
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect β-selectivity of α,β-unsaturated γ-butyrolactam for asymmetric conjugate additions in an organocatalytic manner†
Yuan Zhong‡
ab,
Sihua Hong‡a,
Zhengjun Caia,
Shixiong Mab and
Xianxing Jiang *a
*a
aSchool of Pharmaceutical Sciences, Sun Yat-Sen University, Guangzhou, 510006, China. E-mail: jiangxx5@mail.sysu.edu.cn
bKey Laboratory of Preclinical Study for New Drugs of Gansu Province, School of Basic Medical Sciences, Lanzhou University, Lanzhou, 730000, China
First published on 14th August 2018
Abstract
The β-selective asymmetric addition of γ-butyrolactam with cyclic imino esters catalyzed by a bifunctional chiral tertiary amine has been developed, which provides an efficient access to optically active β-position functionalized pyrrolidin-2-one derivatives in both high yield and enantioselectivity (up to 78% yield and 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 er). This is the first catalytic method to access chiral β-functionalized pyrrolidin-2-one via a direct organocatalytic approach.
:5 er). This is the first catalytic method to access chiral β-functionalized pyrrolidin-2-one via a direct organocatalytic approach.
Metal-free organocatalytic asymmetric transformations have successfully captured considerable enthusiasm of chemists as powerful methods for the synthesis of various kinds of useful chiral compounds ranging from the preparation of biologically important molecules through to novel materials.1 Chiral pyrrolidin-2-ones have been recognized as important structural motifs that are frequently encountered in a variety of biologically active natural and synthetic compounds.2 In particular, the β-position functionalized pyrrolidin-2-one backbones, which can serve as key synthetic precursors for inhibitory neurotransmitters γ-aminobutyric acids (GABA),3 selective GABAB receptor agonists4 as well as antidepressant rolipram analogues,5 have attracted a great deal of attention. Therefore, the development of highly efficient, environmentally friendly and convenient asymmetric synthetic methods to access these versatile frameworks is particularly appealing.
As a direct precursor to pyrrolidin-2-one derivatives, recently, α,β-unsaturated γ-butyrolactam has emerged as the most attractive reactant in asymmetric organometallic or organocatalytic reactions for the synthesis of chiral γ-position functionalized pyrrolidin-2-ones (Scheme 1). These elegant developments have been achieved in the research area of catalytic asymmetric vinylogous aldol,6 Mannich,7 Michael8 and annulation reactions9 in the presence of either metal catalysts or organocatalysts (a, Scheme 1). These well-developed catalytic asymmetric methods have been related to the γ-functionalized α,β-unsaturated γ-butyrolactam to date. However, in sharp contrast, the approaches toward introducing C-3 chirality at the β-position of butyrolactam through a direct catalytic manner are underdeveloped (b, Scheme 1)10 in spite of the fact that β-selective chiral functionalization of butyrolactam can directly build up α,β-functionalized pyrrolidin-2-one frameworks.
