
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrrolidine and oxazolidine ring transformations in proline and serine derivatives of α-hydroxyphosphonates induced by deoxyfluorinating reagents†
Patrycja Kaczmarek,
Magdalena Rapp * and
Henryk Koroniak
* and
Henryk Koroniak
Faculty of Chemistry, Adam Mickiewicz University in Poznań, Umultowska 89b, 61-614 Poznań, Poland. E-mail: magdrapp@amu.edu.pl
First published on 6th July 2018
Abstract
Transformations of α-hydroxyphosphonates derived from proline or serine by treatment with different deoxyfluorinating reagents (DAST, Deoxofluor, PyFluor) are reported. Depending on the applied reagent, as well as the protecting group used (N-Cbz, N-Boc, N-Bn) different types of products are observed. The reaction of N-Cbz or N-Boc prolinols with DAST or Deoxofluor due to aziridinium intermediate participation gave fluorinated amino phosphonates such as piperidine and pyrrolidine derivatives and/or oxazolidine-2-ones. Similarly, the analogous reaction of N-Cbz or N-Boc protected serinol yielded oxazolidine-2-ones or its fluorinated analogues. As the second type of product formed by DAST-induced reaction of serine derivatives, aziridines were obtained. Only in the case of deoxyfluorination of N-benzyl prolinols were both diastereoisomers of β-fluoropiperidine-α-phosphonates formed, while the reaction of protected N-benzyl serinols gave fluorinated oxazolidines. Moreover, application of PyFluor gave sulfonate derivatives.
Introduction
The replacement of the carboxylic groups in amino acids by the phosphonate moiety or related functions resulted in the formation of α- and β-amino phosphonic acid derivatives. Between them, the aminoalkylphosphonate esters are frequently synthesized due to their wide spectrum of biological properties applied in medicine as well as in agriculture.1 Moreover, amino phosphonates represent models of the tetrahedral transition states in activated complexes formed during the hydrolysis of natural peptides2 and were proved to be suitable substrates for some enzymes inhibitions.1a,b As a representative example, dipeptides containing phosphonated proline analogue have been found as specific irreversible inhibitors of dipeptidyl peptidase IV (DPP IV).3 On the other hand, a phosphonic acid analogue of serine as a visualization agent in rat kidney and skeletal bones has been applied.4 The biomedical application of amino phosphonates makes them attractive targets in organic synthesis. Thus, organophosphorus analogues of almost all proteinogenic amino acids have been already obtained. Among them, the preparation of phosphoproline,5 phosphohomoproline6 or synthesis of phosphonic acid analogues of serine have been reported.7 Moreover, since the observation, that group of α-monofluoroalkylaminophosphonates could be applied as a nonhydrolysable isopolar surrogate of naturally occurring phosphates (where C–O–P bridge was replaced by C–CHF–P linkages),8 several syntheses of some monofluorinated alkylphosphonic acid analogues have been reported.9 The introduction of the fluorine atom in organic compounds has a deep electronic and steric impact, affecting interactions between fluorine-containing inhibitors and target enzymes.10 This effect is especially noticeable in a group of fluorinated amino phosphonates.11 Thus, monofluoro- and difluoro phosphoserine analogues as potent inhibitors of alanine racemase have been reported.11a Moreover, dipeptides containing β-fluorinated phosphoproline have been designed as a phosphonate-based transition-state analogue of inhibitors of proline selective serine dipeptidases.11bOne of the common strategy in the synthesis of monofluorinated amino phosphonates has involved nucleophilic fluorination of the hydroxyl group in hydroxyphosphonates and as the most common reagents (diethylamino)sulfur trifluoride (DAST),12 DeoxoFluor13 and PyFluor14 were used. Generally, the mechanism of deoxyfluorination with DAST involves the attack of the hydroxyl group of alcohol substrate to the electrophilic deoxyfluorinating agent (with a generation of activated alcohol –OSF2NEt2 along with fluoride ion).
The latter then displaces of leaving group to produce the corresponding alkyl fluoride. However, β-aminoalcohols such as prolinol derivatives the reaction is frequently going through aziridinium intermediate due to neighbouring group participation leading to ring opening by fluoride ion, resulting in restoration or ring expansion in some cases (Scheme 1).15 In case of phosphonates, depending on the structure and applied reagent, the fluorination proceeds with different regioselectivity. Thus, fluorination with DAST of α-hydroxy-β-aminoalkylphosphonates originated from aliphatic amino acids as well as phenylalanine gave β-fluoro-α-aminoalkylphosphonates as major isomers.16 For comparison, the application of PyFluor and DBU in the same systems resulted in the formation of mainly α-fluoro-β-aminoalkylphosphonates.17 On the other hand, the reactions of proline derived α-hydroxyphosphonates with DAST led to the corresponding α-fluoroalkylphosphonates11b while the fluorination of β-hydroxy-γ-aminoalkylphosphonates gave the appropriate β-fluoroalkylphosphonates.11c Other approaches yielding α-fluoro-β-aminoalkylphosphonates involved electrophilic fluorination of β-ketophosphonates and next enamine formation18 or addition of anion species [C(TMS)FP(O)(OEt)2]− to appropriate iminium salt.8a Moreover, applications of suitable methylphosphonate carbanions in the synthesis of α-fluoro-γ-amino phosphonates11c,19 or γ-monofluoro-β-aminoalkylphosphonates20 were reported.
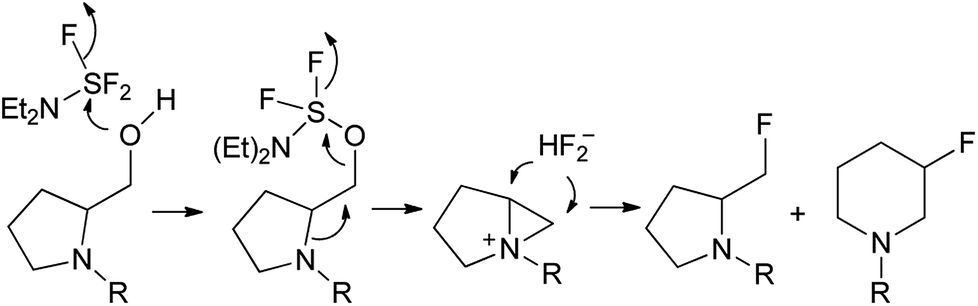 | ||
| Scheme 1 Deoxyfluorination of prolinol with DAST. | ||
Our recent studies revealed that nucleophilic fluorination of α-hydroxyphosphonates derived from O-isopropylidene glyceraldehyde with DAST has led to oxirane formation due to the substitution of DAST-derived leaving group by hydroxyl group from the adjacent stereogenic centre.21 By contrast, stereoselective deoxyfluorination of hydroxyphosphonates derived from an O-isopropylidenepentofuranose gave major fluoride possessing D-glu configuration. Moreover, we found that fluorination of tertiary alcohols derived from di-O-isopropylidenehexofuranose and 1,2-O-isopropylidenepentofuranose have been stereo controlled by the neighbouring bottom-face 1,2-O-isopropylidene group oxygen atom leading preferentially to one diastereoisomer of allylic, phenylacetylene, styryl, and benzylic fluorides.22 Herein, we present our studies evaluating different substrates scope for deoxyfluorination reaction with an emphasis on neighbouring group participation resulting transformation of phosphonate amino acids analogues.
Results and discussion
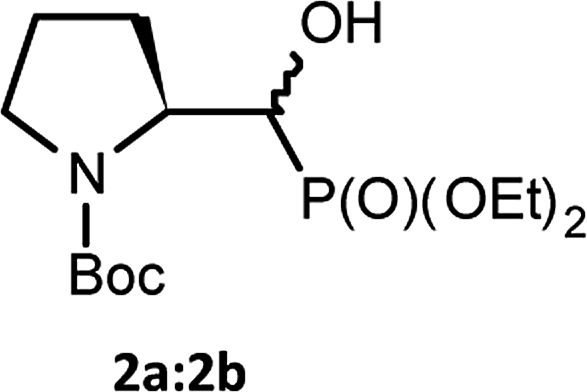
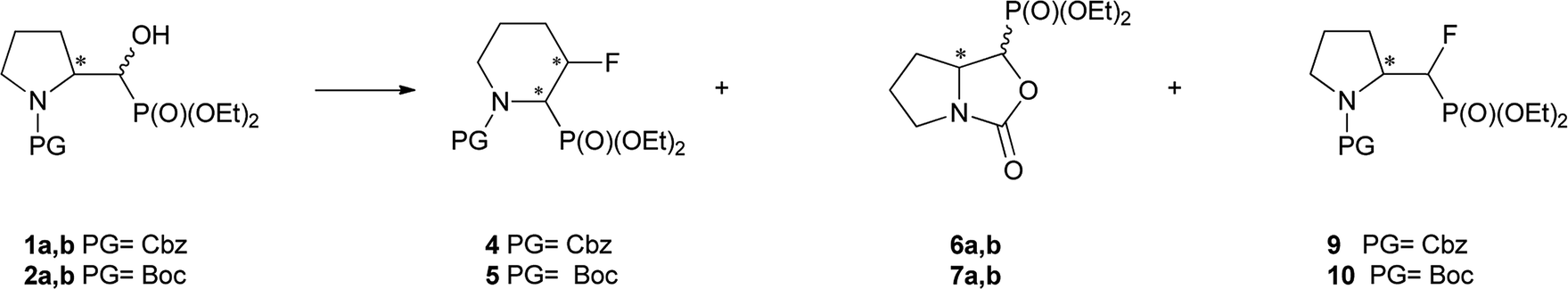
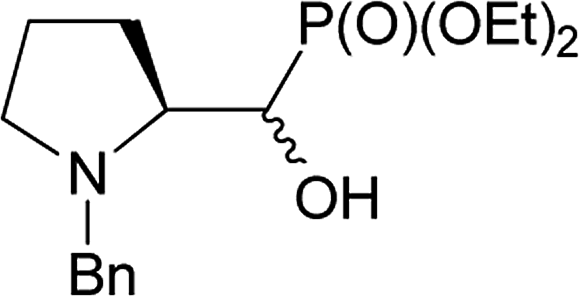
We started with α-hydroxyphosphonates derivatives of proline and serine possessing N-carboxybenzyl, N-tert-butoxycarbonyl, and N-benzyl as protecting groups. Thus the nucleophilic fluorination of prolinols such as 1a,b (Cbz), 2a,b (Boc) and 3a,b (Bn) prepared according to known procedures (see Experimental section) were performed. Predominantly diastereoisomers (2R,1′S)-1a and (2S,1′R) -2a,3a were applied, while minor diastereoisomers possess (2R,1′R)-1b and (2S,1′S)-2b,3b configurations, respectively (Scheme 2). Treatment of α-hydroxyphosphonates 1a,b or 2a,b with DAST gave mainly two type of phosphonates 4–7 (31P NMR)(Scheme 3).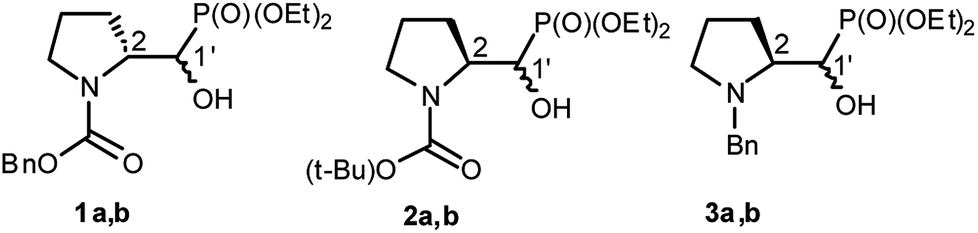 | ||
| Scheme 2 Structures of starting α-hydroxyphosphonates proline derivatives 1–3a,b. | ||
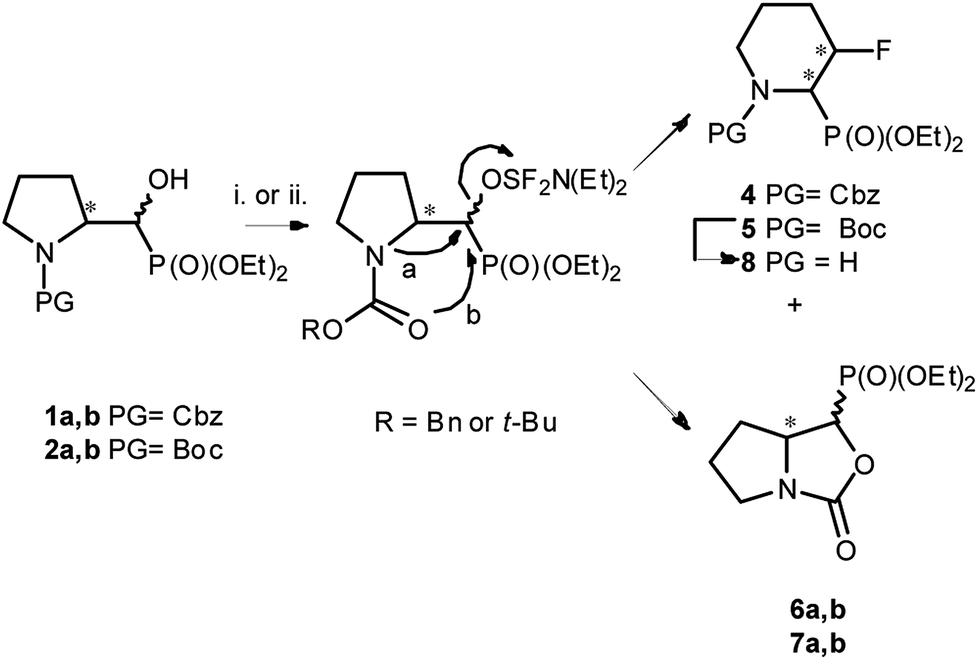 | ||
| Scheme 3 Reaction of 1–2a,b with DAST (Table 1) (4 43% and 6a,b 45%; 5 38% and 7a,b 53%); or DeoxoFluor (CH2Cl2, RT, 24 h)(4 38% and 6a,b 43%; 5 30% and 7a,b 48%); and deprotection of 5 (8 73%); [configurations of stereogenic centres in the text]. | ||
Primary experiments indicated that compounds 4 and 5, as well as non-fluorinated 6a or 7a, arose from major 1a (2R,1′S) or 2a (2S,1′R) diastereoisomers while from 1b (2R,1′R) or 2b (2S,1′S) only bicyclic 6b or 7b were formed (Table 1). Stereochemistry of the fluorination of 1a (or 2a) is a consequence of the transformation of α-hydroxyl moiety into good leaving group (–OSF2N(Et)2) and its substitution by electron pair of pyrrolidine nitrogen atom (SN2, pathway a Scheme 3) yielding aziridinium ion, analogously to the intermediate formed during fluorination of proline derivatives (Scheme 1). A subsequent attack of fluoride ion as HF2−(second SN2 reaction) gave preferentially β-fluoro-α-phosphonate piperidine 4 or 5. Ring expansion during deoxyfluorination of prolinols with DAST has been already reported.15 Moreover, rearrangement of optically active prolinols by treatment with DAST afforded only one optically active diastereoisomer of piperidines.15 Also, Kaźmierczak et al. reported fluorination of α-hydroxyphosphonate analogues of amino acids such as phenylalanine or valine leading to α-amino-β-fluoroalkylphosphonates via aziridinium ion.16 In our case, two signals of main fluorinated product 4 or 5 [appearing as a mixture of rotamers (1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, r.r.), due to presence of N-Cbz or N-Boc protecting group, isolated yields 43% and 38%, respectively] were located in 19F NMR at δ: −180/179 ppm (as m), in the area habitually occupied by signal of secondary alkyl fluoride.23 Moreover, coupling constants values 2JH2P 21 Hz, 2JH3F 46 Hz, 3JH2F 19 Hz as well as the location of signals for CHF at δ: 85 ppm (2JCF 179 Hz), CHP at c.a. δ: 53 ppm (2JCP 150 Hz, 3JCF 22 Hz) in 13C NMR spectra indicated piperidine ring formation with the vicinal arrangement of fluorine substituent and phosphonate moiety. To compare, in case of fluorocyclohexane the values of coupling constants 3JHF equal 44 Hz (for anti) or 3JHF 10 Hz for gauche conformations were reported.23 In our case, we have observed the extremely high value of coupling constants (3JFP 62/63 Hz). Usually, the 3JPF coupling constants range from c.a. 0 Hz to 9 Hz as observed for two stereoisomers of diethyl 2-fluorocyclohexyl phosphonate.24 On the other hand, in case of N,N-dibenzyl-α-amino-β-fluoroalkylphosphonates the values of J for conformations gauche (3JPF 8–10 Hz) and anti (3JPF 15–19 Hz) were reported.16 In our case the most probably, the high value of coupling constants is due to the attempted arrangement of C–F and C–P bonds with equatorially situated fluorine and phosphonate substituents25 in the ring. Thus, piperidine existed as slightly twisted boat conformation forced the most probably by bulky N-protecting group vicinal to phosphonate moiety (compound 4, Scheme 4). Stereochemistry of 4 as (2R,3R) was confirmed by further 19F–1H HOESY experiments showing NOEs between fluorine atom and protons: H-4 and H-5; as well as NOEs between H-2 (CHP) and H-3 (CHF) (1H–1H NOESY) and indicated the additional participation of protecting group (N-Boc) in product formation. Also in β-fluoroethylamide C–F and CN(CO) bonds prefer to adopt the gauche conformation.26 To confirm the influence of bulky N-Cbz or N-Boc groups on a conformation of 4–5, deprotection of 5 (TFA) was carried out to give compound 8 (yield 73%). Thus, piperidine 8 shows 3JFP 5/6 Hz (19F, 31P NMR) indicating the less strained arrangement of fluorine substituent and phosphonate moiety in piperidine ring.
