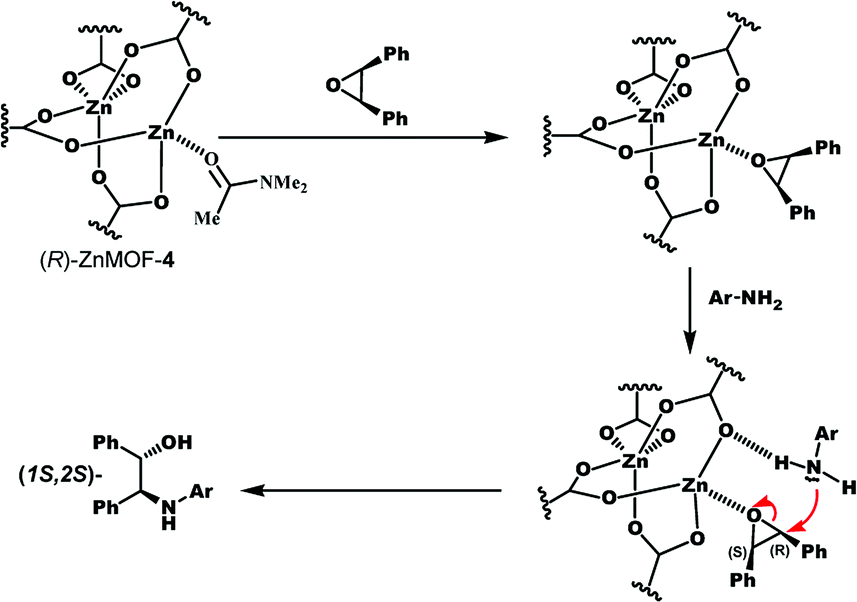 | ||
| Scheme 1 A plausible mechanism of asymmetric ring-opening reaction of cis-stilbene oxide with aromatic amine catalysed by (R)-ZnMOF-4. | ||
Conclusions
In this work, we have developed an efficient heterogeneous chiral MOF catalyst for the synthesis of β-amino alcohols. The enantioselective ring-opening reaction of cyclohexene oxide with N-methylaniline derivatives gave optically active β-amino alcohols in good yields and good to excellent enantioselectivities (up to 95% ee) using (R)-CuMOF-1 as catalyst. On the other hand, (R)-CuMOF-2 and (R)-ZnMOF-4 catalysed ARO reaction of cis-stilbene oxide with 1-naphthylamine furnished the corresponding β-amino alcohol in high yield (up to 95%) and excellent enantioselectivity (up to 97% ee). The MOF catalyst was recyclable with retention of the enantioselectivity. Further study of the mechanism and the scope of this reaction are now underway.Experimental
Synthetic procedures of (R)-ZnMOF-4
Synthesis of (R)-ZnMOF-4
A mixture of (R)-2,2′-diethoxy-1,1′-binaphthyl-4,4′-di(5-isophthalic acid) (4) (15 mg, 0.02 mmol) and Cu(NO3)2·3H2O (15 mg, 0.05 mmol) was dissolved in DMA (2 mL) and EtOH (1 mL). The solution was heated in a screw-capped vial (20 mL) at 110 °C for 48 h to give (R)-ZnMOF-3 (18 mg) as green prisms. IR (KCl, cm−1): 3236, 3067, 2933, 1604, 1508, 1402, 1362, 1265, 1214, 1103, 1029, 918, 862, 816, 769, 715, 593, 475, 457, 419. TGA data for loss of guest solvent: 36%.General procedure for asymmetric ring-opening reaction
A mixture of cis-stilbene oxide (0.042 mmol), aniline derivatives (0.042 mmol) and desolvated (R)-ZnMOF-1 (5 mg) was stirred at indicated temperature and for the indicated time in various solvents (0.3 mL). Then, the solid catalyst was collected by filtration, washed with MeOH, and the solvent of