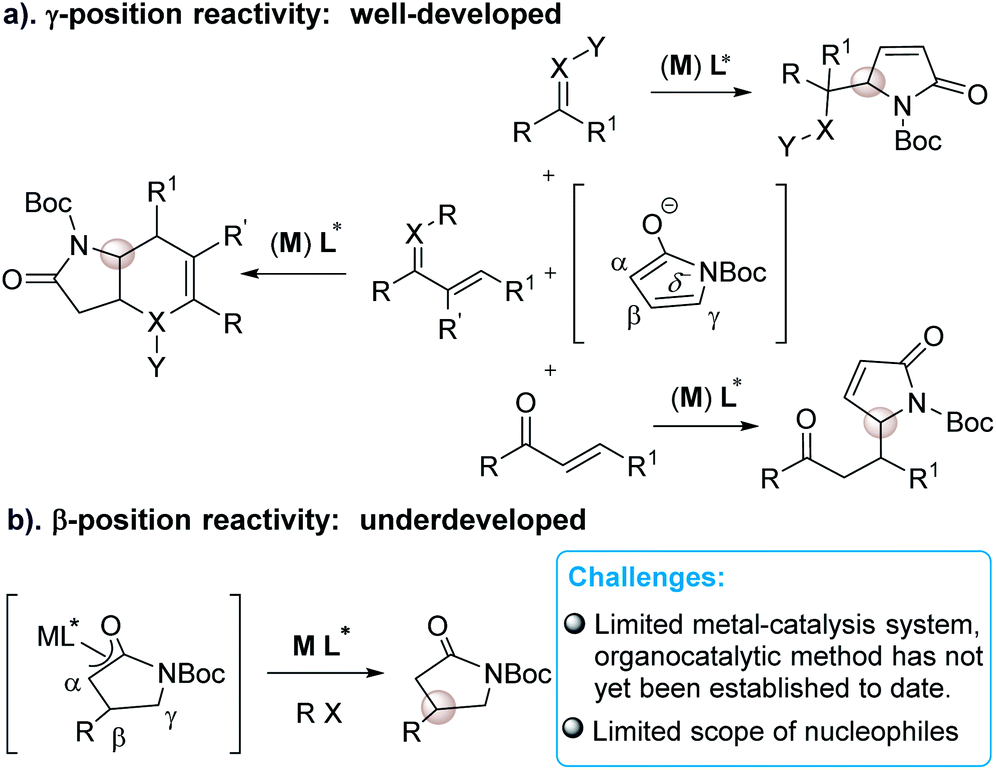 | ||
| Scheme 1 Different reactive position of α,β-unsaturated γ-butyrolactam in catalytic asymmetric reactions. | ||
So far, only a few metal-catalytic enantioselective β-selective functionalized reactions have been reported. For examples, a rhodium/diene complex catalyzed efficient asymmetric β-selective arylation10a and alkenylation10b have been reported by Lin group (a, Scheme 2). Procter and co-workers reported an efficient Cu(I)–NHC-catalyzed asymmetric silylation of unsaturated lactams (b, Scheme 2).10c Despite these creative works, considerable challenges still exist in the catalytic asymmetric β-selective functionalization of γ-butyrolactam. First, the scope of nucleophiles is limited to arylboronic acids, potassium alkenyltrifluoroborates and PhMe2SiBpin reagents. Second, the catalytic system and activation mode is restricted to metal/chiral ligands. To our knowledge, an efficient catalytic method to access chiral β-functionalized pyrrolidin-2-one via a direct organocatalytic approach has not yet been established. Therefore, the development of organocatalytic asymmetric β-selective functionalization of γ-butyrolactam are highly desirable. In conjunction with our continuing efforts in building upon chiral precedents by using chiral tertiary amine catalytic system,11 we rationalized that the activated α,β-unsaturated γ-butyrolactam might serve as a β-position electron-deficient electrophile. This γ-butyrolactam may react with a properly designed electron-rich nucleophile to conduct an expected β-selective functionalized reaction of γ-butyrolactam under a bifunctional organocatalytic fashion, while avoiding the direct γ-selective vinylogous addition reaction or β,γ-selective annulation as outlined in Scheme 2. Herein we report the β-selective asymmetric addition of γ-butyrolactam with cyclic imino esters12 catalyzed by a bifunctional chiral tertiary amine, which provides an efficient and facile access to optically active β-position functionalized pyrrolidin-2-one derivatives with both high diastereoselectivity and enantioselectivity.