:1, r.r.), due to presence of N-Cbz or N-Boc protecting group, isolated yields 43% and 38%, respectively] were located in 19F NMR at δ: −180/179 ppm (as m), in the area habitually occupied by signal of secondary alkyl fluoride.23 Moreover, coupling constants values 2JH2P 21 Hz, 2JH3F 46 Hz, 3JH2F 19 Hz as well as the location of signals for CHF at δ: 85 ppm (2JCF 179 Hz), CHP at c.a. δ: 53 ppm (2JCP 150 Hz, 3JCF 22 Hz) in 13C NMR spectra indicated piperidine ring formation with the vicinal arrangement of fluorine substituent and phosphonate moiety. To compare, in case of fluorocyclohexane the values of coupling constants 3JHF equal 44 Hz (for anti) or 3JHF 10 Hz for gauche conformations were reported.23 In our case, we have observed the extremely high value of coupling constants (3JFP 62/63 Hz). Usually, the 3JPF coupling constants range from c.a. 0 Hz to 9 Hz as observed for two stereoisomers of diethyl 2-fluorocyclohexyl phosphonate.24 On the other hand, in case of N,N-dibenzyl-α-amino-β-fluoroalkylphosphonates the values of J for conformations gauche (3JPF 8–10 Hz) and anti (3JPF 15–19 Hz) were reported.16 In our case the most probably, the high value of coupling constants is due to the attempted arrangement of C–F and C–P bonds with equatorially situated fluorine and phosphonate substituents25 in the ring. Thus, piperidine existed as slightly twisted boat conformation forced the most probably by bulky N-protecting group vicinal to phosphonate moiety (compound 4, Scheme 4). Stereochemistry of 4 as (2R,3R) was confirmed by further 19F–1H HOESY experiments showing NOEs between fluorine atom and protons: H-4 and H-5; as well as NOEs between H-2 (CHP) and H-3 (CHF) (1H–1H NOESY) and indicated the additional participation of protecting group (N-Boc) in product formation. Also in β-fluoroethylamide C–F and CN(CO) bonds prefer to adopt the gauche conformation.26 To confirm the influence of bulky N-Cbz or N-Boc groups on a conformation of 4–5, deprotection of 5 (TFA) was carried out to give compound 8 (yield 73%). Thus, piperidine 8 shows 3JFP 5/6 Hz (19F, 31P NMR) indicating the less strained arrangement of fluorine substituent and phosphonate moiety in piperidine ring.
|
||||||
|---|---|---|---|---|---|---|
| Substrate | d.r. | Cond. | Reagent | Products ratiob (isolated yield%) | ||
| 4 or 5 | 6a:6b or 7a;7b |
9 or 10 | ||||
| a (i) −78 °C → 0 °C (1.5 h); (ii) 0 °C → 40 °C (1 h); (iii) −78 °C, 3 h, RT (1 h); (iv) RT (24 h).b Ratio of products in crude reaction mixture, 31P NMR. | ||||||
 |
22.8:1 |
i | DAST | 11 (43) | 9.5:1 (45) |
2 (8) |
| 22.8:1 |
ii | DAST | 10 (41) | 9.2:1 (45) |
2.6 (7) | |
| 2.6:1 |
iii | DAST | 5.3 (36) | 6.8:5.1 (56) |
1 (—) | |
| 1:1.4 |
i | DAST | 1 (10) | 1:2.9 (40) |
— | |
| 20:1 |
iv | DeoxoFluor | 8.8 (38) | 9.1:1 (43) |
2 (traces) | |
 |
2.7:1 |
iii | DAST | 1.3 (23) | 1.4:1 (44) |
Traces |
| 3.8:1 |
iii | DAST | 1.8 (38) | 2.0:1 (53) |
Traces | |
| 36:1 |
iv | DeoxoFluor | 16 (30) | 23:1 (48) |
Traces | |
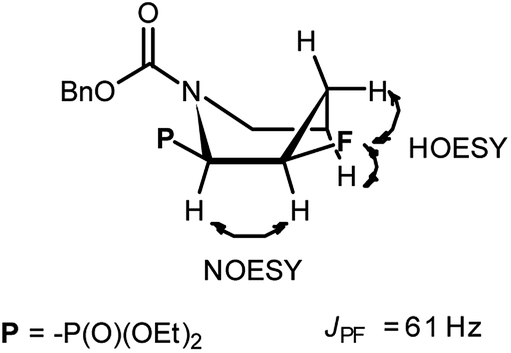 | ||
| Scheme 4 The slightly twisted boat conformation of 4 with observed correlations and values of coupling constants. | ||
The second main type of products – phosphonates 6a,b or its enantiomers 7a,b possess oxazolidine-2-one fragments and have been formed as a result of substitution of DAST derived leaving group in 1–2a,b by carbonyl electron pair from N-Cbz (N-Boc) moieties (pathway b, Scheme 3), analogously to the reaction N-Boc protected β-aminoalcohol with DAST.27 Configuration at stereogenic centers in CHP of both diastereoisomers of 6 or 7 was reversed compared to appropriate diastereoisomer of starting 1–2a,b. Thus, the structure of major and minor diastereoisomers of non-fluorinated oxazolidine-2-ones 6a,b arising from 1a,b were determined on the base of the NMR spectra and NOESY experiments and were determined as the trans 6a for major and cis 6b for minor isomers, and respectively their enantiomers trans 7a and cis 7b if starting material was 2a,b. The stereochemical assignment of trans/cis oxazolidinones was applied already for determination of stereochemistry in L-phenylalaninal derived hydroxyphosphonates, serving as suitable substrates for aspartyl protease renin inhibitors.28 Moreover, careful inspection of 19F NMR spectra led to observation of other fluorinated products 9 or 10 visible as traces at δ: −226/227 ppm with two-bond H–F coupling constants value being about 2JFH 47 Hz and 2JFP 77 ± 2 Hz, analogically to β-amino-α-fluoroalkylphosphonates.16 Compound 9 was formed solely from 1a as a second regioisomer of aziridinium ring opening the most probably (pathway a, Schemes 3 and 1). Analogically, compound 10 derived from 2a. The resulting ratio of products of the reaction of 1a,b as well as 2a,b with various fluorinating agents (crude, 31P NMR), with reaction conditions are presented below (Table 1). The presented experiments indicated, that in both cases from 1a,b as well as from 2a,b three analogous type of compounds were formed and in different temperatures ranges similar ratio of products was observed (DCM, 4 equiv. of DAST). We have determined that compounds 4 and 5, as well as 6a or 7a, were formed from 1a or 2a while from 1b or 2b only bicyclic 6b or 7b were produced. Compound 9 arising from major diastereoisomer of 1a was visible as traces in 31P NMR while in the reaction carried out at a higher temperature slightly higher contribution of fluoride 9 was detected. On the other hand, the reactions of 1a,b or 2a,b (with a different ratio of stereoisomers) carried out with Deoxofluor (RT, 24 h) gave the same products 4–7, 9–10 with a parallel ratio to reaction with DAST. Moreover, the reactions of 1a,b (3.3:1, d.r.) or 2a,b (37:1, d.r.) with PyFluor (DBU, toluene, RT, 5 d) gave alkylphosphoryl pyridine-2-sulfonates 11a,b or 12a,b (3:1 or 74:1, d.r.) with yields 78% and 74%, respectively (Scheme 5). Thus, the reaction of 1a,b with PyFluor gave 11a,b as a mixture of two appropriate diastereoisomers without any configuration changes analogically to starting materials. The positions of signals in 31P NMR were shifted toward higher field (δP 15.1/14.7 ppm for 11a or δP 15.4/15.5 ppm for 12a), comparing with α-hydroxyphosphonates 1–2a,b. The formation of sulfonates instead of fluorides was also already reported.29 Also, Kaźmierczak et al. reported the sulfonates formation during fluorination of α-hydroxyphosphonate analogues of amino acids such as phenylalanine possessing phthaloyl protecting group.17
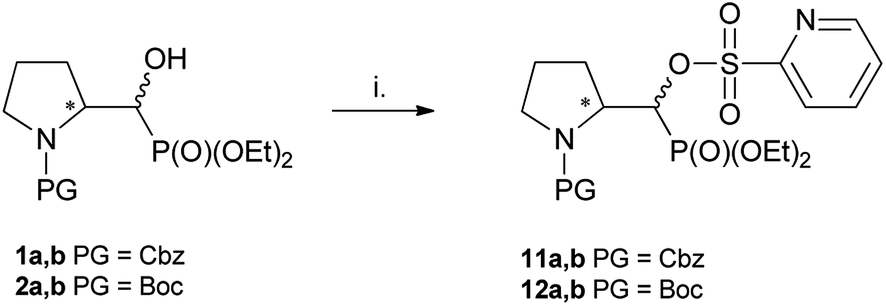 | ||
| Scheme 5 Reaction of 1–2a,b with PyFluor (PyFluor, DBU, MePh, RT, 5d; 11a,b 78%, 12a,b 74%). | ||
On the base of the results described for prolinols 1–2a,b we next examined the fluorination of diastereoisomeric mixture of 3a,b having benzyl as N-protecting group (Table 2).
|
||||||
|---|---|---|---|---|---|---|
| Substrate 3a:3b |
d.r. | Cond. | 13a | 13b | 14 | Yields [%]b (d.r.) |
| a (i) DAST, 0 °C → 45 °C (1 h); (ii) DAST, −78 °C (1 h) → 45 °C (0.75 h); (iii) DAST, −78 °C (1 h) → 45 °C (0.75 h), RT (3 h); (iv) Deoxofluor, RT (24 h); (v) PyFluor, DBU, MePh, RT, 5 d.b Isolated yields (31P NMR).c 27% unreacted 3b.d 44% unreacted 3b. | ||||||
 |
1.1:1 |
i | 3.2 | 1 | — | 59 (3.2:1) |
| 2.1:1 |
iic | 9.8 | 1 | — | 49 (11.2:1) |
|
| 2.1:1 |
iii | 34 | 1 | — | 48 (20:1) |
|
| 2.1:1 |
iv | 5.7 | 1 | — | 58 (6.3:1) |
|
| 1.9:1 |
vd | 2.2 | — | 1 | 19 (—) | |
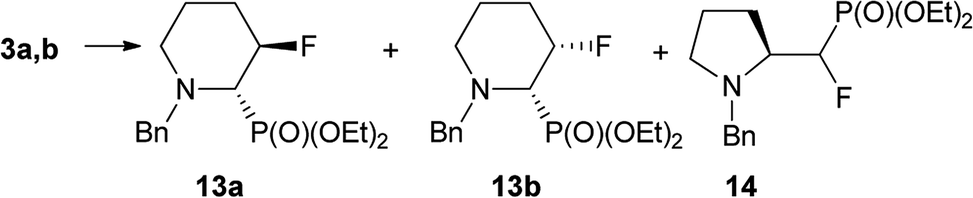
When the reaction of 3a,b (2.1:1 d.r.) with DAST was set up at −78 °C, next was carried out at 45 °C for 0.75 h we observed mainly transformation of 3a into fluorinated product 13a. In addition, while both isomers 3a and 3b were consumed, ratio 13a:13b was not corresponding to starting d.r. ratio, presumably due to the presence of other product, not isolated (δP: 10 ppm, in the crude reaction mixture). On the other hand, the reaction condition −78 °C (1 h) → 45 °C for 1 h followed by treatment at RT for 3 h gave two fluorinated phosphonates 13a,b without by-product (31P NMR) with the isolated yield 59%. The separate experiment indicated that compound 13b is formed from 3b, and for its formation the higher temperature (45 °C, 0.75 h) was necessary. To compare, the reaction of 3a,b with Deoxofluor gave 13a,b with lower diastereoselectivity comparing to reaction with DAST. Surprisingly, the down-field shifted two sets of signals corresponded to compounds 13a and 13b and located around δ: −146 ppm (3JFP 3 Hz) and at δ: −145 ppm (3JFP 64 Hz) in 19F NMR indicated that 13a and 13b have different structures comparing piperidines 4 and 5. Nevertheless, the careful analysis of 13C NMR indicated characteristic signals and coupling constants values for C-3 at δ: 96 ppm (1JCF 180 Hz), C-2 at δ: 65 ppm (1JCP 125/150 Hz, 2JCF 26/22 Hz) confirming that both compounds are diastereoisomers. On the base of 19F–1H HOESY and NOESY experiments, the arrangement of substituents in 13a was determined (Scheme 6). Thus, NOE's between fluorine atom and protons: H-2 as well as H-4 (not shown) and H-5 indicated boat conformation and C–P and C–F in a gauche arrangement, additionally confirmed by the value of 3JH4F 26 Hz indicating coupling of fluorine with equatorial H-4 (syn-periplanar). While compound 13a had 3JFP 4 Hz, analogically to less strained 8, in case of compound 13b we have observed analogous vicinal coupling constants as piperidines 4 and 5 (3JFP 63 Hz) suggesting eclipsed conformation, with dihedral angle value c.a. 0° between C–P and C–F bond (equatorials). Additionally, 19F–1H HOESY experiments showing NOEs between a fluorine atom and protons: H-2, H-4 and H-5, analogically to compound 4. These observations allow us to propose configuration (2S,3R) for 13a and (2S,3S) for piperidine 13b with trans- and cis-arrangements of fluorine and phosphonate group in a six-member ring.25 Analogous reaction of 3a,b with PyFluor (DBU, toluene) led to 13a in addition to β-amino-α-fluoroalkylphosphonates 14 (2.2:1, crude 13a:14 ratio). Moreover, remaining diastereoisomer 3b stayed intact in the reaction mixture, while reaction at a higher temperature (45 °C, 0.75 h) led to decomposition of starting materials. These results are contrary to the reaction of β-amino-α-hydroxyalkylphosphonates with PyFluor where mainly α-fluoroalkylphosphonates were formed,17 although the amount of the second regioisomer in our case was higher compared to the analogous reaction of 3a,b with DAST.