the filtrate were removed in vacuo to give the optically active β-amino alcohol. All known compounds were compared with data reported in the literature. The enantiomeric purity and absolute configuration of the product was determined by comparison with the reported HPLC data.22-(N-Methyl-N-phenylamino)cyclopentanol
1H-NMR (CDCl3, 400 MHz) δ 7.23–7.19 (m, 2H), 6.86 (d, 2H, J = 8.4 Hz), 6.74–6.71 (m, 1H), 4.18–4.12 (m, 1H), 3.96–3.90 (m, 1H), 2.75 (s, 3H), 2.35 (brs, 1H), 1.96–1.54 (m, 6H). 13C-NMR (CDCl3, 100 MHz) δ 150.97, 128.99, 117.30, 114.16, 74.30, 67.98, 32.13, 31.50, 24.35, 19.82. Enantiomeric excess was determined by HPLC with a Chiralcel OD-H column (Daicel) (n-hexane/2-PrOH = 90/10, 1.0 mL min−1, 254 nm).2-(N-Methyl-N-phenylamino)cyclohexanol
1H-NMR (CDCl3, 400 MHz) δ 7.25–7.30 (m, 2H), 6.97 (d, 2H, J = 8.6 Hz), 6.83 (t, 1H, J = 8.1 Hz), 3.65–3.71 (ddd, 1H, J = 10.2, 9.9, 4.3 Hz), 3.42–3.46 (m, 1H), 2.80 (bs, 1H), 2.78 (s, 3H), 2.20–2.24 (m, 1H), 1.70–1.80 (m, 3H), 1.27–1.45 (m, 4H). 13C-NMR (CDCl3, 100 MHz) δ 151.32, 129.01, 118.45, 115.52, 69.94, 66.92, 33.29, 31.04, 25.97, 25.40, 24.26. Enantiomeric excess was determined by HPLC with a Chiralcel OD-H column (Daicel) (n-hexane/2-PrOH = 98/2, 0.3 mL min−1, 254 nm).2-(N-Methyl-N-phenylamino)cycloheptanol
1H-NMR (CDCl3, 400 MHz) δ 7.28–7.24 (m, 2H), 6.96 (d, 2H, J = 7.6 Hz), 6.84 (t, 1H, J = 7.6 Hz), 3.76–3.70 (m, 1H), 3.50–3.44 (m, 1H), 3.08 (brs, 1H), 2.74 (s, 3H), 2.11–2.05 (s, 1H), 1.79–1.37 (m, 9H). 13C-NMR (CDCl3, 100 MHz) δ 151.32, 129.06, 119.01, 116.20, 72.36, 69.49, 32.64, 31.33, 26.67, 25.42, 24.33, 21.72. Enantiomeric excess was determined by HPLC with a Chiralcel OD-H column (Daicel) (eluent: n-hexane/2-PrOH = 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, flow rate: 1.0 mL min−1, 254 nm).
:10, flow rate: 1.0 mL min−1, 254 nm).