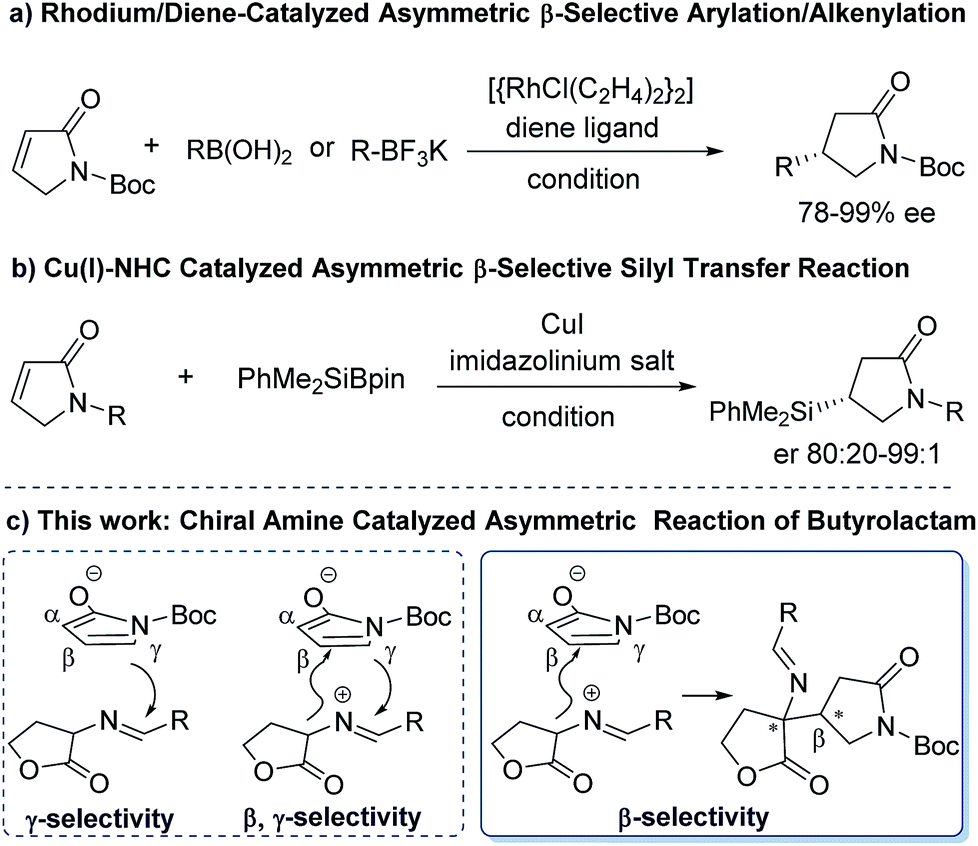 | ||
| Scheme 2 β-Selective functionalization of γ-butyrolactam via metal- (previous work) or organo- (this work) catalytic approach. | ||
To begin our initial investigation, several bifunctional organocatalysts13 were firstly screened to evaluate their ability to promote the β-selective asymmetric addition of γ-butyrolactam 2a with cyclic imino ester 3a in the presence of 15 mol% of catalyst loading at room temperature in CH2Cl2 (entries 1–6, Table 1). These results indicated that chiral catalyst (DHQD)2AQN 1f is the best catalyst in terms of chemical yield and enantioselectivity, furnishing the products with excellent diastereoselectivity in 71% yield and 77:23 er (entry 6). Subsequently, a survey of other solvents was carried out with catalyst 1f (entries 7–12). We found that a substantial change of the solvent has a significant effect on the reaction. Among the solvents tested, dichloroethane appeared to be the most suitable reaction media, giving the product with 80:20 er (entry 7). Screening of solvents revealed a mixture of dichloroethane and methyl tert-butyl ether (volume ratio = 10:1) offered a better yield and enantioselectivity (75% yield, and 87:13 er) (entry 13).