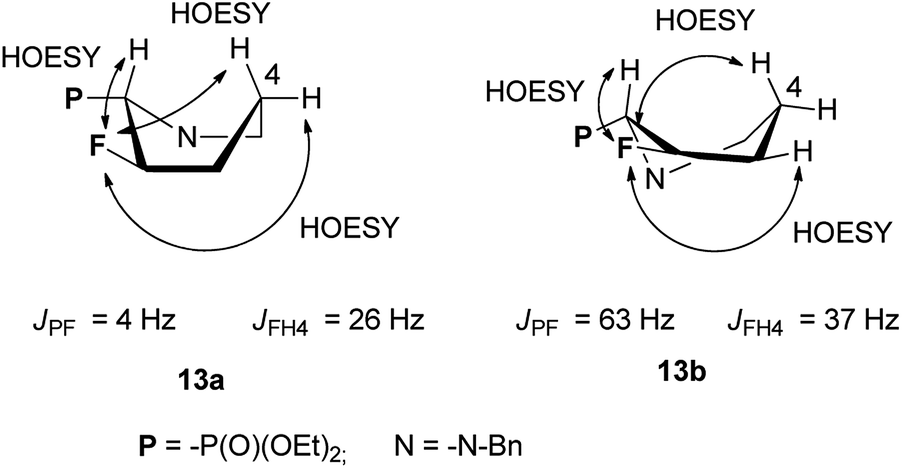 | ||
| Scheme 6 The conformations of 13a and 13b with observed 19F–1H NOEs correlations and some values of coupling constants. | ||
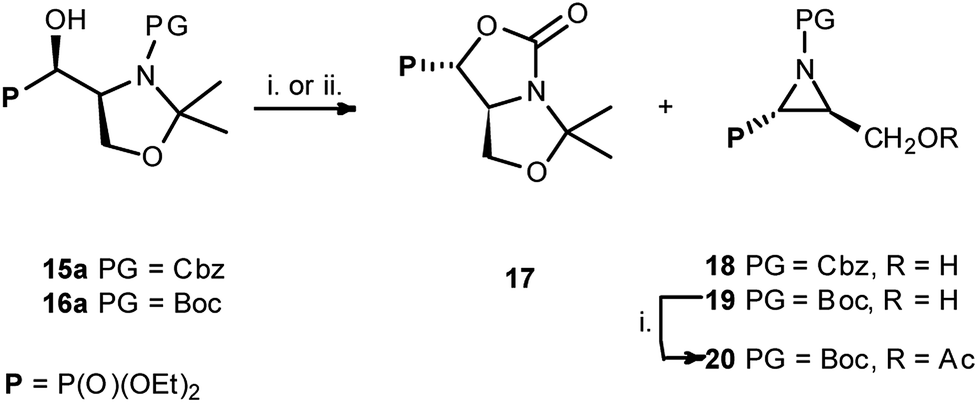
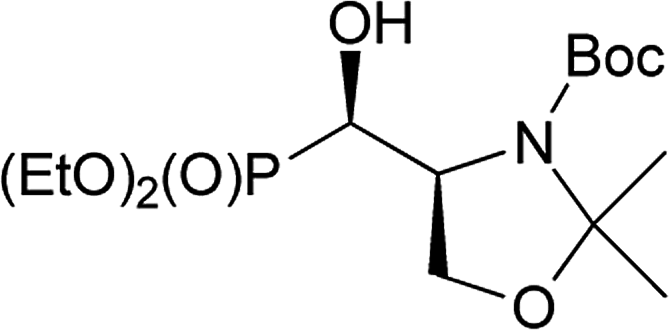
Taking into account the neighbouring groups participation in deoxyfluorination of α-hydroxyphosphonate proline derivatives, we have decided to investigate similar reaction on serine derivatives having N-Cbz, N-Boc, and N-Bn protecting groups. Thus, the reaction with DAST [−78 °C (3 h) → RT (16 h)] carried on 15a,b or 16a,b [99:1 d.r., (4S,1′R,:4R,1′S)] gave two type of products: bicyclic 17 and aziridines 18 with yields 32% and 17% (or 17 and 19 with yields 40% and 56%, respectively) (Scheme 7).
 | ||
| Scheme 7 Reaction of 15–16a with DAST (i) or DeoxoFluor (ii) from 15a: DAST: 17 32% and 18a 17%; from 16a (Table 3), and preparation of 20 ((i) Ac2O, K2CO3, AcOEt, 20 82%). | ||
Similarly, to the formation of 6a,b, bicyclic oxazolidine-2-one 17 arose by the attack of carbamate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O electron pair (from Cbz or Boc) on leaving group (–OSF2N(Et)2) coming from the reaction of an alcohol moiety with DAST (Scheme 7). On the base of NMR analysis, we were able to assign stereochemistry of compound 17. Thus, diagnostic signals located at δH 4.38 ppm (dd, J 6 Hz, CHP) and at δH 4.54 ppm (ddt, J 15, 7, 6 Hz, CHCHP) indicated (1S,7aS) diastereoisomer of 17, and additional NOESY experiments confirmed that both protons are on the opposite side of oxazolidinone ring. Moreover, coupling constants value 3JPH 15 Hz in case of 17 corresponded to dihedral-angle dependence in phosphonates.30 Similar value 3JPH 11 Hz was also reported by De La Cruz et al. and confirmed trans oxazolidine-2-one formation.31 Second isolated type of products, aziridines 18 or 19 were formed by attack of electrons from neighbouring nitrogen atom (N-Boc, Cbz) on hydroxyl derived leaving group (-OSF2N(Et)2), as in case of first step of 4 and 5 formation (pathway a, Scheme 3), with subsequent removal of N,O-isopropylidene protecting group. These assumptions were confirmed by NMR spectroscopy, as well as the transformation of 19, to known acetyl derivatives 2032 additionally proving configuration (2S,3S) of compound 19. Moreover, aziridine 19 existed as a mixture of two rotamers that could be separated by the chromatography techniques.
O electron pair (from Cbz or Boc) on leaving group (–OSF2N(Et)2) coming from the reaction of an alcohol moiety with DAST (Scheme 7). On the base of NMR analysis, we were able to assign stereochemistry of compound 17. Thus, diagnostic signals located at δH 4.38 ppm (dd, J 6 Hz, CHP) and at δH 4.54 ppm (ddt, J 15, 7, 6 Hz, CHCHP) indicated (1S,7aS) diastereoisomer of 17, and additional NOESY experiments confirmed that both protons are on the opposite side of oxazolidinone ring. Moreover, coupling constants value 3JPH 15 Hz in case of 17 corresponded to dihedral-angle dependence in phosphonates.30 Similar value 3JPH 11 Hz was also reported by De La Cruz et al. and confirmed trans oxazolidine-2-one formation.31 Second isolated type of products, aziridines 18 or 19 were formed by attack of electrons from neighbouring nitrogen atom (N-Boc, Cbz) on hydroxyl derived leaving group (-OSF2N(Et)2), as in case of first step of 4 and 5 formation (pathway a, Scheme 3), with subsequent removal of N,O-isopropylidene protecting group. These assumptions were confirmed by NMR spectroscopy, as well as the transformation of 19, to known acetyl derivatives 2032 additionally proving configuration (2S,3S) of compound 19. Moreover, aziridine 19 existed as a mixture of two rotamers that could be separated by the chromatography techniques.
The formation of aziridine from aziridinium ion by DAST treatment is contrary to known ring-expansion reactions observed for hydroxyphosphonate derivatives of prolinal 1a,b–3a,b. However, treatment of hydroxy diazepan-2-ones33 or indolizine34 derivatives with DAST followed by the nitrogen participation yielded ring contractions as well. On the other hand, application of DAST or Deoxofluor with 16a under varied conditions gave aziridine 19 and phosphonates 17 and/or 21, 22a,b respectively (Table 3).
|
|||||
|---|---|---|---|---|---|
| Substrate 16a | Cond. | Ratio (isolated yield%) | |||
| 17 | 21 | 22a,b | 19 | ||
| a (i) DAST, −78 °C (3 h) → RT (16 h); (ii) DAST, −78 °C → 0 °C (1 h); (iii) DAST, 0 °C → RT (0.5 h); (iv) DAST, −78 °C (3 h) → 0 °C (0.5 h) → RT (16 h); (v) Deoxofluor, RT (30 h). | |||||
 |
i | 2.1 (40) | — | — | 1 (33) |
| ii | 1.8 (38) | 1 (25) | — | 1.2 (10) | |
| iii | 1 (4) | 9.4 (38) | — | 1.9 (8) | |
| iv | — | 1 (—) | 16 (20:1) |
3 (5) | |
| v | 1.6 (34) | — | — | 1 (18) | |
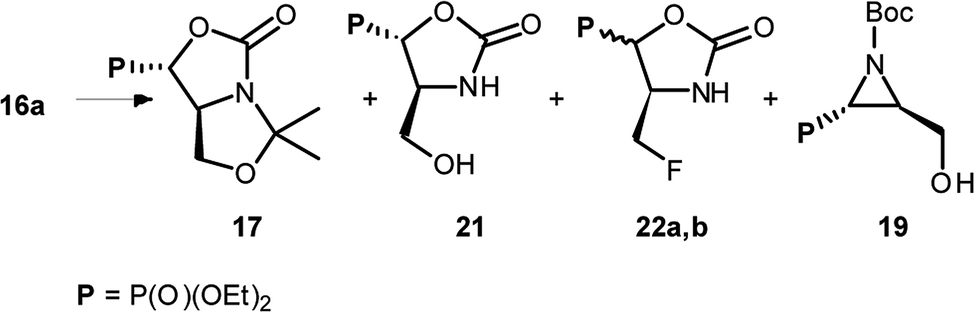
Surprisingly, when the temperature of the reaction mixture was increased (0 °C → RT, 0.75 h) as a major product oxazolidine-2-one 21 (after workup, isolated yield 38%), with a minor aziridine 19, were obtained. Moreover, extended reaction time at RT [0 °C (0.5 h) → RT (16 h)] gave fluoride 22a (isolated yield 37%). Analogous treatment of 16a with DeoxoFluor gave the same results as with DAST. The structure and stereochemistry of compound 21 were assumed to be analogous to 17, since only N,O-isopropylidene protecting group was removed. Thus trans-oxazolidinone ring geometry was confirmed by NMR spectra analysis (3JHP 18 Hz) indicating (4S,5S)-21 configurations. On the base of these observations, we propose the mechanism of DAST-induced transformation of 16a yielding 21 or 22a (Scheme 8).
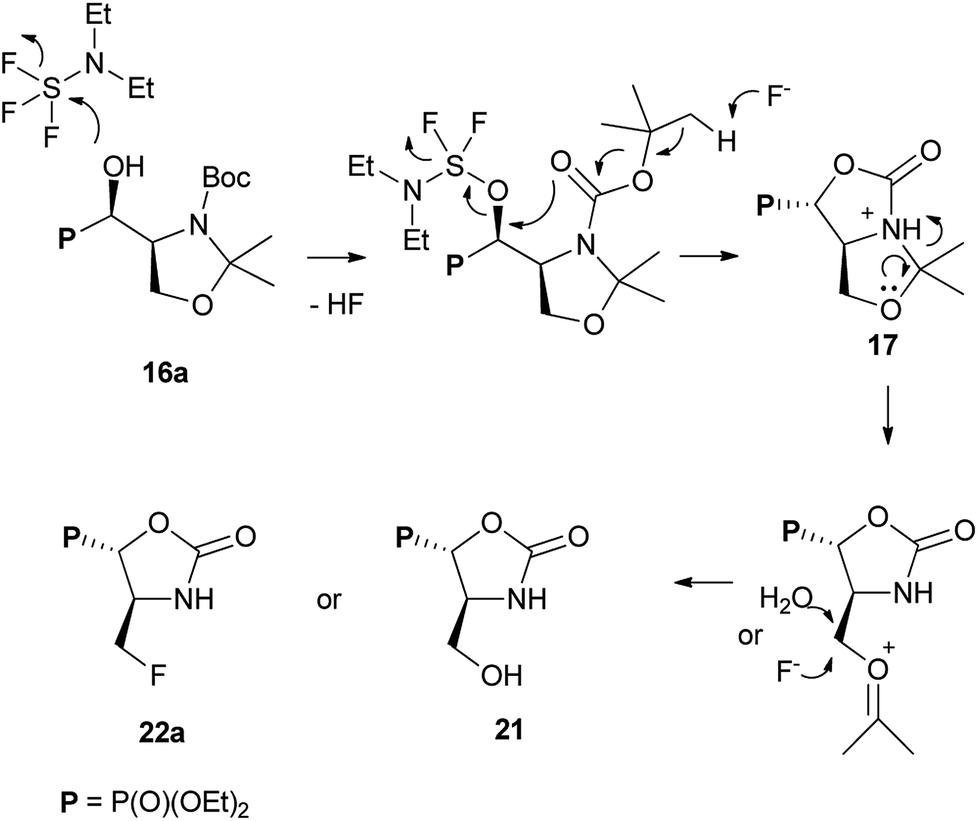 | ||
| Scheme 8 The mechanism of DAST-induced transformation of 16a leading to 21 or 22a. | ||
In the beginning, we observed the formation of bicyclic oxazolidine-2-one 17. Subsequent removal of N,O-isopropylidene protecting group gave after workup hydroxymethyl derivative 21 or due to the attack of fluoride “HF2−” (extended reaction time) led to fluorinated phosphonate oxazolidine-2-one 22a. Similar DAST-mediated removal of O-isopropylidene group has been reported for deoxyfluorination of α-hydroxyphosphonate derivatives of glyceraldehyde yielding fosfomycin analogue.21 On the other hand, during the reaction of 16a with DAST, we have observed the 22b formation, which epimerized during reaction and purification on silica gel to give exclusively 22a (NMR). The presence of fluoride in exocyclic methyl group is confirmed by the high-field chemical shift of CH2F signal at δF ∼ −230/−235 ppm (td, J 47, 19/22 Hz). The configurations of 22a as (4S,5S) has been confirmed by 2D NMR experiments. Thus, 19F–1H HOESY experiment showed NOEs between a fluorine atom and geminal protons in CH2F as well as with H-4 (CHP) while no NOEs between H-4 and H-5 has been detected (1H–1H NOESY) and proved trans arrangement of protons in oxazolidine-2-one ring. Moreover, the reaction of 15a with DAST at RT yielded 21 and 22a. Analogously, the deprotection/deoxyfluorination were applied in case of synthesis of N-protected L-fluoroalanine. Thus, the desired compound has been obtained by a desilylation/deoxofluorination reaction of oxazolidinone analogue of L-serine using XtalFluor-E in the presence of triethylamine trihydrofluoride.35
At the same time, the reaction performed on 15a as well as on 16a with PyFluor gave sulfonates 23 or 24 with 60% and 47% isolated yields (Scheme 9), similarly to the reaction of 1–2a,b with PyFluor leading to compounds 11–12a,b.