2-(N-(2-Methylphenyl)-N-methylamino)cyclohexanol
1H-NMR (CDCl3, 400 MHz) δ 7.19–7.11 (m, 3H), 7.02–6.98 (m, 1H), 3.68–3.62 (m, 2H), 2.72–2.66 (m, 4H), 2.35 (s, 3H), 2.18–2.14 (m, 1H), 1.75–1.11 (m, 7H). 13C-NMR (CDCl3, 100 MHz) δ 151.15, 132.98, 131.60, 126.29, 123.61, 122.71, 70.46, 67.86, 33.47, 33.15, 25.38, 24.38, 23.33, 19.09. Enantiomeric excess was determined by HPLC with a Chiralcel OD-H column (Daicel) (n-hexane/2-PrOH = 98/2, 0.3 mL min−1, 254 nm).2-(N-(3-Methylphenyl)-N-methylamino)cyclohexanol
1H-NMR (CDCl3, 400 MHz) δ 7.14–7.12 (m, 1H), 6.76–6.63 (3, 3H), 3.68–3.62 (m, 1H), 3.42–3.35 (m, 1H), 2.74 (s, 3H), 2.32 (s, 3H), 2.21–2.17 (m, 1H), 1.76–1.67 (m, 4H), 1.42–1.24 (m, 4H). 13C-NMR (CDCl3, 100 MHz) δ 151.44, 138.76, 128.87, 119.46, 116.44, 112.72, 69.95, 67.02, 33.26, 31.07, 25.99, 25.44, 24.30, 21.77. Enantiomeric excess was determined by HPLC with a Chiralcel OD-H column (Daicel) (n-hexane/2-PrOH = 98/2, 0.3 mL min−1, 254 nm).2-(N-(4-Methylphenyl)-N-methylamino)cyclohexanol
1H-NMR (CDCl3, 400 MHz) δ 7.06 (d, 2H, J = 8.4 Hz), 6.86 (d, 2H, J = 8.8 Hz), 3.66–3.59 (m, 1H), 3.33–3.27 (m, 1H), 2.71 (s, 3H), 2.26 (s, 3H), 2.21–2.16 (m, 1H), 1.77–1.63 (m, 3H). 13C-NMR (CDCl3, 100 MHz) δ 149.20, 129.50, 128.11, 116.22, 69.82, 67.66, 33.23, 31.26, 25.45, 25.43, 24.26, 20.26. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (Daicel) (n-hexane/2-PrOH = 95/5, 1.0 mL min−1, 254 nm).2-(N-(4-Methoxyphenyl)-N-methylamino)cyclohexanol
1H-NMR (CDCl3, 400 MHz) δ 6.93 (d, 2H, J = 9.2 Hz) 6.85 (d, 2H, J = 9.6 Hz), 3.77 (s, 3H), 3.64–3.57 (m, 1H), 3.18–3.12 (m, 2H), 2.69 (s, 3H), 2.21–2.16 (m, 1H), 1.75–1.63 (m, 3H), 1.42–1.19 (m, 4H). 13C-NMR (CDCl3, 100 MHz) δ 153.22, 145.51, 118.59, 114.29, 69.76, 68.94, 55.57, 33.20, 31.91, 25.42, 24.80, 24.25. Enantiomeric excess was determined by HPLC with a Chiralcel OB-H column (Daicel) (n-hexane/2-PrOH = 95:5, flow rate : 1.0 mL min−1, 254 nm).
2-(N-(4-Chlorophenyl)-N-methylamino)cyclohexanol
1H-NMR (CDCl3, 400 MHz) δ 7.15 (d, 2H, J = 9.2 Hz), 6.81 (d, 2H, J = 9.2Hz), 3.65–3.58 (m, 1H), 3.44–3.28 (m, 1H), 2.7 (s, 1H), 2.18–2.13 (m, 3H), 1.75–1.63 (m, 3H), 1.42–1.16 (m, 5H). 13C-NMR (CDCl3, 100 MHz) δ 149.89, 128.74, 122.96, 116.40, 69.93, 66.96, 33.34, 31.08, 26.07, 25.31, 24.19. Enantiomeric excess was determined by HPLC with a Chiralcel OB-H column (Daicel) (n-hexane/2-PrOH = 95/5, 1.0 mL min−1, 254 nm).2-Phenylamino-1,2-diphenylethanol
1H-NMR (CDCl3, 400 MHz) δ 7.20–7.09 (m, 10H), 7.04–7.00 (t, J = 8.0 Hz, 2H), 6.62 (t, J = 7.6 Hz, 1H), 6.48 (d, J = 8.0 Hz, 2H), 4.68 (d, J = 6.0 Hz, 1H), 4.42 (d, J = 6.0 Hz, 1H), 2.87 (brs, 1H). 13C-NMR (CDCl3, 100 MHz) δ 147.25, 140.54, 140.22, 129.05, 128.57, 128.26, 127.89, 127.52, 127.26, 126.54, 117.89, 114.11, 78.05, 64.70. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (Daicel) (n-hexane/2-PrOH = 95/5, 1.0 mL min−1, 254 nm).2-(N-Methyl-N-phenylamino)-1,2-diphenylethanol
1H-NMR (CDCl3, 400 MHz) δ 7.37 (d, J = 7.2 Hz, 2H), 7.27–7.07 (m, 8H), 6.99–6.93 (m, 4H), 6.48 (d, J = 8.0 Hz, 2H), 6.90–6.86 (m, 1H), 5.25 (d, J = 10.0 Hz, 1H), 4.86 (d, J = 10.4 Hz, 1H), 4.04 (brs, 1H), 2.64 (s, 3H). 13C-NMR (CDCl3, 100 MHz) δ 151.20, 140.59, 134.60, 129.03, 128.66, 128.11, 127.86, 127.66, 127.55, 127.50, 120.15, 117.53, 73.51, 71.40, 32.56. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (Daicel) (n-hexane/2-PrOH = 95/5, 1.0 mL min−1, 254 nm).2-(Naphtalene-1-ylamino)-1,2-diphenylethanol