| Entry | Cat. | Solvent | Yielde | erf |
|---|---|---|---|---|
| a Reaction conditions: unless specified, a mixture of 2a (0.2 mmol), 3a (0.3 mmol) and a catalyst (15 mmol%) in a solvent (2.0 mL) was stirred at rt. for 48 h.b The reaction was carried out in 2.2 mL a mixture of dichloroethane and methyl tert-butyl ether (volume ratio = 10:1).c The reaction was carried out in 2.2 mL a mixture of dichloroethane and methyl tert-butyl ether (volume ratio = 10:1) for 24 h.d The reaction was carried out in 2.2 mL a mixture of dichloroethane and methyl tert-butyl ether (volume ratio = 10:1) and 10 mol% of catalyst was used.e Isolated yields.f Determined by chiral HPLC, the product was observed with >99:1 dr by 1H NMR and HPLC. Configuration was assigned by X-ray crystal data of 4a. |
||||
| 1 | 1a | CH2Cl2 | 70% | 40:60 |
| 2 | 1b | CH2Cl2 | <5% | 57:43 |
| 3 | 1c | CH2Cl2 | 70% | 65:35 |
| 4 | 1d | CH2Cl2 | 68% | 70:30 |
| 5 | 1e | CH2Cl2 | 58% | 63:47 |
| 6 | 1f | CH2Cl2 | 71% | 77:23 |
| 7 | 1f | DCE | 72% | 80:20 |
| 8 | 1f | CHCl3 | 70% | 80:20 |
| 9 | 1f | MTBE | 68% | 79:21 |
| 10 | 1f | Toluene | 63% | 78:22 |
| 11 | 1f | THF | 45% | 76:24 |
| 12 | 1f | MeOH | 32% | 62:38 |
| 13b | 1f | DCE:MTBE |
75% | 87:13 |
| 14c | 1f | DCE:MTBE |
72% | 87:13 |
| 15d | 1f | DCE:MTBE |
70% | 85:15 |
The results of experiments under the optimized conditions that probed the scope of the reaction are summarized in Scheme 3. The catalytic β-selective asymmetric addition of γ-butyrolactam 2a with cyclic imino esters 3a in the presence of 15 mol% (DHQD)2AQN 1f was performed. A variety of phenyl-substituted cyclic imino esters including those bearing electron-withdrawing and electron-donating substituents on the aryl ring, heterocyclic were also examined. The electron-neutral, electron-rich, or electron-deficient groups on the para-position of phenyl ring of the cyclic imino esters afforded the products 4a–4m in 57–75% yields and 82:18 to 95:5 er values. It appears that either an electron-withdrawing or an electron-donating at the meta- or ortho-position of the aromatic ring had little influence on the yield and stereoselectivity. Similar results on the yield and enantioselectivities were obtained with 3,5-dimethoxyl substituted cyclic imino ester (71% yield and 91:9 er). It was notable that the system also demonstrated a good