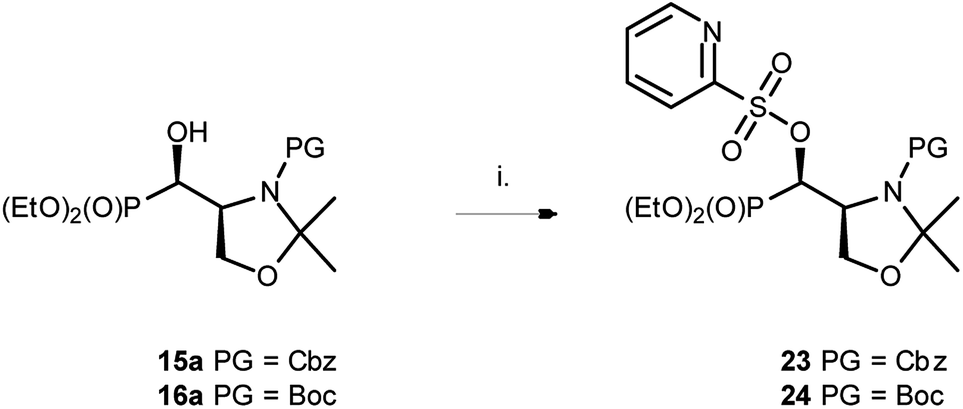 | ||
| Scheme 9 Reaction of 15–16a with PyFluor. (i) PyFluor, DBU, MePh, RT, 5d; 23a 60%, 24a (47%). | ||
To compare, the reaction of 25a,b [12.5:1 or 3.4:1, d.r., (4S,1′R:4R,1′S)]) with DAST at RT (0.5 h) or at 0 °C (2 h) → RT (2 h) gave compound 26a with a traces of 26b, while conditions starting from −78 °C (3 h) → 0 °C (1 h) led to the compound 26a only (as two rotamers in ratio 98:2, 58% of yield). Moreover, the reaction of 25a,b (3.4:1) with PyFluor (PyFluor (2.4 eq.), DBU (4 eq.), MePh, RT, 5 d) gave only 26a (37%) (Scheme 10). The mechanism of the formation of 26a relied on the attack of fluoride during removal of O-isopropylidene protection (as depicted on Scheme 8) followed by substitution of leaving group (–OSF2N(Et)2) by oxygen atom derived from just created carbonyl group. While 1H, 13C and 19F NMR spectra of 26a are similar to 22a and indicated trans arrangement of protons in oxazolidine-2-one ring,28 as well as presence of CH2F group, the chemical shifts of signals in 31P NMR are distinctively different from 22a (δP 15.7) but fit to structure of oxazolidine ring in 26a (δP 21.6).30 Additionally, NOESY experiments indicating correlations between CHP–CHHF, while other relationship for CHN and CHHF as well as NOE between a fluorine atom and CHN (1H–1H NOESY, 1H–19F HOESY) confirmed the structure of 26a as trans oxazolidine.
 | ||
| Scheme 10 Reaction of 25a,b with DAST or PyFluor. (i) DAST, RT 0.5 h (26a 58%); (ii) PyFluor, DBU, MePh, RT, 5d; 26a (37%). | ||
Conclusions
In summary, we have discussed the DAST/DeoxoFluor induced transformation of proline or serine derived hydroxyphosphonates having -Cbz, -Boc and -Bn moieties as N- protecting groups. It seems that diastereoselective course of deoxyfluorination depends on the participation of the neighbouring group and applied reagent. Thus, the reaction of N-Cbz or N-Boc prolinols 1–2a with DAST or Deoxofluor, through aziridinium intermediate and ring opening gave fluorinated piperidine phosphonates 4 or 5 and minors pyrrolidine fluorides 9 or 10, respectively. In addition, due to the participation of N-protecting group oxazolidine-2-ones 6–7a were formed. Analogically DAST/DeoxoFluor treatment of the second diastereoisomer of 1–2b led only to oxazolidine-2-ones 6–7b. Similarly, the reaction of N-Cbz or N-Boc protected serinols 15,16a with DAST or Deoxyfluor yielded analogous oxazolidine-2-one 17 transforming during workup to 21 or by fluorination to 22a,b, as presented in proposed mechanism. As a second path of the reaction, aziridines 18 and 19 were isolated as the ring contraction products. Only in case of deoxyfluorination of N-benzyl prolinols 3a,b both diastereoisomers of β-fluoropiperidine-α-phosphonates 13a,b were formed, while the reaction of protected N-benzyl serinol 25a,b gave fluorinated oxazolidines 26a,b. Moreover, application of PyFluor in the reactions with 1–2a,b and 15–16a,b gave sulfonates 11–12a,b and 23–24. These studies gave an example of the synthesis of valuable building blocks for the asymmetric synthesis of peptide analogues as well as versatile substrates in the synthesis of biologically active species since amino phosphonates mimic naturally occurring α-amino acids.Experimental part
General information
1H NMR, 13C NMR, 19F NMR and 31P NMR spectra were performed on Bruker ASCEND 400 (400 MHz), Bruker ASCEND 600 (600 MHz) spectrometers in CDCl3 solution. All 2D NMR spectra were recorded on Bruker ASCEND 600 (600 MHz) spectrometer. Chemical shifts of 1H NMR were expressed in parts per million downfield from tetramethylsilane (TMS) as an internal standard (δ = 0) in CDCl3 or CDCl3 (δ = 7.26). Chemical shifts of 13C NMR were expressed in parts per million downfield and upfield from CDCl3 as an internal standard (δ = 77.16). Chemical shifts of 19F NMR were expressed in parts per million upfield from CFCl3 as an internal standard (δ = 0) in CDCl3. Chemical shifts of 31P NMR were expressed in parts per million in CDCl3. All d.r. ratios were evaluated on the basis of 31P NMR in crude reaction mixture. High-resolution mass spectra were recorded by electron spray (MS-ESI) techniques using QToF Impact HD Bruker spectrometer. Reagent grade chemicals were used and solvents were dried by refluxing with sodium metal (toluene), with CaH2 (DCM) and distilled under an argon atmosphere. All moisture sensitive reactions were carried out under an argon atmosphere using oven-dried glassware. Reaction temperatures below 0 °C were performed using a cooling bath (liquid N2/i-PrOH). TLC was performed on Merck Kieselgel 60-F254 with EtOAc/hexane, EtOAc//EtOAc/i-PrOH/H2O (4:1:2, upper layer; SSE) or CHCl3/MeOH as developing systems, and products were detected by inspection under UV light (254 nm) and with a solution of potassium permanganate. Merck Kieselgel 60 (230–400 mesh) was used for column chromatography. DAST was supplied by Sigma Aldrich or Apollo Scientific. All remaining starting materials were supplied by Sigma Aldrich. Substrates have to be well dried prior to use. Compounds 1a,b, 3a,b, 15a,b, 25a,b;36 2a,b;6c 16a,b37 were prepared as described.
1.1. Procedure A. Reactions of α-hydroxyphosphonates with DAST. To a solution of DAST (4 eq.) in dry CH2Cl2 (7 mL) in a cooling bath (liquid N2/i-PrOH, or ice), α-hydroxyphosphonates (1 eq.) in dry CH2Cl2 (3 mL) was added slowly and a reaction mixture was kept accordingly to notes below. Then the reaction mixture was diluted with water (5 mL), extracted with CH2Cl2 (3 × 15 mL), dried (Na2SO4), filtered and concentrated. The products were isolated using column chromatography (CHCl3/MeOH or EtOAc/hexane).
1.2. Procedure B. Reactions of α-hydroxyphosphonates with DeoxoFluor. To a solution of α-hydroxyphosphonates (1 eq.) in dry CH2Cl2 (5 mL), DeoxoFluor (2 eq.) was added and reaction mixture was stirred at room temperature for 24 h under ambient atmosphere. Then the reaction mixture was diluted with water (5 mL), extracted with CH2Cl2 (3 × 5 mL), dried over Na2SO4, and filtered. Removal of solvent at reduced pressure gave a residue, which was then purified using column chromatography (CHCl3/MeOH or EtOAc/hexane).
1.3. Procedure C. Reactions of α-hydroxyphosphonate with PyFluor. To a solution of α-hydroxyphosphonates (1 eq.) in dry CH2Cl2 (5 mL), PyFluor (2.4 eq.) and DBU (4 eq.) was added and reaction mixture was stirred at room temperature for 4 days under ambient atmosphere (monitored by TLC). After reaction was completed, the solvent was removed at reduced pressure, and the products were isolated using column chromatography (CHCl3/MeOH).
Note A1: treatment of 1a:1b (22.8:1 d.r.) according to procedure A [−78 °C → 0 °C (1.5 h)] gave 4 as a mixture with 9 (60 mg, 43% and 8%, respectively, 5.8:1) and 6a,6b (38 mg, 45%, 9.5:1 d.r.).
Note A2: treatment of 1a:1b (2.7:1 d.r.) according to procedure A [−78 °C (3 h); RT (1 h)] gave 4 (43 mg, 36%) and 6a,6b (47 mg, 56%, 1.86:1 d.r.).
Note A3: treatment of 2a:2b (3.8:1 d.r.) according to procedure A [−78 °C (3 h); RT (1 h)] gave 5 (60 mg, 38%) and 7a,7b (65 mg, 53%, 1.9:1 d.r.).
Note A4: treatment of 3a:3b (1.1:1 d.r.) according to procedure A [0 °C → 45 °C (1 h)] gave 13a,b in few fractions containing different ratio of diastereoisomers (125 mg, 59%, 6.2:1 d.r.).
Note A5: treatment of 15a according to procedure A [0 °C (0.5 h); RT (18 h)] gave 17 (33 mg, 32%) and 18 (21 mg, 17%).
Note A6: treatment of 16a according to procedure A [−78 °C (3 h); RT (16 h)] gave 17 (18 mg, 40%) and 19 (27 mg, 33%).
Note A7: treatment of 16a according to procedure A [0 °C → RT (0.5 h)] gave 17 (5 mg, 4%), 19 (12 mg, 8%) and 21 (41 mg, 38%).
Note A8: treatment of 16a according to procedure A [−78 °C (3 h) → 0 °C (0.5 h); RT (16 h)]; gave 19 (9 mg, 5%) and 22a,b (59 mg, 37%).
Note A9: treatment of 25a,b (3.4:1 d.r.) according to procedure A (RT, 0.5 h) gave compounds 26a (19 mg, 58%).
Note B1: treatment of 3a:3b (2.1:1 d.r.) according to procedure B gave compounds 13a,b (48 mg, 5.7:1, d.r., 74%).
Note C1: treatment of 1a,b (3.3:1 d.r.) according to procedure C, gave 11a,b (72 mg, 3:1, 78%)
Note C2: treatment of 1a,b (24:1:1 d.r.) according to procedure C, gave 11a,b (36 mg, 18:1, 69%)
Note C3: treatment of 2a,b (37:1 d.r.) according to procedure C, gave 12a,b (42 mg, 74:1 d.r., 74%)
Note C4: treatment of 3a:3b (1.9:1 d.r.) according to procedure C gave compounds 13a (10 mg, 19%)
Note C5: treatment of 15a according to procedure C gave compounds 23 (31 mg, 60%).
Note C6: treatment of 16a according to procedure C gave compounds 24 (32 mg, 47%).
Note C7: treatment of 25a,b (3.4:1 d.r.) according to procedure C gave compounds 26a (24 mg, 37%).
(2R,3R)-benzyl 2-(diethoxyphosphoryl)-3-fluoropiperidine-1-carboxylate (4). Isolated with a yield 36% (Note A2) or with 43% as a mixture with 9 (Note A1) as slightly yellow oil, mixture of two rotamers (1.1
:1). Major rotamer had: 1H NMR (400 MHz) δ = 7.39–7.33 (m, 4H, Ph), 7.33–7.30 (m, 1H, Ph), 5.21 (d, J = 12.3 Hz, 1H, CHHPh), 5.12 (d, J = 12.3 Hz, 1H, CHHPh), 5.03 (dd, J = 21.3, 19.4 Hz, 1H, CHP), 5.02 (dd, J = 46.6, 12.2 Hz, 1H, CHF), 4.17–4.10 (m, 3H, NCHH, OCH2CH3), 4.09–3.99 (m, 2H, OCH2CH3), 3.37 (td, J = 13.2, 2.6 Hz, 1H, NCHH), 2.07–2.02 (m, 2H, CH2CHF), 1.91–1.76 (m, 1H, NCH2CHH), 1.57–1.48 (m, 1H, NCH2CHH), 1.32 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.24 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz) δ = 155.37 (d, J = 3.0 Hz, CO), 136.45, 128.50, 128.11, 127.96 (4 × s, Ph), 85.45 (dd, J = 178.9, 19.9 Hz, CHF), 67.72 (s, CH2Ph), 63.06 (d, J = 7.3 Hz, OCH2CH3), 62.48 (d, J = 6.8 Hz, OCH2CH3), 53.23 (dd, J = 150.2, 22.5 Hz, CHP), 41.21 (s, NCH2), 26.36 (d, J = 6.0 Hz, CH2CHF), 19.00 (s, NCH2CH2), 16.36 (d, J = 5.6 Hz, OCH2CH3), 16.34 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (377 MHz) δ = −179.55 to −180.05 (m). 31P {/1H} NMR (162 MHz) δ = 19.24 (d, J = 61.7 Hz). Minor rotamer had: 1H NMR (400 MHz) δ = 7.39–7.31 (m, 4H, Ph), 7.33–7.30 (m, 1H, Ph), 5.20 (d, J = 12.3 Hz, 1H, CHHPh), 5.12 (d, J = 12.3 Hz, 1H, CHHPh), 5.09 (dd, J = 44.8, 17.7 Hz, 1H, CHF), 4.86 (dd, J = 21.0, 18.8 Hz, 1H, CHP), 4.26 (d, J = 13.6 Hz, 1H, NCHH), 4.17–4.10 (m, 2H, OCH2CH3), 4.09–3.99 (m, 2H, OCH2CH3), 3.26 (td, J = 13.3, 2.7 Hz, 1H, NCHH), 2.21–2.12 (m, 1H, CHHCHF), 2.11–2.07 (m, 1H, CHHCHF), 1.91–1.76 (m, 1H, NCH2CHH), 1.57–1.48 (m, 1H, NCH2CHH), 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.22 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz) δ = 155.73 (d, J = 3.8 Hz, CO), 136.32, 128.48, 128.11, 127.98 (4 × s, Ph), 85.31 (dd, J = 179.0, 20.2 Hz, CHF), 67.70 (s, CH2Ph), 63.01 (d, J = 7.1 Hz, OCH2CH3), 62.34 (d, J = 7.1 Hz, OCH2CH3), 54.11 (dd, J = 150.4, 22.3 Hz, CHP), 40.85 (s, NCH2), 26.15 (d, J = 5.9 Hz, CH2CHF), 18.82 (s, NCH2CH2), 16.44 (d, J = 5.7 Hz, OCH2CH3), 16.36 (d, J = 5.6 Hz, OCH2CH3). 19F NMR (377 MHz) δ = −178.86 to −179.39 (m). 31P {/1H} NMR (162 MHz) δ = 18.96 (d, J = 62.0 Hz). HRMS (ESI) calcd for C17H25FNNaO5P+ ([M + Na]+): 396.1347, found: 396.1361.
(2S,3S)-tert-butyl 2-(diethoxyphosphoryl)-3-fluoropiperidine-1-carboxylate (5). Isolated with a yield 38% (Note A3), as slightly yellow oil, mixture of two rotamers (1.1
:1).