1H-NMR (CDCl3, 400 MHz) δ 8.01–7.95 (m, 1H), 7.78–7.70 (m, 1H), 7.52–7.42 (m, 2H), 7.39–7.26 (m, 8H), 7.26–7.07 (m, 3H), 6.29 (d, J = 7.3 Hz 1H), 5.55 (s, 1H), 5.05 (d, J = 5.5 Hz, 1H), 4.72 (d, J = 5.5 Hz, 1H), 2.44 (s, 1H). 13C-NMR (CDCl3, 100 MHz) δ 142.11, 140.65, 139.94, 134.19, 128.64, 128.59, 128.34, 127.99, 127.58, 127.19, 126.49, 126.35, 125.64, 124.80, 123.92, 120.00, 117.64, 106.54, 78.28, 64.36. Enantiomeric excess was determined by HPLC with a Chiralpak AD-H column (Daicel) (n-hexane/2-PrOH = 95/5, 1.0 mL min−1, 254 nm).X-ray crystallographic data collection, data processing, structure determination and refinement, thermal analysis, Zn-coordination
A yellow crystal of (R)-ZnMOF-1 of dimensions 0.196 × 0.191 × 0.145 mm was mounted in a cryoloop and transferred to a diffractometer. Intensity data for the single crystal was obtained by collecting reflections on a Bruker X8 diffractometer furnished with an APEX II CCD detector and CuKα (λ = 1.54178 Å) radiation at 296 K using 0.5° ω scans. Integration was done using the SAINT software package16 that is a part of the APEX II software suite and absorption corrections were conducted using SADABS. Structures were solved via direct methods SHELXS-2014.17 The APEX II program package was used to determine the unit-cell parameters and for data collection (60 sec per frame scan time for a sphere of diffraction data). The raw frame data was processed using SAINT and S4 SADABS to yield the reflection data file. Subsequent calculations were carried out using the SHELXTL program. The diffraction symmetry was consistent with the monoclinic space groups P21. The structure was solved by direct methods and refined on F2 by full-matrix least-squares techniques. The analytical scattering factors8 for neutral atoms were used throughout the analysis.Asymmetric unit contains (R)-ZnMOF-4 ligand, two Zn(II) cations and three N,N-dimethylacetamide molecules – one coordinated to Zn1 via carbonyl O-atom and two other with half occupancy included into the void space of the crystal by CH⋯O interactions. There were 1.5 molecules of water present in the asymmetric unit – one coordinated to Zn1 atom and the second was distributed over two positions with 0.5 total occupancy. Additionally one ethanol molecule with site occupancy 0.5 is also present in this crystal. Moreover, crystallographic refinement strongly suggests that there are two slightly different ligand molecules present in the studied crystals: one with full ethylation of both hydroxy groups and second, where OH groups was substituted in ca. 50% (hard to separate mixture by any of chromatographic methods). Disordered ethyl groups and all solvent molecules were refined in isotropic mode. All non-hydrogen atoms were located via difference Fourier maps and refined anisotropically as an inversion twin. Aromatic and methyl/methylene groups hydrogen atom positions were placed at their idealized positions and allowed to ride on the coordinates of the parent atom with isotropic