tolerance to naphthyl substituted imino ester (78% yield and 92:8 er value). The 2-thienyl substituted cyclic imino ester proceeded smoothly under standard conditions as well, which gave the desired product 4p in good enantioselectivity (88:12 er), although yield was slightly lower. However, attempts to extend this methodology to aliphatic-substituted product proved unsuccessful due to the low reactivity of the substrate 3q. It is worth noting that the replacement of Boc group with 9-fluorenylmethyl, tosyl or benzyl group as the protection, no reaction occurred. The absolute and relative configurations of the products were unambiguously determined by X-ray crystallography (4a, see the ESI†).
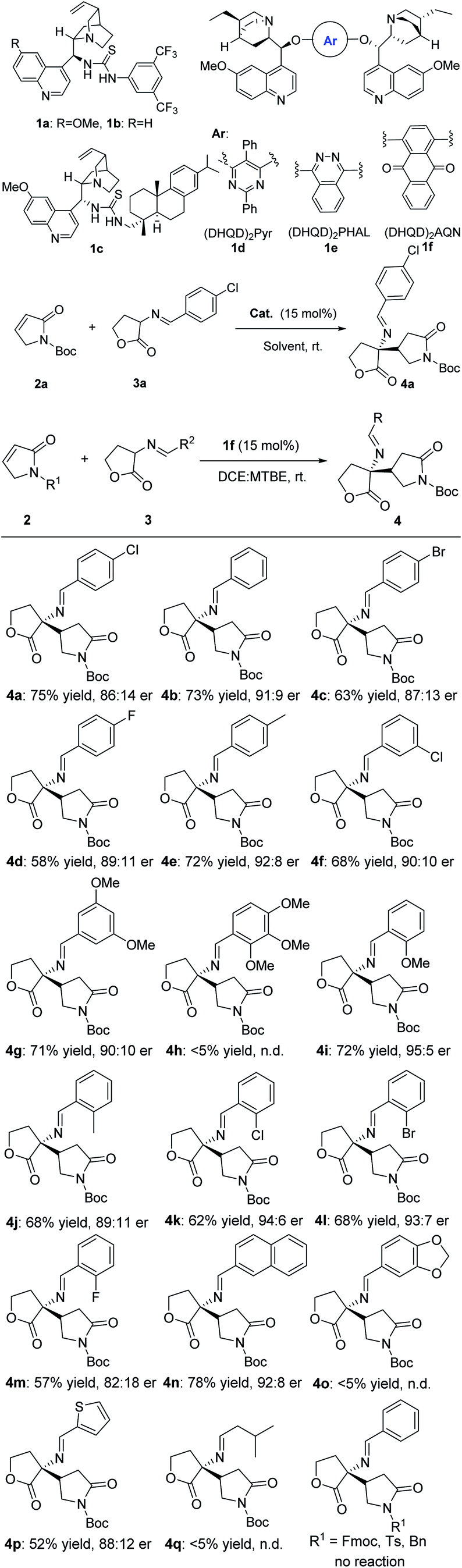 | ||
| Scheme 3 Substrate scope of the asymmetric reaction of α,β-unsaturated γ-butyrolactam 2 to cyclic imino esters 3.a aReaction conditions: unless specified, a mixture of 2 (0.2 mmol), 3 (0.3 mmol) and 1f (15.0 mmol%) in 2.2 mL a mixture of dichloroethane and methyl tert-butyl ether (volume ratio = 10:1) was stirred at rt. bIsolated yields. cDetermined by chiral HPLC, all products were observed with >99:1 dr by 1H NMR and HPLC. Configuration was assigned by comparison of HPLC data and X-ray crystal data of 4a. | ||
We then examined the substrate scope of the imide derivatives (Scheme 4). Investigations with maleimides 4r–4u gave 48–61% yield of corresponding products as lower er and dr values than most of γ-butyrolactams. As for methyl substituted maleimides, the reaction failed to give any product.
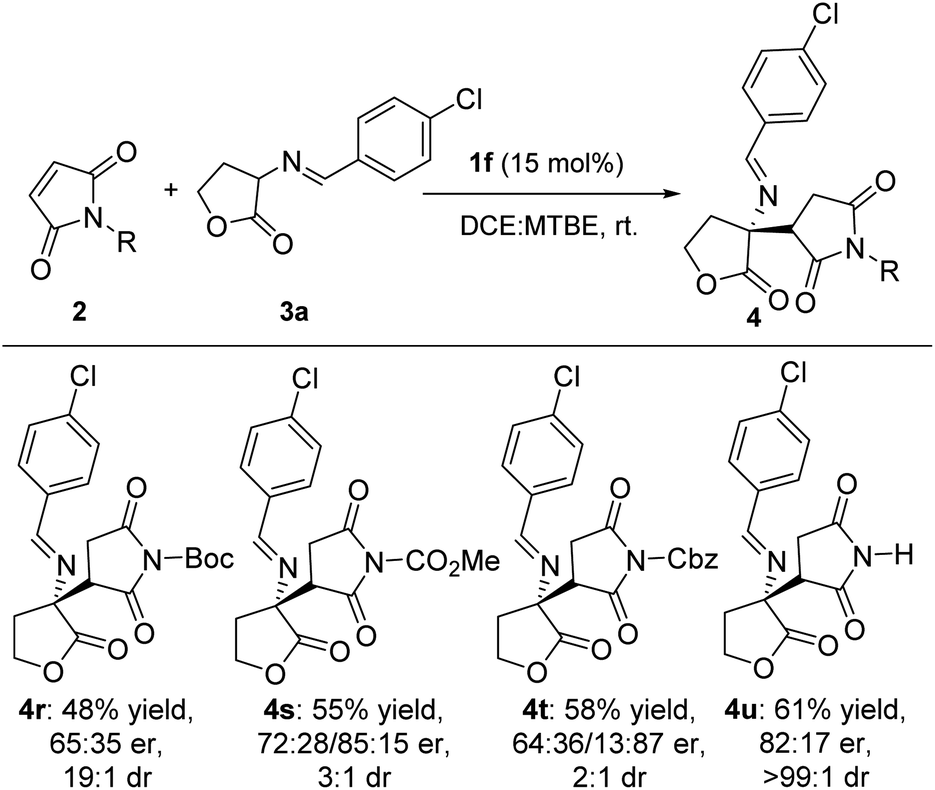 | ||
| Scheme 4 Substrate scope of the asymmetric reaction of maleimides to cyclic imino esters.a aReaction conditions: unless specified, a mixture of 2 (0.2 mmol), 3 (0.3 mmol) and 1f (15.0 mmol%) in 2.2 mL a mixture of dichloroethane and methyl tert-butyl ether (volume ratio = 10:1) was stirred at rt. bIsolated yields. cDetermined by 1H NMR and chiral HPLC. | ||
The chloride product 4a ((R)-tert-butyl 4-((R)-3-((E)-(4-chlorobenzylidene)amino)-2-oxotetra hydrofuran-3-yl)-2-oxopyrrolidine-1-carboxylate) was recrystallized and the corresponding single crystal was subjected to X-ray analysis to determine the absolute structure. Based on this result and our previous work, a plausible catalytic mechanism involving multisite interactions was