Major rotamer had: 1H NMR (400 MHz) δ = 5.07 (br d, J = 42.0 Hz, 1H, CHF), 5.00 (dd, J = 21.5, 18.1 Hz, 1H, CHP), 4.24–4.09 (m, 4H, 2 × OCH2CH3), 4.04 (br d, J = 13.6 Hz, 1H, NCHH), 3.30 (td, J = 13.3, 3.0 Hz, 1H, NCHH), 2.16 (tdd, J = 13.6, 4.7, 2.3 Hz, 1H, CHHCHF), 2.08–2.02 (m, 1H, CHHCHF), 1.87–1.75 (m, 1H, NCH2CHH), 1.53–1.50 (m, 1H, NCH2CHH), 1.48 (s, 9H, C(CH3)3), 1.34 (t, J = 6.1 Hz, 3H, OCH2CH3), 1.32 (t, J = 6.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz) δ = 154.63 (d, J = 2.9 Hz, CO), 85.56 (dd, J = 178.8, 2.2 Hz, CHF), 80.58 (s, C(CH3)3), 62.31 (d, J = 6.8 Hz, OCH2CH3), 62.17 (d, J = 7.1 Hz, OCH2CH3), 52.35 (dd, J = 149.7, 22.5 Hz, CHP), 41.28 (s, NCH2), 28.30 (s, C(CH3)3), 26.31 (d, J = 21.4 Hz, CH2CHF), 19.03 (s, NCH2CH2), 16.42 (d, J = 5.9 Hz, OCH2CH3), 16.31 (d, J = 6.2 Hz, OCH2CH3). 19F {/1H} NMR (376 MHz) δ = −179.53 (d, J = 63.3 Hz). 19F NMR (376 MHz) δ = −179.25 to −179.78 (m). 31P {/1H} NMR (162 MHz) δ = 19.78 (d, J = 62.9 Hz). Minor rotamer: 1H NMR (400 MHz) δ = 4.99 (br d, J = 42.5 Hz, 1H, CHF), 4.80 (dd, J = 24.0, 20.4 Hz, 1H, CHP), 4.23–4.21 (m, 1H, NCHH), 4.24–4.09 (m, 4H, 2 × OCH2CH3), 3.15 (td, J = 13.3, 2.9 Hz, 1H, NCHH), 2.08–2.02 (m, 2H, CH2CHF), 1.87–1.75 (m, 1H, NCH2CHH), 1.53–1.50 (m, 1H, NCH2CHH), 1.48 (s, 9H, C(CH3)3), 1.36 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.33 (t, J = 6.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz) δ = 154.83 (d, J = 3.5 Hz, CO), 85.35 (dd, J = 178.8, 2.4 Hz, CHF), 80.36 (s, C(CH3)3), 62.92 (d, J = 7.1 Hz, OCH2CH3), 62.85 (d, J = 7.1 Hz, OCH2CH3), 54.35 (dd, J = 150.1, 22.5 Hz, CHP), 39.96 (s, NCH2), 28.27 (s, C(CH3)3), 26.39 (d, J = 21.6 Hz, CH2CHF), 18.85 (s, NCH2CH2), 16.55 (d, J = 5.6 Hz, OCH2CH3), 16.42 (d, J = 5.9 Hz, OCH2CH3). 19F {/1H} NMR (376 MHz) δ = −179.99 (d, J = 63.0 Hz). 19F NMR (376 MHz) δ = −179.74 to −180.25 (m). 31P {/1H} NMR (162 MHz) δ = 19.70 (d, J = 63.1 Hz). HRMS (ESI) calcd for C14H27FNNaO5P+ ([M + Na]+): 362.1503, found: 362.1511.
Diethyl ((1R,7aR)-3-oxohexahydropyrrolo[1,2-c]oxazol-1-yl)phosphonate and diethyl ((1S,7aR)-3-oxohexahydropyrrolo[1,2-c]oxazol-1-yl)phosphonate (6a,6b) and diethyl ((1S,7aS)-3-oxohexahydropyrrolo[1,2-c]oxazol-1-yl)phosphonate and diethyl ((1R,7aS)-3-oxohexahydropyrrolo[1,2-c]oxazol-1-yl)phosphonate(7a,7b). Isolated with a yield 45% (9.5
:1 d.r., Note A1) or 56% (1.86:1 d.r., Note A2) or 53% (1.9:1 d.r., Note A3) as a transparent oil, mixture of two diastereoisomers.
Major diastereoisomer 6a/7a had: 1H NMR (400 MHz) δ = 4.44 (d, J = 4.4 Hz, 1H, CHP), 4.27–4.20 (m, 4H, 2 × OCH2CH3), 4.10 (tdd, J = 11.3, 6.2, 2.7 Hz, 1H, CHCHP), 3.66–3.54 (m, 1H, NCHH), 3.25–3.16 (m, 1H, NCHH), 2.21–2.13 (m, 1H, CHHCH), 2.15–2.06 (m, 1H, NCH2CHH), 2.02–1.92 (m, 1H, NCH2CHH), 1.59–1.48 (m, 1H, CHHCH), 1.39–1.33 (m, 6H, 2 × OCH2CH3). 13C NMR (101 MHz) δ = 159.82 (d, J = 3.9 Hz, CO), 73.80 (d, J = 173.6 Hz, CHP), 63.95 (d, J = 6.9 Hz, OCH2CH3), 63.47 (d, J = 6.7 Hz, OCH2CH3), 60.30 (s, CHCHP), 45.80 (s, NCH2), 31.49 (d, J = 11.0 Hz, CH2CH), 25.51 (s, NCH2CH2), 16.48 (d, J = 5.4 Hz, 2 × OCH2CH3). 31P {/1H} NMR (162 MHz) δ = 16.49 (s). Minor diastereoisomer 6b/7b had: 1H NMR (400 MHz) δ = 4.80 (ddd, J = 8.3, 3.6, 2.1 Hz, 1H, CHP), 4.27–4.20 (m, 4H, 2 × OCH2CH3), 4.09–3.99 (m, 1H, CHCHP), 3.66–3.54 (m, 1H, NCHH), 3.25–3.16 (m, 1H, NCHH), 2.15–2.06 (m, 1H, NCH2CHH), 2.02–1.92 (m, 2H, CH2CH), 1.95–1.86 (m, 1H, NCH2CHH), 1.39–1.33 (m, 6H, 2 × OCH2CH3). 13C NMR (101 MHz) δ = 160.17 (d, J = 9.3 Hz, CO), 70.62 (d, J = 172.3 Hz, CHP), 63.83 (d, J = 7.0 Hz, OCH2CH3), 63.15 (d, J = 6.8 Hz, OCH2CH3), 61.01 (s, CHCHP), 45.61 (s, NCH2), 26.91 (d, J = 5.7 Hz, CH2CH), 25.70 (s, NCH2CH2), 16.44 (br d, J = 5.4 Hz, 2 × OCH2CH3). 31P {/1H} NMR (162 MHz) δ = 14.56 (s). HRMS (ESI) calcd for C10H18NNaO5P+ ([M + Na]+): 286.0815, found: 286.0829.
(2R)-benzyl 2-((diethoxyphosphoryl)fluoromethyl)pyrrolidine-1-carboxylate9. Isolated with a yield 8% as a mixture with 4 (Note A1); slightly yellow oil, a mixture of two rotamers 1.46
:1. Major rotamer had: 19F NMR (565 MHz) δ = −226.78 (ddd, J = 78.6, 46.9, 39.8 Hz). 31P {/1H} NMR (243 MHz) δ = 15.71 (d, J = 76.5 Hz). Minor rotamer had: 19F NMR (565 MHz) δ = −225.83 (ddd, J = 75.1, 47.1, 33.8 Hz). 31P {/1H} NMR (243 MHz) δ = 15.36 (d, J = 75.7 Hz).
(2S)-tert-butyl 2-((diethoxyphosphoryl)fluoromethyl)pyrrolidine-1-carboxylate (10). Observed in a crude reaction mixture as two rotamers 1.08
:1. Major rotamer had: 19F NMR (565 MHz) δ = −226.96 (ddd, J = 79.1, 47.0, 34.2 Hz). 31P {/1H} NMR (243 MHz) δ = 16.14 (d, J = 77.6 Hz). Minor rotamer had: 19F NMR (565 MHz) δ = −226.08 (ddd, J = 79.9, 47.2, 34.4 Hz). 31P {/1H} NMR (243 MHz) δ = 15.94 (d, J = 77.5 Hz).
Benzyl (R)-2-((S)-(diethoxyphosphoryl)((pyridin-2-ylsulfonyl)oxy)methyl)pyrrolidine-1-carboxylate (11a) and benzyl (R)-2-((R)-(diethoxyphosphoryl)((pyridin-2-ylsulfonyl)oxy)methyl)pyrrolidine-1-carboxylate (11b). Isolated with yield 78% (3
:1, d.r., Note C1), or 69% (18:1 d.r.; Note C2) as a transparent oil, mixture of two diastereoisomers. Major diastereoisomer 11a exist as a mixture of two rotamers (1.1:1). Major diastereoisomer 11a (major rotamer) had: 1H NMR (600 MHz) δ = 8.68 (d, J = 4.0 Hz, 1H, Ar), 7.92 (d, J = 7.9 Hz, 1H, Ar), 7.74 (m, 1H, Ar), 7.47 (br d, J = 7.6 Hz, 1H, Ph), 7.44 (dd, J = 7.6, 4.8 Hz, 1H, Ar), 7.41–7.33 (m, 3H, Ph), 7.35–7.29 (m, 1H, Ph), 5.77 (dd, J = 11.9, 1.7 Hz, 1H, CHP), 5.14–5.08 (m, 2H, OCH2Ph), 4.24 (dd, J = 9.1, 4.8 Hz, 1H, CHCHP), 4.18–4.06 (m, 4H, 2 × OCH2CH3), 3.28 (q, J = 7.7 Hz, 1H, NCHH), 2.96 (ddd, J = 10.4, 7.5, 5.5 Hz, 1H, NCHH), 2.34–2.22 (m, 1H, CHHCH), 2.07–1.97 (m, 1H, CHHCH), 1,97–1.87 (m, 1H, NCH2CHH), 1.78–1.66 (m, 1H, NCH2CHH), 1.33–1.26 (m, 3H, OCH2CH3), 1.23 (t, J = 7.0 Hz, 3H, OCH2CH3). 13C NMR (101 MHz) δ = 154.92 (s, CO), 154.20 (s, Ar), 150.38 (s, Ar), 138.06, 136.75, 128.63, 127.96 (4 × s, Ph), 127.68 (s, Ar), 123.36 (s, Ar), 77.51 (d, J = 159.1 Hz, CHP), 66.86 (s, OCH2Ph), 63.81 (d, J = 6.9 Hz, OCH2CH3), 63.47 (d, J = 6.6 Hz, OCH2CH3), 57.99 (d, J = 10.2 Hz, CHCHP), 46.59 (s, NCH2), 26.04 (s, CH2CH), 24.53 (s, NCH2CH2), 16.50 (d, J = 5.6 Hz, 2 × OCH2CH3)·31P {/1H} NMR (243 MHz) δ = 15.15 (s). Minor rotamer 11a: 1H NMR (600 MHz) δ = 8.72 (d, J = 4.4 Hz, 1H, Ar), 7.86 (m, 1H, Ar), 7.74 (d, J = 7.8 Hz, 1H, Ar), 7.54 (dd, J = 7.7, 4.7 Hz, 1H, Ar), 7.47 (br d, J = 7.6 Hz, 1H, Ph), 7.41–7.33 (m, 3H, Ph), 7.35–7.29 (m, 1H, Ph), 5.58 (dd, J = 12.1, 1.7 Hz, 1H, CHP), 5.21 (d, J = 12.1 Hz, 1H, OCH2Ph), 5.14–5.08 (m, 1H, OCH2Ph), 4.20–4.15 (m, 1H, CHCHP), 4.18–4.06 (m, 2H, OCH2CH3), 4.06–3.95 (m, 2H, OCH2CH3), 3.19 (dt, J = 10.8, 7.4 Hz, 1H, NCHH), 2.49 (ddd, J = 10.7, 7.4, 5.6 Hz, 1H, NCHH), 2.34–2.22 (m, 1H, CHHCH), 2.07–1.97 (m, 1H, CHHCH), 1.78–1.66 (m, 1H, NCH2CHH), 1.64–1.57 (m, 1H, NCH2CHH), 1.33–1.26 (m, 6H, 2 × OCH2CH3). 13C NMR (101 MHz) δ = 154.45 (s, CO), 154.36, 150.67, 138.38 (3 × s, Ar), 136.41, 128.66, 128.11, 127.76 (4 × s, Ph), 123.03 (s, Ar), 76.44 (d, J = 160.9 Hz, CHP), 67.56 (s, OCH2Ph), 64.12 (d, J = 7.2 Hz, OCH2CH3), 63.32 (d, J = 6.4 Hz, OCH2CH3), 57.13 (d, J = 10.7 Hz, CHCHP), 47.02 (s, NCH2), 27.13 (s, CH2CH), 23.88 (s, NCH2CH2), 16.43–16.29 (m, 2 × OCH2CH3). 31P {/1H} NMR (243 MHz) δ = 14.72 (s). HRMS (ESI) calcd for C22H29KN2O8PS+ ([M + K]+): 551.1014, found: 551.1022.
Minor diastereoisomer 11b exist as a mixture of two rotamers (1:1). Both rotamers 11b had: 1H NMR (600 MHz) δ = 8.72–8.69 (m, 1H, Ar), 8.68–8.65 (m, 1H, Ar), 8.05 (d, J = 7.9 Hz, 1H, Ar), 7.88–7.79 (m, 2H, Ar), 7.76 (d, J = 8.0 Hz, 1H, Ar), 7.55–7.51 (m, 1H, Ph), 7.49 (dd, J = 7.6, 4.6 Hz, 1H, Ar), 7.46–7.40 (m, 1H, Ph), 7.40 (d, J = 7.4 Hz, 1H, Ar), 7.38–7.33 (m, 2H, Ph), 7.33–7.28 (m, 6H, Ph), 5.34 (t, J = 8.8 Hz, 1H, CHP), 5.14–5.11 (m, 1H, CHP), 5.14–5.11 (m, 1H, OCH2Ph), 5.12–5.04 (m, 1H, OCH2Ph), 5.00–4.97 (m, 2H, OCH2Ph), 4.40–4.29 (m, 2H, 2 × CHCHP), 4.19–3.95 (m, 8H, 4 × OCH2CH3), 3.50–3.41 (m, 2H, NCH2), 3.40–3.34 (m, 1H, NCHH), 3.30–3.22 (m, 1H, NCHH), 2.35–2.30 (m, 2H, CH2CH), 2.06–1.97 (m, 1H, CH2CH), 1.95–1.87 (m, 2H, NCH2CH2), 1.82–1.76 (m, 1H, NCH2CH2), 1.33–1.24 (m, 9H, OCH2CH3), 1.24–1.17 (m, 3H, OCH2CH3). 13C NMR (151 MHz) δ = 155.24 (s, CO), 154.69 (s, CO), 154.65, 154.51, 150.40, 150.25, 138.26, 138.20 (6 × s, Ar), 136.68, 136.43, 128.66, 128.48, 128.42, 128.19, 128.06, 127.98 (8 × s, Ph), 127.84 (s, Ar), 127.81 (s, Ar), 123.55 (s, Ar), 123.14 (s, Ar), 76.62 (d, J = 160.4 Hz, CHP), 76.19 (d, J = 161.6 Hz, CHP), 67.23 (s, OCH2Ph), 66.86 (s, OCH2Ph), 64.01 (d, J = 6.3 Hz, 2 × OCH2CH3), 63.27–63.10 (m, 2 × OCH2CH3), 57.33 (d, J = 3.5 Hz, CHCHP), 56.54 (d, J = 5.1 Hz, CHCHP), 47.04 (s, NCH2), 46.83 (s, NCH2), 28.13 (s, CH2CH), 27.20 (s, CH2CH), 23.69 (s, NCH2CH2), 22.91 (s, NCH2CH2), 16.40 (d, J = 6.3 Hz, 2 × OCH2CH3), 16.29 (d, J = 6.0 Hz, 2 × OCH2CH3). 31P {/1H} NMR (243 MHz) δ = 15.44 (s), 15.36 (s).