thermal parameters (Uiso) fixed at 1.2 Ueq of the carbon atom to which they are attached. All solvent molecules were refined in isotropic mode with geometric restraints for all three N,N-dimethylacetamide molecules. Presence of disorder in the methoxy groups generates close contacts between protons. Refinement converged at wR2 = 0.1811 refined against 11706 data (0.80 Å), and R1 = 0.0664 for 10606 data with I > 2.0σ(I) with GOF = 1.08. There was a single residual density 0.84 e present in the final difference-Fourier map located around Zn atoms (min −0.63 e). crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as CCDC 1832085.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research (C) (No. 26410126) from The Ministry of Education, Culture, Sports, Science and Technology (MEXT). The authors acknowledge the measurement of the BET surface area by the nitrogen adsorption isotherms of (R)-ZnMOF-4 by MicrotracBEL Corp. Osaka, Japan.Notes and references
- (a) G. A. Rogers, S. M. Parsons, D. C. Anderson, L. M. Nilsson, B. A. Bahr, W. D. Kornreich, R. Kaufman, R. S. Jacobs and B. Kirtman, J. Med. Chem., 1989, 32, 1217–1230 CrossRef PubMed; (b) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561–2576 CrossRef.
- D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835–876 CrossRef PubMed.
- (a) E. J. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef PubMed; (b) C. Wang, L. Luo and H. Yamamoto, Acc. Chem. Res., 2016, 49, 193–204 CrossRef PubMed; (c) S. Meninno and A. Lattanzi, Chem.–Eur. J., 2016, 22, 3632–3642 CrossRef PubMed.
- (a) A. Sekine, T. Ohshima and M. Shibasaki, Tetrahedron, 2002, 58, 75–82 CrossRef; (b) G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, M. Massaccesi, P. Melchiorre and L. Sambri, Org. Lett., 2004, 6, 2173–2176 CrossRef PubMed; (c) F. Carree, R. Gill and J. Collin, Org. Lett., 2005, 7, 1023–1026 CrossRef PubMed; (d) S. Azoulay, K. Manabe and S. Kobayashi, Org. Lett., 2005, 7, 4593–4595 CrossRef PubMed; (e) K. Arai, S. Lucarini, M. M. Salter, K. Ohta, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2007, 129, 8103–8111 CrossRef PubMed; (f) E. Mai and C. Schneider, Chem.–Eur. J., 2007, 13, 2729–2741 CrossRef PubMed; (g) B. Gao, Y. Wen, Z. Yang, X. Huang, X. Liu and X. Fen, Adv. Synth. Catal., 2008, 350, 385–390 CrossRef; (h) M. Kokubo, T. Naito and S. Kobayashi, Chem. Lett., 2009, 38, 904–905 CrossRef; (i) D. R. Boyd, N. D. Sharma, L. Sbircea, D. Murphy, J. F. Malone, S. L. James, C. C. R. Allen and J. T. G. Hamilton, Org. Biomol. Chem., 2010, 8, 1081–1090 RSC; (j) M. Kokubo, T. Naito and S. Kobayashi, Tetrahedron, 2010, 66, 1111–1118 CrossRef; (k) R. I. Kureshy, K. J. Prathap, M. Kumar, P. K. Bera, N. H. Khan, S. H. R. Abdi and H. C. Bajaj, Tetrahedron, 2011, 67, 8300–8307 CrossRef; (l) B. Gao, M. Xie, A. Sun, X. Hu, X. Ding, X. Liu, L. Lin and X. Feng, Adv. Synth. Catal., 2012, 354, 1509–1518 CrossRef; (m) G. V. More and B. M. Bhanage, Eur. J. Org. Chem., 2013, 6900–6906 CrossRef; (n) M. Kumar, R. I. Kureshy, S. Saravanan, S. Verma, A. Jakhar, N. H. Khan, S. H. R. Abdi and H. C. Baja, Org. Lett., 2014, 16, 2798–2801 CrossRef PubMed; (o) A. Sharma, J. Agarwal and R. K. Peddinti, Org. Biomol. Chem., 2017, 15, 1913–1920 RSC.