assumed to explain the high stereoselectivity of this process (Fig. 1). Similar to the conformation reported for the dihydroxylation and the asymmetric direct aldol reaction, the transition state structure of the substrate/catalyst complexes might be presumably in the open conformation. The acidic α-carbon atom of cyclic imino ester 3a could be activated by interaction between the tertiary amine moiety of the catalyst and the enol of 3a via a hydrogen bonding. Moreover, the enolate of 3a in the transition state might be in part stabilized through the π–π stacking between the phenyl ring of 3a and the quinoline moiety. Consequently, the Re-face of the enolate is blocked by the left half of the quinidine moiety. The steric hindrance between the Boc group of 2a and the right half of the quinidine moiety make the Re-face of 2a face to the enolate of 3a. Subsequently, the attack of the incoming nucleophiles forms the Si-face of enolate of 3a to Re-face of 2a takes place, which is consistent with the experimental results.
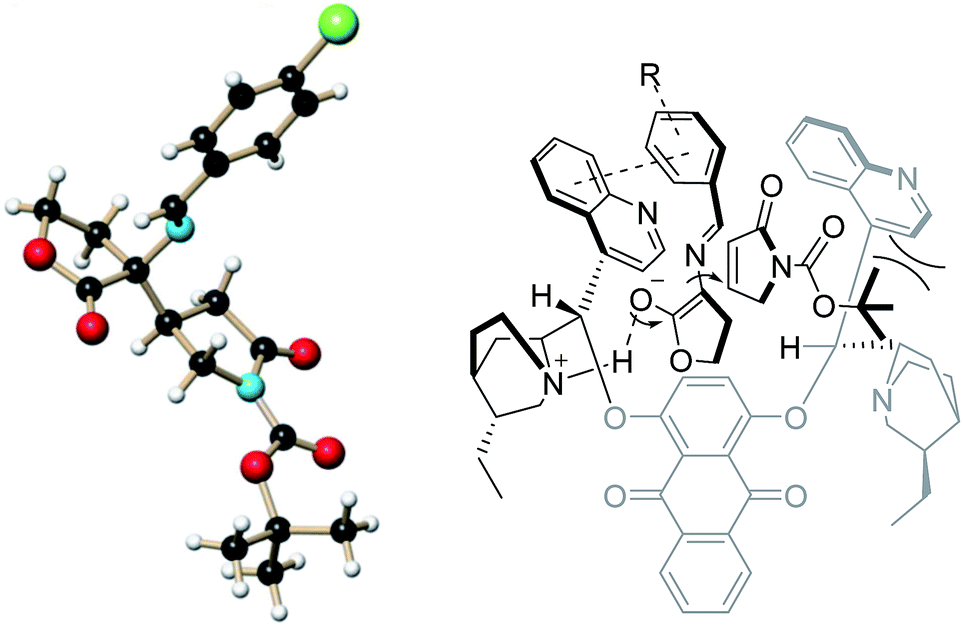 | ||
| Fig. 1 Proposed transition state for the reaction. | ||
In conclusion, we have disclosed the β-selective asymmetric addition of γ-butyrolactam with cyclic imino esters catalyzed by a bifunctional chiral tertiary amine, which provides an efficient and facile access to optically active β-position functionalized pyrrolidin-2-one derivatives with high diastereoselectivity and enantioselectivity. To our knowledge, this is the first catalytic method to access chiral β-functionalized pyrrolidin-2-one via a direct organocatalytic approach. Current efforts are in progress to apply this new methodology to synthesize biologically active products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We greatly appreciate the financial support from the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2016ZT06Y337), and X. J. thanks the Thousand Young Talents Program for financial support.Notes and references
- For selected reviews: (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 CrossRef; (b) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef PubMed; (c) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef PubMed; (d) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef PubMed; (e) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef PubMed.