tert-butyl (S)-2-((R)-(diethoxyphosphoryl)((pyridin-2ylsulfonyl)oxy)methyl)pyrrolidine-1-carboxylate (12a) and tert-butyl (S)-2-((S)-(diethoxyphosphoryl)((pyridin-2-ylsulfonyl)oxy)methyl)pyrrolidine-1-carboxylate (12b). Isolated with a yield 74% (74
:1 d.r., Note C3) as a slightly yellow oil. Major diastereoisomer 12a exist as a mixture of two rotamers (1.7:1). Major diastereoisomer 12a (major rotamer) had: 1H NMR (400 MHz) δ = 8.77–8.70 (m, 1H, Ar), 7.98–7.87 (m, 2H, Ar), 7.57–7.51 (m, 1H, Ar), 5.63 (dd, J = 12.3, 1.6 Hz, 1H, CHP), 4.27–4.12 (m, 4H, 2 × OCH2CH3), 4.12–4.06 (m, 1H, CHCHP), 3.05 (q, J = 7.9 Hz, 1H, NCHH), 2.30–2.15 (m, 2H, NCHH, CHHCH), 2.04–1.87 (m, 1H, CHHCH), 1.72–1.52 (m, 2H, NCH2CH2), 1.52 (br s, 9H, C(CH3)3), 1.39–1.29 (m, 6H, 2 × OCH2CH3). 13C NMR (101 MHz) δ = 154.58 (s, CO), 153.69 (s, Ar), 150.64 (s, Ar), 138.38 (s, Ar), 127.67 (s, Ar), 122.96 (s, Ar), 80.49 (s, C(CH3)3), 78.06 (d, J = 140.2 Hz, CHP), 64.12 (d, J = 7.3 Hz, OCH2CH3), 63.14 (d, J = 6.6 Hz, OCH2CH3), 57.14 (d, J = 11.0 Hz, CHCHP), 46.25 (s, NCH2), 28.52 (s, C(CH3)3), 27.01 (s, CH2CH), 23.83 (s, NCH2CH2), 16.46 (d, J = 5.9 Hz, 2 × OCH2CH3). 31P {/1H} NMR (162 MHz) δ = 15.38 (s). Minor rotamer 12a had: 1H NMR (400 MHz) δ = 8.77–8.70 (m, 1H, Ar), 8.01 (d, J = 7.9 Hz, 1H, Ar), 7.98–7.87 (m, 1H, Ar), 7.57–7.51 (m, 1H, Ar), 5.75 (d, J = 11.7 Hz, 1H, CHP), 4.27–4.12 (m, 1H, CHCHP), 4.12–4.06 (m, 2H, OCH2CH3), 4.06–3.97 (m, 2H, OCH2CH3), 3.29–3.17 (m, 1H, NCHH), 3.05 (q, J = 7.9 Hz, 1H, NCHH), 2.30–2.15 (m, 1H, CHHCH), 2.04–1.87 (m, 3H, CHHCH, NCH2CH2), 1.47 (br s, 9H, C(CH3)3), 1.27 (br t, J = 7.1 Hz, 3H, OCH2CH3), 1.23 (br t, J = 6.8 Hz, 3H, OCH2CH3). 13C NMR (101 MHz) δ = 154.76 (s, CO), 153.69 (s, Ar), 150.30 (s, Ar), 138.05 (s, Ar), 127.63 (s, Ar), 123.40 (s, Ar), 79.79 (s, C(CH3)3), 76.64 (d, J = 144.5 Hz, CHP), 63.42 (d, J = 6.9 Hz, OCH2CH3), 63.37 (d, J = 5.6 Hz, OCH2CH3), 57.38 (d, J = 9.9 Hz, CHCHP), 46.75 (s, NCH2), 28.63 (s, C(CH3)3), 26.04 (s, CH2CH), 24.50 (s, NCH2CH2), 16.57 (d, J = 5.8 Hz, 2 × OCH2CH3). 31P {/1H} NMR (162 MHz) δ = 15.55 (s). Minor diastereoisomer 12b was present in crude reaction mixture as a mixture of two rotamers 3.9:1. Major rotamer had: 31P {/1H} NMR (243 MHz) δ = 15.98 (s). Minor rotamer had: 31P {/1H} NMR (243 MHz) δ = 15.89 (s). HRMS (ESI) calcd for C19H32N2O8PS+ ([M + H]+): 479.1611, found: 479.1606.
Diethyl ((2S,3R)-1-benzyl-3-fluoropiperidin-2-yl)phosphonate (13a) and diethyl ((2S,3S)-1-benzyl-3-fluoropiperidin-2-yl)phosphonate (13b). Isolated with a yield 59% (6.2
:1 d.r., Note A4) or 74% (5.7:1, d.r., Note B1) or 30% (99:1, d.r., Note C4), as slightly yellow oil, mixture of two diastereoisomers. Major diastereoisomer 13a had: 1H NMR (600 MHz) δ = 7.42–7.37 (m, 3H, Ph), 7.32–7.27 (m, 2H, Ph), 4.15–4.07 (m, 4H, 2 × OCH2CH3), 4.06 (dd, J = 14.4, 4.8 Hz, 1H, NCHHPh), 3.89 (d, J = 14.0 Hz, 1H, NCHHPh), 3.34 (t, J = 14.5 Hz, 1H, CHHCHF), 3.30 (d, J = 9.0 Hz, 1H, CHP), 3.26 (ddd, J = 26.1, 14.2, 2.0 Hz, 1H, CHHCHF), 2.97–2.92 (m, 1H, NCHH), 2.62 (br dd, J = 12.1, 3.5 Hz, 1H, NCHH), 2.56–2.48 (m, 1H, NCH2CHH), 1.75 (br d, J = 13.0 Hz, 1H, NCH2CHH), 1.31 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.26 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (151 MHz) δ = 139.90, 128.46, 128.25, 127.14 (4 × s, Ph), 95.87 (dd, J = 177.6, 3.1 Hz, CHF), 64.40 (dd, J = 125.5, 25.9 Hz, CHP), 61.79 (dd, J = 7.3, 4.1 Hz, OCH2CH3) 60.78 (d, J = 7.2 Hz, OCH2CH3), 60.22 (m, CH2Ph), 45.86 (d, J = 3.2 Hz, NCH2), 43.28 (dd, J = 21.8, 10.0 Hz, CH2CHF), 30.20 (d, J = 20.5 Hz, NCH2CH2), 16.64 (d, J = 5.8 Hz, OCH2CH3), 16.54 (d, J = 5.7 Hz, OCH2CH3). 19F NMR (565 MHz) δ = −146.12 (br dt, J = 27.4, 14.2 Hz). 19F {/1H} NMR (565 MHz) δ = −146.11 (d, J = 3.7 Hz). 31P {/1H} NMR (243 MHz) δ = 26.96 (d, J = 4.1 Hz).
Minor diastereoisomer 13b had: 1H NMR (600 MHz) δ = 7.37–7.32 (m, 3H, Ph), 7.31–7.29 (m, 2H, Ph), 4.27–4.23 (m, 1H, OCHHCH3), 4.22–4.18 (m, 1H, OCHHCH3), 4.18–4.07 (m, 4H, OCH2CH3, NCH2Ph) 3.43 (dd, J = 21.9, 13.5 Hz, 1H, CHP), 3.19 (d, J = 14.6 Hz, 1H, NCHH), 3.16 (d, J = 13.1 Hz, 1H, CHHCHF), 3.09 (dd, J = 37.3, 14.2 Hz, 1H, CHHCHF), 2.69 (br d, J = 13.5 Hz, 1H, NCHH), 2.08 (dtd, J = 43.5, 13.6, 4.8 Hz, 1H, NCH2CHH), 1.92–1.86 (m, 1H, NCH2CHH), 1.38 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.32 (t, J = 7.0 Hz, 3H, OCH2CH3). 13C NMR (151 MHz) δ = 135.63, 128.96, 128.27, 128.01 (4 × s, Ph), 95.95 (dd, J = 180.3, 26.3 Hz, CHF), 65.36 (dd, J = 146.2, 21.4 Hz, CHP), 63.12 (d, J = 7.2 Hz, OCH2CH3), 61.25 (d, J = 7.7 Hz, OCH2CH3), 60.56 (dd, J = 12.0, 3.6 Hz, CH2Ph), 46.02 (s, NCH2), 44.34 (d, J = 20.8 Hz, CH2CHF), 29.74 (d, J = 22.4 Hz, NCH2CH2), 16.75 (d, J = 6.2 Hz, OCH2CH3), 16.47 (d, J = 6.2 Hz, OCH2CH3). 19F NMR (565 MHz) δ = −144.73 to −145.09 (m). 31P{/1H} (243 MHz) δ = 23.22 (d, J = 63.4 Hz). HRMS (ESI) calcd for C16H26FNO3P+ ([M + H]+): 330.1629, found: 330.1626, major peak: C23H32FNO3P+ ([M + Bn]+): 420.2098, found: 420.2095.
Diethyl (((S)-1-benzylpyrrolidin-2-yl)fluoromethyl)phosphonate (14). Observed in a crude reaction mixture. Diagnostic signals: 19F NMR (565 MHz) δ = −207.63 (dd, J = 81.6, 44.8 Hz). 31P{/1H} (243 MHz) δ = 16.51 (d, J = 82.0 Hz).
Diethyl (1S,7aS)-5,5-dimethyl-3-oxotetrahydro-1H-oxazolo[3,4-c]oxazol-1-ylphosphonate (17). Isolated with a yield 32% (Note A5) or 40% (Note A6) as a slightly yellow oil. Compound 17 had: 1H NMR (400 MHz) δ = 4.54 (ddt, J = 15.3, 7.3, 6.2 Hz, 1H, CHP), 4.38 (dd, J = 6.2, 1.5 Hz, 1H, CHCHP), 4.30–4.21 (m, 4H, 2 × OCH2CH3), 4.18 (dd, J = 8.6, 6.2 Hz, 1H, OCHH), 3.67 (dd, J = 8.6, 7.4 Hz, 1H, OCHH), 1.72 (s, 3H, C(CH3)), 1.45 (s, 3H, C(CH3)), 1.37 (t, J = 7.0 Hz, 6H, 2 × OCH2CH3). 13C NMR (101 MHz) δ = 155.91 (d, J = 6.4 Hz, C
O), 95.45 (s, C(CH3)2), 71.82 (d, J = 176.3 Hz, CHP), 68.73 (d, J = 10.6 Hz, OCH2), 64.29 (d, J = 6.9 Hz, OCH2CH3), 63.81 (d, J = 6.7 Hz, OCH2CH3), 59.56 (s, CHCHP), 27.48 (s, C(CH3)), 23.35 (s, C(CH3)), 16.64 (d, J = 5.3 Hz, OCH2CH3), 16.59 (d, J = 5.7 Hz, OCH2CH3). 31P {/1H} NMR (162 MHz) δ = 15.70 (s). HRMS (ESI) calcd for C11H20NNaO6P+ ([M + Na]+): 316.0920, found: 316.0929.
(2S,3S)-benzyl 2-(diethoxyphosphoryl)-3-(hydroxymethyl)aziridine-1-carboxylate (18). Isolated with a yield 17% (Note A5) as a slightly yellow oil. Major rotamer had: 1H NMR (400 MHz) δ = 7.43–7.34 (m, 5H, Ph), 5.21 (d, J = 12.1 Hz, 1H, CHHPh), 5.17 (d, J = 12.1 Hz, 1H, CHHPh), 4.24–4.10 (m, 5H, 2 × OCH2CH3, OCHH), 3.73 (dd, J = 13.0, 4.1 Hz, 1H, OCHH), 3.04 (dtd, J = 7.7, 4.0, 2.3 Hz, 1H, CHCP), 2.76–2.70 (m, 1H, CHP), 1.36–1.30 (m, 6H, 2 × OCH2CH3). 13C NMR (151 MHz) δ = 160.98 (d, J = 7.1 Hz, C
O), 135.42, 128.77, 128.73, 128.59 (4 × s, Ph), 68.99 (s, CH2Ph), 63.41 (d, J = 6.2 Hz, OCH2CH3), 62.92 (d, J = 6.1 Hz, OCH2CH3), 59.24 (s, OCH2), 41.84 (d, J = 3.2 Hz, CHCP), 31.84 (d, J = 201.2 Hz, CHP), 16.55 (d, J = 6.6 Hz, OCH2CH3), 16.48 (d, J = 6.8 Hz, OCH2CH3). 31P NMR {/1H} NMR (162 MHz) δ = 18.43 (s). Minor rotamer (traces) had: 31P {/1H} NMR (162 MHz) δ = 19.2 (s). HRMS (ESI) calcd for C15H23NO6P+ ([M + H]+): 344.1258, found: 344.1252.
(2S,3S)-tert-butyl 2-(diethoxyphosphoryl)-3-(hydroxymethyl)aziridine-1-carboxylate (19). Isolated with a yield 56% (Note A6) or 24% (Note A7) or 5% (Note A8) as a transparent oil. Major rotamer had: 1H NMR (400 MHz) δ = 4.21–4.14 (m, 4H, 2 × OCH2CH3), 4.08 (br d, J = 12.8 Hz, 1H, OCHH), 3.67 (dd, J = 12.9, 5.0 Hz, 1H, OCHH), 2.96 (dddd, J = 7.5, 5.0, 3.5, 2.6 Hz, 1H, CHCHP), 2.60 (dd, J = 18.7, 3.6 Hz, 1H, CHP), 1.47 (s, 9H, C(CH3)3), 1.35 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.34 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (75 MHz) δ = 159.79 (d, J = 6.9 Hz, C
O), 82.61 (s, C(CH3)3), 63.29 (d, J = 6.4 Hz, OCH2CH3), 62.81 (d, J = 6.0 Hz, OCH2CH3), 60.16 (s, OCH2), 41.78 (d, J = 3.3 Hz, CHCHP), 31.94 (d, J = 201.4 Hz, CHP), 28.00 (s, C(CH3)3), 16.54 (br d, J = 6.2 Hz, 2 × OCH2CH3). 31P{/1H} NMR (162 MHz) δ = 18.89 (s). Minor rotamer had: 1H NMR (400 MHz) δ = 4.23–4.14 (m, 4H, 2 × OCH2CH3), 4.14–4.09 (m, 1H, OCHH), 3.64 (dt, J = 12.6, 4.8 Hz, 1H, OCHH), 2.98 (dddd, J = 7.6, 5.7, 3.6, 2.4 Hz, 1H, CHCHP), 2.60 (dd, J = 18.4, 3.6 Hz, 1H, CHP), 2.30 (t, J = 6.7 Hz, 1H, OH), 1.48 (s, 9H, C(CH3)3), 1.36 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3). 13C NMR (101 MHz) δ = 159.88 (d, J = 6.9 Hz, CO), 82.77 (s, C(CH3)3), 63.32 (d, J = 6.4 Hz, OCH2CH3), 62.81 (d, J = 6.2 Hz, OCH2CH3), 60.58 (d, J = 2.0 Hz, OCH2), 41.70 (d, J = 3.3 Hz, CHCHP), 32.11 (d, J = 201.9 Hz, CHP), 28.03 (s, C(CH3)3), 16.58 (d, J = 6.1 Hz, OCH2CH3), 16.57 (d, J = 6.1 Hz, OCH2CH3). 31P {/1H} NMR (162 MHz) δ = 18.74 (s). HRMS (ESI) calcd for C12H24NNaO6P+ ([M + Na]+): 332.1233, found: 332.1240.
Diethyl ((4S,5S)-4-(hydroxymethyl)-2-oxooxazolidin-5-yl)phosphonate (21). Isolated with a yield 38% (Note A7) as slightly pink oil. Compound 21 had: 1H NMR (400 MHz) δ = 7.12 (br s, 1H, NH), 4.63 (d, J = 5.8 Hz, 1H, CHP), 4.25–4.16 (m, 4H, 2 × OCH2CH3), 4.11 (ddt, J = 18.4, 5.8, 3.7 Hz, 1H, CHCHP), 3.71 (dd, J = 12.0, 3.2 Hz, 1H, OCHH), 3.54 (dd, J = 12.0, 4.3 Hz, 1H, OCHH), 1.34 (t, J = 7.0 Hz, 6H, 2 × OCH2CH3)·13C NMR (101 MHz) δ = 159.00 (d, J = 4.4 Hz, C
O), 71.74 (d, J = 173.3 Hz, CHP), 64.34 (d, J = 6.9 Hz, OCH2CH3), 63.86 (d, J = 6.9 Hz, OCH2CH3), 63.04 (d, J = 10.2 Hz, OCH2), 55.35 (s, CHCHP), 16.55 (d, J = 5.0 Hz, OCH2CH3), 16.47 (d, J = 5.0 Hz, OCH2CH3). 31P{/1H} NMR (121 MHz) δ = 17.82 (s). HRMS (ESI) calcd for C8H16NNaO6P+ ([M + Na]+): 278.0564, found: 278.0564.
Diethyl ((4S,5S)-4-(fluoromethyl)-2-oxooxazolidin-5-yl)phosphonate (22a) and diethyl ((4S,5R)-4-(fluoromethyl)-2-oxooxazolidin-5-yl)phosphonate (22b). Isolated with a yield 37% (Note A8) as transparent oil, a mixture of two invertomers 12.6
:1. Major invertomer had: 1H NMR (600 MHz) δ = 6.39 (s, 1H, NH), 4.51 (d, J = 6.1 Hz, 1H, CHP), 4.50 (ddd, J = 47.1, 10.1, 3.4 Hz, 1H, CHHF), 4.40 (ddd, J = 46.6, 9.7, 4.7 Hz, 1H, CHHF), 4.29–4.26 (m, 1H, CHCHP), 4.26–4.21 (m, 4H, 2 × OCH2CH3), 1.37 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3). 13C NMR (151 MHz) δ = 157.82 (d, J = 4.7 Hz, CO), 82.82 (dd, J = 176.6, 10.2 Hz, CH2F), 70.85 (dd, J = 174.6, 6.1 Hz, CHP), 64.44 (d, J = 7.0 Hz, OCH2CH3), 63.92 (d, J = 6.8 Hz, OCH2CH3), 53.44 (d, J = 20.7 Hz, NCH), 16.59 (d, J = 5.7 Hz, OCH2CH3), 16.54 (d, J = 5.9 Hz, OCH2CH3). 19F NMR (565 MHz) δ = −230.26 (td, J = 46.6, 18.6 Hz). 31P {/1H} NMR (243 MHz) δ = 15.70 (s). Minor invertomer had: 19F NMR (565 MHz) δ = −230.66 (td, J = 46.8, 19.2 Hz). 31P {/1H} NMR (243 MHz) δ = 15.88 (s). HRMS (ESI) calcd for C8H15FNNaO5P+ ([M + Na]+): 278.0564, found: 278.0576. Compound 22b, epimerized during reaction or purification on silica gel yielding 22a. Major invertomer had: 31P NMR (162 MHz) δ = 15.87 (d, J = 1.2 Hz). 19F NMR (377 MHz) δ = −235.11 (tdd, J = 46.1, 22.8, 1.3 Hz). Minor invertomer had: 31P NMR (162 MHz) δ = 16.84 (s). 19F NMR (377 MHz) δ = −233.90 (td, J = 46.8, 22.6 Hz).
Benzyl (S)-4-((R)-(diethoxyphosphoryl)((pyridin-2-ylsulfonyl)oxy)methyl)-2,2-dimethyloxazolidine-3-carboxylate (23). Isolated with a yield 60% (Note C5) as a slightly yellow oil, mixture of two rotamers (1.5
:1). Major rotamer had: 1H NMR (400 MHz) δ = 8.71 (br d, J = 4.6 Hz, 1H, Ar), 7.95 (d, J = 7.8 Hz, 1H, Ar), 7.94–7.85 (m, 1H, Ar), 7.56–7.49 (m, 2H, Ar), 7.43–7.34 (m, 3H, Ph), 7.36–7.31 (m, 1H, Ph), 5.73 (dd, J = 11.0, 1.5 Hz, 1H, CHP), 5.33–5.10 (m, 2H, OCH2Ph), 4.33–4.28 (m, 1H, CHCHP), 4.28–4.25 (m, 1H, OCHH), 4.16–4.02 (m, 1H, OCHH), 4.16–4.02 (m, 2H, OCH2CH3), 3.95–3.88 (m, 2H, OCH2CH3), 1.47 (s, 3H, C(CH3)2), 1.22 (s, 3H, C(CH3)2), 1.19 (t, J = 7.1 Hz, OCH2CH3), 1.18 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (151 MHz) δ = 154.84 (s, CO), 152.27 (s, Ar), 150.58 (s, Ar), 138.13 (s, Ar), 136.12 (s, Ph), 128.91 (s, Ph), 128.70 (s, Ph), 128.37 (s, Ph), 128.36 (s, Ph), 128.28 (s, Ph), 127.81 (s, Ar), 123.40 (s, Ar), 95.13 (s, C(CH3)2), 75.37 (d, J = 159.0 Hz, CHP), 67.69 (s, OCH2Ph), 63.75–63.53 (m, OCH2CH3), 63.44 (d, J = 6.6 Hz, OCH2CH3), 63.64 (s, OCH2), 56.78 (d, J = 10.7 Hz, CHCHP), 24.73 (s, C(CH3)2), 23.70 (s, C(CH3)2), 16.43 (d, J = 5.6 Hz, OCH2CH3), 16.32 (d, J = 5.8 Hz, OCH2CH3). 31P {/1H} NMR (162 MHz) δ = 15.08 (s). Minor rotamer had: 1H NMR (400 MHz) δ = 8.71 (br d, J = 4.6 Hz, 1H, Ar), 8.03 (d, J = 7.9 Hz, 1H, Ar), 7.94–7.85 (m, 1H, Ar), 7.56–7.49 (m, 2H, Ar), 7.43–7.34 (m, 3H, Ph), 7.36–7.31 (m, 1H, Ph), 5.88 (dd, J = 11.1, 1.6 Hz, 1H, CHP), 5.33–5.10 (m, 2H, OCH2Ph), 4.41–4.36 (m, 1H, CHCHP), 4.33–4.28 (m, 1H, OCHH), 4.16–4.02 (m, 1H, OCHH), 4.16–4.02 (m, 4H, 2 × OCH2CH3), 1.49 (s, 3H, C(CH3)2), 1.44 (s, 3H, C(CH3)2), 1.33–1.22 (m, 6H, 2 × OCH2CH3). 13C NMR (151 MHz) δ = 154.65 (s, CO), 153.51 (s, Ar), 150.29 (s, Ar), 138.05 (s, Ar), 136.15 (s, Ph), 128.91 (s, Ph), 128.70 (s, Ph), 128.37 (s, Ph), 128.30 (s, Ph), 128.27 (s, Ph), 128.16 (s, Ar), 123.70 (s, Ar), 94.67 (s, C(CH3)2), 74.05 (d, J = 161.5 Hz, CHP), 67.59 (s, OCH2Ph), 63.89 (d, J = 6.9 Hz, OCH2CH3), 63.75–63.53 (m, OCH2CH3), 63.01 (s, OCH2), 58.02 (d, J = 10.0 Hz, CHCHP), 25.93 (s, C(CH3)2), 25.43 (s, C(CH3)2), 16.46 (d, J = 5.7 Hz, OCH2CH3), 16.33 (d, J = 5.7 Hz, OCH2CH3). 31P {/1H} NMR (162 MHz) δ = 15.36 (s). HRMS (ESI) calcd for C23H31N2NaO9PS+ ([M + Na]+): 565.1380, found: 565.1394.
tert-Butyl (S)-4-((R)-(diethoxyphosphoryl)((pyridin-2-ylsulfonyl)oxy)methyl)-2,2-dimethyloxazolidine-3-carboxylate (24). Isolated with a yield 47% (Note C7) as a transparent oil, mixture of two rotamers (1
:1). Both rotamers had: 1H NMR (400 MHz) δ = 8.74 (br t, J = 4.8 Hz, 2H, Ar), 8.05 (dd, J = 7.5, 4.5 Hz, 2H, Ar), 7.92 (t, J = 7.8 Hz, 2H, Ar), 7.57–7.51 (m, 2H, Ar), 5.82 (dd, J = 11.0, 1.6 Hz, 1H, CHP), 5.77 (d, J = 11.1 Hz, 1H, CHP), 4.35–4.30 (m, 1H, CHCHP), 4.28–4.23 (m, 3H, 2 × OCHH, CHCHP), 4.22–3.94 (m, 10H, 2 × OCHH, 4 × OCH2CH3), 4.11–4.03 (m, 4H, 2 × OCH2CH3), 1.58 (s, 12H, 4 × CH3), 1.53 (s, 9H, 3 × CH3), 1.47 (s, 3H, CH3), 1.41 (s, 3H, CH3), 1.33 (t, J = 7.0 Hz, 3H, OCH2CH3), 1.30 (t, J = 7.0 Hz, 3H, OCH2CH3), 1.30–1.22 (m, 3H, OCH2CH3), 1.25 (s, 3H, CH3), 1.22 (t, J = 7.0 Hz, 3H, OCH2CH3). 13C NMR (101 MHz) δ = 155.05 (s, CO), 154.81 (s, CO), 152.75 (s, Ar), 151.77 (s, Ar), 150.48 (s, Ar), 150.23 (s, Ar), 138.09 (s, Ar), 137.98 (s, Ar), 127.78 (s, Ar), 127.75 (s, Ar), 123.73 (s, Ar), 123.34 (s, Ar), 94.69 (s, C(CH3)2), 94.33 (s, C(CH3)2), 81.07 (s, C(CH3)3), 81.02 (s, C(CH3)3), 75.93 (d, J = 161.2 Hz, CHP), 74.29 (d, J = 161.4 Hz, CHP), 63.84 (d, J = 7.4 Hz, OCH2CH3), 63.61 (d, J = 6.7 Hz, OCH2CH3), 63.54–63.23 (m, 2 × OCH2CH3), 63.35 (s, OCH2), 62.82 (s, OCH2), 57.61 (d, J = 10.2 Hz, CHCHP), 56.98 (d, J = 10.9 Hz, CHCHP), 29.83 (s, CH3), 28.55 (s, 9 × CH3), 16.48 (d, J = 5.6 Hz, 4 × OCH2CH3). 31P NMR (162 MHz) δ = 15.86 (s), 15.62 (s). HRMS (ESI) calcd for C20H34N2O9PS+ ([M + H]+): 509.1717, found: 509.1711.
Diethyl ((4S,5S)-3-benzyl-4-(fluoromethyl)-2,2-dimethyloxazolidin-5-yl)phosphonate (26a) and diethyl ((4S,5R)-3-benzyl-4-(fluoromethyl)-2,2-dimethyloxazolidin-5-yl)phosphonate (26b). Isolated with a yield 58% (Note A9) as transparent oil. Major diastereoisomer 26a (major invertomer) had: 1H NMR (400 MHz) δ = 7.40–7.37 (m, 2H, Ph), 7.33–7.28 (m, 2H, Ph), 7.26–7.21 (m, 1H, Ph), 4.25 (dd, J = 47.2, 4.0 Hz, 1H, CHHF), 4.23 (d, J = 47.4, 4.0 Hz, 1H, CHHF), 4.29–4.14 (m, 4H, 2 × OCH2CH3), 4.14 (dd, J = 8.7, 2.3 Hz, 1H, CHP), 3.94 (d, J = 14.6 Hz, 1H, CHHPh), 3.74 (d, J = 14.6 Hz, 1H, CHHPh), 3.62–3.46 (m, 1H, CHCHP), 1.40 (s, 3H, CH3), 1.40–1.30 (m, 6H, 2 × OCH2CH3), 1.29 (s, 3H, CH3). 13C NMR (101 MHz) δ = 139.88, 128.41, 128.20, 127.24 (4 × s, Ph), 98.68 (d, J = 6.6 Hz, C(CH3)2), 82.05 (dd, J = 174.6, 4.4 Hz, CH2F), 71.09 (dd, J = 172.9, 6.1 Hz, CHP), 64.18 (dd, J = 19.7, 3.5 Hz, NCH), 63.17 (d, J = 6.8 Hz, OCH2CH3), 62.76 (d, J = 6.9 Hz, OCH2CH3), 52.95 (CH2Ph), 28.17 (s, CH3), 22.84 (s, CH3), 16.62 (d, J = 5.7 Hz, 2 × OCH2CH3). 19F NMR (376 MHz) δ = −227.50 (td, J = 47.5, 22.2 Hz). 31P {/1H} NMR (162 MHz) δ = 21.56 (s). Minor invertomer 26a (observed in a crude reaction mixture) had: 19F NMR (376 MHz) δ = −227.50 (td, J = 47.5, 22.2 Hz). 31P {/1H} NMR (162 MHz) δ = 21.56 (s). Minor diastereoisomer 26b was observed in a crude reaction mixture. Major inwertomer had: 19F NMR (565 MHz) δ = −230.84 (td, J = 47.1, 27.4 Hz). 31P NMR (243 MHz) δ = 18.90 (s). Minor invertomer had: 19F NMR (565 MHz) δ = −229.67 (td, J = 47.3, 28.1 Hz). 31P {/1H} NMR (162 MHz) δ = 20.09 (s). HRMS (ESI) calcd for C17H27FNNaO4P+ ([M + Na]+): 382.1557, found: 382.1554.
:5) compound 8 as transparent oil (32 mg, 73%): 1H NMR (600 MHz) δ = 4.62 (dqd, J = 48.0, 8.3, 4.4 Hz, 1H, CHF), 4.17 (′′pd′′, J = 7.1, 2.2 Hz, 4H, 2 × OCH2CH3), 3.02 (dt, J = 11.7, 3.5 Hz, 1H, NCHH), 2.97 (ddd, J = 10.4, 8.5, 5.4 Hz, 1H, CHP), 2.50 (t, J = 11.2 Hz, 1H, NCHH), 2.26–2.18 (m, 1H, CHHCHF), 1.99 (br s, 1H, NH), 1.74 (tt, J = 7.0, 3.9 Hz, 1H, NCH2CHH), 1.60–1.48 (m, 2H, CHHCHF, NCH2CHH), 1.33 (t, J = 7.0 Hz, 3H, OCH2CH3), 1.32 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (151 MHz) δ = 88.28 (dd, J = 177.3, 3.7 Hz, CHF), 62.76 (d, J = 6.7 Hz, OCH2CH3), 62.59 (d, J = 6.7 Hz, OCH2CH3), 58.09 (dd, J = 156.6, 23.2 Hz, CHP), 45.62 (d, J = 13.8 Hz, NCH2), 31.12 (dd, J = 19.0, 11.0 Hz, CH2CHF), 24.12 (d, J = 9.3 Hz, NCH2CH2), 16.58 (d, J = 5.7 Hz, 2 × OCH2CH3). 19F NMR (565 MHz) δ = −176.49 to −176.61 (m). 31P {/1H} NMR (243 MHz) δ = 23.79 (d, J = 6.0 Hz). HRMS (ESI) calcd for C9H20FNO3P+ ([M + H]+): 240.1159, found: 240.1148.:1). Major rotamer had: 1H NMR (400 MHz) δ = 4.31 (dd, J = 12.1, 4.3 Hz, 1H, OCHH), 4.23–4.09 (m, 5H, OCHH, 2 × OCH2CH3), 3.18–3.12 (m, 1H, CHCHP), 2.62 (dd, J = 18.4, 3.2 Hz, 1H, CHP), 2.18 (s, 3H, CH3), 1.45 (s, 9H, C(CH3)3), 1.33 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3). 13C NMR (101 MHz) δ = 179.19 (d, J = 6.0 Hz, CO), 152.90 (s, CO), 83.06 (s, C(CH3)3), 64.63 (s, OCH2), 62.90 (d, J = 6.2 Hz, OCH2CH3), 62.78 (d, J = 6.0 Hz, OCH2CH3), 38.14 (d, J = 2.6 Hz, CHCHP), 32.97 (d, J = 195.6 Hz, CHP), 27.78 (s, C(CH3)3), 23.80 (s, CH3), 16.56 (d, J = 5.6 Hz, OCH2CH3), 16.52 (d, J = 6.1 Hz, OCH2CH3). 31P {/1H} NMR (162 MHz) δ = 18.33 (s). Minor rotamer had: 1H NMR (400 MHz) δ = 4.27 (dd, J = 4.3, 0.9 Hz, 2H, OCH2), 4.23–4.09 (m, 4H, 2 × OCH2CH3), 3.03 (dq, J = 7.9, 4.1 Hz, 1H, CHCHP), 2.54 (dd, J = 17.8, 3.5 Hz, 1H, CHP), 2.04 (s, 3H, CH3), 1.46 (s, 9H, C(CH3)3), 1.34 (t, J = 6.7 Hz, 6H, 2 × OCH2CH3). 13C NMR (101 MHz) δ = 170.19 (s, CO), 158.50 (d, J = 7.6 Hz, CO), 82.47 (s, C(CH3)3), 63.48 (d, J = 6.3 Hz, OCH2CH3), 63.32 (d, J = 6.2 Hz, OCH2CH3), 61.57 (s, OCH2), 38.37 (d, J = 2.7 Hz, CHCHP), 32.79 (d, J = 197.8 Hz, CHP), 27.97 (s, C(CH3)3), 20.82 (s, CH3), 16.56 (d, J = 5.6 Hz, OCH2CH3), 16.52 (d, J = 6.1 Hz, OCH2CH3). 31P {/1H} NMR (162 MHz) δ = 18.63 (s).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank to National Science Centre (grant HARMONIA/2017/26/M/ST5/00437) for financial support.References
- (a) Aminophosphonic and Aminophosphinic Acids, ed. V. P. Kukhar and H. R. Hudson, John Wiley & Sons Ltd, Chichester, 2000; For some review see Search PubMed; (b) P. Kafarski and B. Lejczak, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 301–312 CrossRef PubMed; (c) J. Grembecka and P. Kafarski, Mini-Rev. Med. Chem., 2001, 1, 133–144 CrossRef PubMed; (d) M. Ordóñez, H. Rojas-Cabrera and C. Cativiela, Tetrahedron, 2009, 65, 17–49 CrossRef PubMed; (e) S. Van der Jeught and Ch. S. Stevens, Chem. Rev., 2009, 109, 2672–2702 CrossRef PubMed; (f) F. Orsini, G. Sello and M. Sisti, Curr. Med. Chem., 2010, 17, 264–289 CrossRef PubMed; (g) E. D. Naydenova, P. T. Todorov and K. D. Troev, Amino Acids, 2010, 38, 23–30 CrossRef PubMed; (h) M. Ordóñez, J. L. Viveros-Ceballos, C. Cativiela and F. J. Sayago, Tetrahedron, 2015, 71, 1745–1784 CrossRef.
- G. P. Horsman and D. L. Zechel, Chem. Rev., 2017, 117, 5704–5783 CrossRef PubMed.
- (a) B. Boduszek, J. Oleksyszyn, C.-M. Kam, J. Selzler, R. E. Smith and J. C. Powers, J. Med. Chem., 1994, 37, 3969–3976 CrossRef PubMed; (b) A. M. Lambeir, M. Borloo, I. De Meester, A. Belyaev, K. Augustyns, D. Hendriks, S. Scharpé and A. Haemers, Biochim. Biophys. Acta, 1996, 1290, 76–82 CrossRef PubMed.
- Y. Surh, R. P. Spencer, L. A. Spitznagle, F. Hosain and B. Lejczak, J. Nucl. Med., 1986, 27, 847–849 Search PubMed.
- (a) A. R. Katritzky, X.-L. Cui, B. Yang and P. J. Steel, J. Org. Chem., 1999, 64, 1979–1985 CrossRef PubMed; (b) P. Dinér and M. Amedjkouh, Org. Biomol. Chem., 2006, 4, 2091–2096 RSC; (c) Q. Tao, G. Tang, K. Lin and Y.-F. Zhao, Chirality, 2008, 20, 833–838 CrossRef PubMed; (d) S. Hirata, M. Kuriyama and O. Onomura, Tetrahedron, 2011, 67, 9411–9416 CrossRef; (e) T. Ma, X. Fu, C. W. Kee, L. Zong, Y. Pan, K.-W. Huang and C.-H. Tan, J. Am. Chem. Soc., 2011, 133, 2828–2831 CrossRef PubMed; (f) F. Wuggenig, A. Schweifer, K. Mereiter and F. Hammerschmidt, Eur. J. Org. Chem., 2011, 1870–1879 CrossRef; (g) B. Kaboudin, J.-Y. Kato, H. Aoyama and T. Yokomatsu, Tetrahedron: Asymmetry, 2013, 24, 1562–1566 CrossRef; (h) M. H. Kudzin, Z. H. Kudzin and J. Drabowicz, ARKIVOC, 2011, vi, 227–269 Search PubMed.
- (a) G. T. Anderson, M. D. Alexander, S. D. Taylor, D. B. Smithrud, S. J. Benkovic and S. M. Weinreb, J. Org. Chem., 1996, 61, 125–132 CrossRef; (b) T. Widianti, Y. Hiraga, S. Kojima and M. Abe, Tetrahedron: Asymmetry, 2010, 21, 1861–1868 CrossRef; (c) O. O. Kolodyazhnaya, A. O. Kolodyazhnaya and O. I. Kolodyazhnyi, Russ. J. Gen. Chem., 2014, 84, 169–170 CrossRef.
- (a) A. B. Smith, III, K. M. Yager, B. W. Phillips and C. M. Taylor, Org. Synth., 1998, 75, 19–29 CrossRef; (b) F. Hammerschmidt, W. Lindner, F. Wuggenig and E. Zarbl, Tetrahedron: Asymmetry, 2000, 11, 2955–2964 CrossRef; (c) E. K. Dolence, G. Mayer and B. D. Kelly, Tetrahedron: Asymmetry, 2005, 16, 1583–1594 CrossRef.
- (a) J. Nieschalk, A. S. Batsanov, D. O'Hagan and J. A. K. Howard, Tetrahedron, 1996, 52, 165–176 CrossRef; (b) D. B. Berkowitz, M. Bose, T. J. Pfannenstiel and T. Doukov, J. Org. Chem., 2000, 65, 4498–4508 CrossRef PubMed; (c) D. O'Hagan and H. S. Rzepa, Chem. Commun., 1997, 645–652 RSC; (d) S. M. Forget, D. Bhattasali, V. C. Hart, T. S. Cameron, R. T. Syvitskiad and D. L. Jakeman, Chem. Sci., 2012, 3, 1866–1878 RSC.
- For some review see: (a) V. D. Romanenko and V. P. Kukhar, Chem. Rev., 2006, 106, 3868–3935 CrossRef PubMed; (b) K. V. Turcheniuk, V. P. Kukhar, G.-V. Röschenthaler, J. L. Aceña, V. A. Soloshonok and A. E. Sorochinsky, RSC Adv., 2013, 3, 6693–6716 RSC; (c) M. Rapp, T. Cytlak, M. Z. Szewczyk and H. Koroniak, Curr. Green Chem., 2015, 2, 237–253 CrossRef; (d) T. Cytlak, M. Kaźmierczak, M. Skibińska and H. Koroniak, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 602–620 CrossRef.
- (a) J. T. Welch, Tetrahedron, 1987, 43, 3123–3197 CrossRef; (b) J. Mann, Chem. Soc. Rev., 1987, 16, 381–436 RSC; (c) J.-P. Bégué and D. Bonnet-Delpon, Bioorganic and Medicinal Chemistry of Fluorine, Wiley & Sons: New Jersey, 2008 CrossRef; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (e) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- For some examples see: (a) G. A. Flynn, D. W. Beight, E. H. W. Bohme and B. W. Metcalf, Tetrahedron Lett., 1985, 26, 285–288 CrossRef; (b) P. Van der Veken, K. Senten, I. Kertèsz, A. Haemers and K. Augustyns, Tetrahedron Lett., 2003, 44, 969–972 CrossRef; (c) P. Cui, W. F. McCalmont, J. L. Tomsig, K. R. Lynch and T. L. Macdonald, Bioorg. Med. Chem., 2008, 16, 2212–2225 CrossRef PubMed; (d) J. E. East, A. J. Kennedy, J. L. Tomsig, A. R. De Leon, K. R. Lynch and T. L. Macdonald, Bioorg. Med. Chem. Lett., 2010, 20, 7132–7136 CrossRef PubMed.
- (a) A. H. Fauq, N,N-diethylaminosulfur trifluoride, in Encyclopedia of Reagents for Organic Synthesis, ed. L. Paquette, John Wiley & Sons, New York, 2004 Search PubMed; (b) W. J. Middleton, J. Org. Chem., 1975, 40, 574–578 CrossRef.
- (a) G. S. Lal, G. P. Pez, R. J. Pesaresi and F. M. Prozonic, Chem. Commun., 1999, 215–216 RSC; (b) G. S. Lal, G. P. Pez, R. J. Pesaresi, F. M. Prozonic and H. Cheng, J. Org. Chem., 1999, 64, 7048–7054 CrossRef.
- (a) M. K. Nielsen, C. R. Ugaz, W. Li and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 9571–9574 CrossRef PubMed; (b) M. K. Nielsen, D. T. Ahneman, O. Riera and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 5004–5008 CrossRef PubMed.
- (a) S. B. D. Jarvis and A. B. Charette, Org. Lett., 2011, 13, 3830–3833 CrossRef PubMed; (b) C. Ye and J. M. Shreeve, J. Fluorine Chem., 2004, 125, 1869–1872 CrossRef; (c) T. Rosen, D. T. W. Chu, I. M. Lico, P. B. Fernandes, K. Marsh, L. Shen, V. G. Cepa and A. G. Pernet, J. Med. Chem., 1988, 31, 1598–1611 CrossRef PubMed; (d) I. Déchamps, D. Gomez Pardo and J. Cossy, Synlett, 2007, 2, 263–267 Search PubMed; (e) I. Déchamps, D. Gomez Pardo and J. Cossy, Eur. J. Org. Chem., 2007, 4224–4234 CrossRef; (f) P. K. Mykhailiuk, S. V. Shishkina, O. V. Shishkin, O. A. Zaporozhec and I. V. Komarov, Tetrahedron, 2011, 67, 3091–3097 CrossRef; (g) H. Ferret, I. Déchamps, D. Gomez Pardo, L. Van Hijfte and J. Cossy, ARKIVOC, 2010, 8, 126–159 Search PubMed; (h) T.-X. Métro, B. Duthion, D. Gomez Pardo and J. Cossy, Chem. Soc. Rev., 2010, 39, 89–102 RSC.
- M. Kaźmierczak and H. Koroniak, J. Fluorine Chem., 2012, 139, 23–27 CrossRef.
- M. Kaźmierczak, M. Kubicki and H. Koroniak, Eur. J. Org. Chem. DOI:10.1002/ejoc.201800631.
- K. Radwan-Olszewska, F. Palacios and P. Kafarski, J. Org. Chem., 2011, 76, 1170–1173 CrossRef PubMed.
- M. Kaźmierczak, M. Kubicki and H. Koroniak, J. Fluorine Chem., 2014, 167, 128–134 CrossRef.
- F. Palacios, A. M. Ochoa de Retana, J. Oyarzabal, S. Pascual and G. F. de Trocóniz, J. Org. Chem., 2008, 73, 4568–4574 CrossRef PubMed.
- M. Rapp, P. Mrowiec and H. Koroniak, Phosphorus, Sulfur Silicon Relat. Elem., 2017, 192, 745–751 CrossRef.
- M. Rapp, M. Bilska and H. Koroniak, J. Fluorine Chem., 2011, 132, 1232–1240 CrossRef.
- W. R. Dolbier Jr, Guide to Fluorine NMR for Organic Chemists, Wiley Interscience, 2009 Search PubMed.
- C. Zhang, Z. Li, L. Zhu, L. Yu, Z. Wang and C. Li, J. Am. Chem. Soc., 2013, 135, 14082–14085 CrossRef PubMed.
- T. Wu, G. Yin and G. Liu, J. Am. Chem. Soc., 2009, 131, 16354–16355 CrossRef PubMed.
- (a) C. R. S. Briggs, D. O'Hagan, J. A. K. Howard and D. S. Yufit, J. Fluorine Chem., 2003, 119, 9–13 CrossRef; (b) D. O'Hagan, C. Bilton, J. A. K. Howard, L. Knight and D. J. Tozer, J. Chem. Soc., Perkin Trans. 2, 2000, 605–607 RSC.
- (a) H. Zhao and A. Thurkauf, Synlett, 1999, 8, 1280–1282 CrossRef; (b) S. De Jonghe, I. Van Overmeire, S. Van Calenbergh, C. Hendrix, R. Busson, D. De Keukeleire and P. Herdewijn, Eur. J. Org. Chem., 2000, 3177–3183 CrossRef.
- (a) D. V. Patel, K. Rielly-Gauvin and D. E. Ryono, Tetrahedron Lett., 1990, 31, 5587–5590 CrossRef; (b) D. V. Patel, K. Rielly-Gauvin, D. E. Ryono, C. A. Free, W. L. Rogers, S. A. Smith, J. M. DeForrest, R. S. Oehl and E. W. Petrillo Jr, J. Med. Chem., 1995, 38, 4557–4569 CrossRef PubMed.
- D. Willén, D. Bengtsson, S. Clementson, E. Tykesson, S. Manner and U. Ellervik, J. Org. Chem., 2018, 83, 1259–1277 CrossRef PubMed.
- Phosphorus-31 NMR. Principles and Applications, ed. D. G. Gorenstein, Academic Press. Inc., Orlando, 1984 Search PubMed.
- A. De La Cruz, A. He, A. Thanavaro, B. Yan, C. D. Spilling and N. P. Rath, J. Organomet. Chem., 2005, 690, 2577–2592 CrossRef.
- A. E. Wróblewski and J. Drozd, Tetrahedron: Asymmetry, 2011, 22, 200–206 CrossRef.
- G. V. De Lucca, J. Org. Chem., 1998, 63, 4755–4766 CrossRef.
- R. H. Furneaux, G. J. Gainsford, J. M. Mason and P. C. Tyler, Tetrahedron, 1994, 50, 2131–2160 CrossRef.
- C. Carpentier, R. Godbout, F. Otis and N. Voyer, Tetrahedron Lett., 2015, 56, 1244–1246 CrossRef.
- T. Cytlak, M. Skibińska, P. Kaczmarek, M. Kaźmierczak, M. Rapp, M. Kubicki and H. Koroniak, RSC Adv., 2018, 8, 11957–11974 RSC.
- A. E. Wróblewski and K. B. Balcerzak, Tetrahedron: Asymmetry, 2001, 12, 427–431 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compounds characterization, 1H, 13C, 31P and 2D NMR spectra of compounds. See DOI: 10.1039/c8ra05186k |
| This journal is © The Royal Society of Chemistry 2018 |