- (a) A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef PubMed; (b) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef PubMed; (c) B. Van de Voorde, B. Bueken, J. Denayer and D. De Vos, Chem. Soc. Rev., 2014, 43, 5766–5788 RSC; (d) I. Cota and M. F. Fernandez, Coord. Chem. Rev., 2017, 351, 189–204 CrossRef.
- (a) P. Peluso, V. Mamane and S. Cossu, J. Chromatogr. A, 2014, 1363, 11–26 CrossRef PubMed; (b) D. Banerjee, A. J. Cairns, J. Liu, R. K. Motkuri, S. K. Nune, C. A. Fernandez, R. Krishna, D. M. Strachan and P. K. Thallapally, Acc. Chem. Res., 2015, 48, 211–219 CrossRef PubMed; (c) L. Heinke, M. Tu, S. Wannapaiboon, R. Fischer and C. Woell, Microporous Mesoporous Mater., 2015, 216, 200–215 CrossRef.
- (a) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef PubMed; (b) D. Liu, K. Lu, C. Poon and W. Lin, Inorg. Chem., 2014, 53, 1916–1924 CrossRef PubMed; (c) A. Karmakar, P. Samanta, A. V. Desai and S. K. Ghosh, Acc. Chem. Res., 2017, 50, 2457–2469 CrossRef PubMed.
- (a) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC; (b) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (c) L. Ma and W. Lin, Top. Curr. Chem., 2010, 293, 175–205 CrossRef PubMed; (d) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC; (e) A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC; (f) Y. Zhang, J. Guo, L. Shi, Y. Zhu, K. Hou, Y. Zheng and Z. Tang, Sci. Adv., 2017, 3, e1701162 CrossRef PubMed; (g) W. Shi, L. Cao, H. Zhang, X. Zhou, B. An, Z. Lin, R. Dai, J. Li, C. Wang and W. Lin, Angew. Chem., Int. Ed., 2017, 56, 9704–9709 CrossRef PubMed.
- (a) K. Tanaka, S. Oda and M. Shiro, Chem. Commun., 2008, 820–822 RSC; (b) K. Doitomi, K. Xua and H. Hirao, Dalton Trans., 2017, 46, 3470–3481 RSC.
- K. Tanaka, S. Nagase, T. Anami, M. Wierzbickib and Z. Urbanczyk-Lipkowska, RSC Adv., 2016, 6, 111436–111439 RSC.
- K. Tanaka, D. Yanamoto, K. Yoshimura, T. Anamia and Z. Urbanczyk-Lipkowska, CrystEngComm, 2015, 17, 1291–1295 RSC.
- M. W. A. MacLean, T. K. Wood, G. Wu, R. P. Lemieux and C. M. Crudden, Chem. Mater., 2014, 26, 5852–5859 CrossRef.
- M. L. Connolly, Science, 1983, 221, 709 CrossRef PubMed.
- R. I. Kureshy, S. Singh, N. H. Khana, S. H. R. Abdi, E. Suresh and R. V. Jasra, J. Mol. Catal. A: Chem., 2007, 264, 162–169 CrossRef.
- S. Regati, Y. He, M. Thimmaiah, R. Li, S. Xiang, B. Chen and J. C. G. Zhao, Chem. Commun., 2013, 49, 9836–9838 RSC.
- Bruker, APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA, 2005 Search PubMed.
- G. M. Sheldrick, Program for the Refinement of Crystal Structures from Diffraction Data, SHELXL-2014, University of Göttingen, Germany, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis of (R)-ZnMOF-1, catalytic asymmetric ring opening reactions, IR, TG, NMR spectra, HPLC chromatographic data. CCDC 1832085. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra05163a |
| This journal is © The Royal Society of Chemistry 2018 |