- Reviews: (a) Y. Cheng, Z.-T. Huang and M. X. Wang, Curr. Org. Chem., 2004, 8, 325 CrossRef; (b) S. G. Pyne, A. S. Davis, N. J. Gates, J. P. Hartley, K. B. Lindsay, T. Machan and M. Tang, Synlett, 2004, 2670 CrossRef; (c) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC.
- For reviews, see: (a) A. Trabocchi, F. Guarna and A. Guarna, Curr. Org. Chem., 2005, 9, 1127 CrossRef; (b) M. Ordóñez and C. Cativiela, Tetrahedron: Asymmetry, 2007, 18, 3 CrossRef.
- (a) B. Bettler, K. Kaupmann, J. Mosbacher and M. Gassmann, Physiol. Rev., 2004, 84, 835 CrossRef PubMed; (b) L. A. Blackshaw, E. Staunton, A. Lehmann and J. Dent, Am. J. Physiol., 1999, 277, 867 Search PubMed; (c) P. Berthelot, C. Vaccher, N. Flouquet, M. Debaert, M. Luyckx and C. Brunet, J. Med. Chem., 1991, 34, 2557 CrossRef PubMed; (d) D. I. B. Kerr and J. Ong, Med. Res. Rev., 1992, 12, 593 CrossRef PubMed.
- (a) P. W. Baures, D. S. Eggleston, K. F. Erhard, L. B. Cieslinski, T. J. Torphy and S. B. Christensen, J. Med. Chem., 1993, 36, 3274 CrossRef PubMed; (b) N. Sommer, P. A. Loschmann, G. H. Northoff, M. Weller, A. Steinbrecher, J. P. Steinbach, R. Lichtenfels, R. Meyermann, A. Riethmuller, A. Fontana, J. Dichgans and R. Martin, Nat. Med., 1995, 1, 244 CrossRef PubMed.
- J. Zhang, X. Liu, X. Ma and R. Wang, Chem. Commun., 2013, 49, 3300 RSC.
- N. E. Shepherd, H. Tanabe, Y. Xu, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 3666 CrossRef PubMed.
- For selected examples, see: (a) H. Huang, Z. Jin, K. Zhu, X. Liang and J. Ye, Angew. Chem., Int. Ed., 2011, 50, 3232 CrossRef PubMed; (b) L. Lin, J. Zhang, X. Ma, X. Fu and R. Wang, Org. Lett., 2011, 13, 6410 CrossRef PubMed; (c) Y. Yang, S. Dong, X. Liu, L. Lin and X. Feng, Chem. Commun., 2012, 48, 5040 RSC; (d) J. Zhang, X. Liu, X. Ma and R. Wang, Chem. Commun., 2013, 49, 9329 RSC; (e) X. Gu, T. Guo, Y. Dai, A. Franchino, J. Fei, C. Zou, D. J. Dixon and J. Ye, Angew. Chem., Int. Ed., 2015, 54, 10249 CrossRef PubMed; (f) Y.-R. Chen, U. Das, M.-H. Liu and W. Lin, J. Org. Chem., 2015, 80, 1985 CrossRef PubMed.
- X. Jiang, L. Liu, P. Zhang, Y. Zhong and R. Wang, Angew. Chem., Int. Ed., 2013, 52, 11329 CrossRef PubMed.
- (a) C. Shao, H.-J. Yu, N.-Y. Wu, P. Tian, R. Wang, C.-G. Feng and G.-Q. Lin, Org. Lett., 2011, 13, 788 CrossRef PubMed; (b) H.-J. Yu, C. Shao, Z. Cui, C.-G. Feng and G.-Q. Lin, Chem.–Eur. J., 2012, 18, 13274 CrossRef PubMed; (c) V. Pace, J. P. Rae and D. J. Procter, Org. Lett., 2014, 16, 476 CrossRef PubMed; (d) J.-H. Fang, C.-A. Chang, B. Gopula, T.-S. Kuo, P.-Y. Wu, J. P. Henschke and H.-L. Wu, Asian J. Org. Chem., 2016, 5, 481 CrossRef.
- For recent reviews on organocatalytic asymmetric conjugate additions: (a) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703 CrossRef PubMed; (b) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626 CrossRef PubMed; (c) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 11, 1701 CrossRef.
- (a) L. Wang, X.-M. Shi, W.-P. Dong, L.-P. Zhu and R. Wang, Chem. Commun., 2013, 49, 3458 RSC; (b) X. Fang, X.-Q. Dong and C.-J. Wang, Tetrahedron Lett., 2014, 55, 5660 CrossRef; (c) A. Cayuelas, R. Ortiz, C. Nájera, J. M. Sansano, O. Larrañaga, A. de Cózar and F. P. Cossío, Org. Lett., 2016, 18, 2926 CrossRef PubMed; (d) Y. Wu, H. Liu, L. Zhang, Z. Sun, Y. Xiao, J. Huang, M. Wang and H. Guo, RSC Adv., 2016, 6, 73547 RSC; (e) L.-W. Tang, C. Li, B.-J. Zhao, L. Lan, M. Zhang and Z.-M. Zhou, Tetrahedron, 2017, 73, 923 CrossRef.
- For the selected examples of bifunctional amine catalysts: reviews, see: (a) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef PubMed; (b) S. J. Connon, Chem.–Eur. J., 2006, 12, 5418 CrossRef PubMed; (c) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef PubMed; (d) Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299 RSCFor selected recent examples, see: (e) B. Tan, N. R. Candeias and C. F. Barbas III, J. Am. Chem. Soc., 2011, 133, 4672 CrossRef PubMed; (f) B. Tan, N. R. Candeias and C. F. Barbas III, Nat. Chem., 2011, 3, 473 CrossRef PubMed; (g) X.-X. Jiang, Y.-F. Zhang, A. S.-C. Chan and R. Wang, Org. Lett., 2009, 11, 153 CrossRef PubMed; (h) S.-C. Pan, J. Zhou and B. List, Angew. Chem., Int. Ed., 2007, 46, 612 CrossRef PubMed; (i) Y. Yamaoka, H. Miyabe and Y. Takemoto, J. Am. Chem. Soc., 2007, 129, 6686 CrossRef PubMed; (j) L.-S. Zu, J. Wang, H. Li, H.-X. Xie, W. Jiang and W. Wang, J. Am. Chem. Soc., 2007, 129, 1036 CrossRef PubMed; (k) S. B. Tsogoeva and S. Wei, Chem. Commun., 2006, 1451 RSC; (l) D. A. Yalalov, S. B. Tsogoeva and S. Schmatz, Adv. Synth. Catal., 2006, 348, 826 CrossRef; (m) R. P. Herrera, V. Sgarzani, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 6576 CrossRef PubMed; (n) S. H. McCooey and S. J. Connon, Angew. Chem., Int. Ed., 2005, 44, 6367 CrossRef PubMed; (o) X. X. Jiang, Y. M. Cao, Y. Q. Wang, L. P. Liu, F. F. Shen and R. Wang, J. Am. Chem. Soc., 2010, 132, 15328 CrossRef PubMed; (p) X. X. Jiang, D. Fu, G. Zhang, Y. M. Cao, L. P. Liu and R. Wang, Chem. Commun., 2010, 46, 4294 RSC; (q) X. X. Jiang, D. Fu, X. M. Shi, S. L. Wang and R. Wang, Chem. Commun., 2011, 47, 8289 RSC; (r) L.-J. Zhou, Y.-C. Zhang, F. Jiang, G. He, J. Yan, H. Lu, S. Zhang and F. Shi, Adv. Synth. Catal., 2016, 358, 3069 CrossRef; (s) F. Jiang, D. Zhao, X. Yang, F.-R. Yuan, G.-J. Mei and F. Shi, ACS Catal., 2017, 7, 6984 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1475321. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra05264f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |