
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced photocarriers separation of novel CdS/pt/Mo2C heterostructure for visible-light-driven hydrogen evolution†
Xinbo Jing a,
Xueying Penga,
Youzhi Caoa,
Wei Wang*a and
Shufen Wang*b
a,
Xueying Penga,
Youzhi Caoa,
Wei Wang*a and
Shufen Wang*b
aSchool of Chemistry and Chemical Engineering, Key Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan, Shihezi University, Shihezi 832000, China
bCollege of Sciences, Shihezi University, Shihezi 832000, China
First published on 3rd October 2018
Abstract
The production of H2 from water using photocatalysts is a promising way of generating clean, renewable and alternative energy. The key issue is to develop active and stable photocatalysts. Here, we report a novel CdS/Pt/Mo2C heterostructure photocatalyst, where Pt nanoparticles are closely supported on CdS/Mo2C. The UV-vis spectrum and EIS Nyquist plots show that Mo2C can boost the absorption in the UV-vis region and improve the separation of the photogenerated electron–hole pairs from CdS. The Pt nanoparticles act as the active co-catalyst that promotes the transient photocurrent response. As a result, the CdS/Pt/Mo2C photocatalyst exhibits an excellent H2 evolution activity up to 1828.82 μmol h−1 g−1 under visible-light irradiation, 8.5 and 16.2 times higher than that of pristine CdS and CdS/Mo2C, respectively. Moreover, a high apparent quantum yield (AQY) of 9.39% is obtained at 400 nm for the CdS/Pt/Mo2C heterostructure photocatalyst.
1. Introduction
The cleanability and renewability of solar energy can be used to solve the problems of energy shortage and the environmental pollution, which are caused by the rapid development of global factories.1–3 One of the most promising means to make full use of solar energy is photocatalytic water decomposition excited by semiconductor photocatalyst. In past decades, there have been considerable efforts to develop perfect semiconductor photocatalysts with high activities for water splitting.4,5 However, there are still many limitations for us to promote the photocatalytic efficiency, for example, the low light absorption and the rapid recombination of photogenerated electron–hole pairs. Moreover, suitable photocatalysts with matched band gap positions and the stability necessary for overall water splitting are still lacking. Many semiconductors, such as TiO2 and ZnO, mainly absorb UV light, which indicates that they use only about 4% of the solar spectrum.6,7 To increase the visible-light response activity of semiconductors, many strategies have been reported, including doping,8 coupling with other photocatalyst,9 structure engineering,10 and so on. Among all these strategies, a heterostructure is one way to simultaneously obtain both efficient charge separation and photo-electrochemical stability.11For heterostructure photocatalysts, it is much important to select proper components. In recent years, many reports have shown that molybdenum carbides are active in a variety of catalytic reactions including desulfurization,12 water–gas shift,13 and hydrogenation reactions.14–16 It has been proven that Mo2C has excellent charge-transfer resistance.17 However, research about molybdenum carbides mostly focus on electrocatalysts for hydrogen production. The investigations of molybdenum carbide applied as photocatalyst are rare and indistinct. At the same time, CdS is popular and promising material as it can be excited in the visible light region.18 It has been proven that Pt is an excellent co-catalyst which can efficiently improve the photocatalytic H2 evolution capacity of semiconductor photocatalysts.19
Here, we designed and synthesized a heterogeneous CdS/Pt/Mo2C photocatalyst where the CdS acted as the photocatalyst, Pt as the photosensitizer20 and Mo2C as the electron-transfer mediator. The preparation process of CdS/Pt/Mo2C is illustrated in Scheme 1. Powder active charcoal (PAC) and ammonium molybdate were used as the charcoal and molybdenum source to synthesize tubular Mo2C in Ar atmosphere. Pt nanoparticles were synthesized by photodeposition method. Then, Pt nanoparticles were deposited on CdS, which was synthesized by a typical chemical bath deposition process. CdS/Pt/Mo2C photocatalyst is an efficient photocatalyst for H2 production under visible-light irradiation. The CdS/Pt/Mo2C samples exhibit enhanced visible-light absorption and excellent H2 evolution performance compared to pristine CdS and CdS/Mo2C. The superior performance of CdS/Pt/Mo2C is outstanding when compared with previously developed photocatalysts.11,21–23 A possible mechanism was also put forward to explain the improved photocatalytic performance of the heterogeneous catalyst.
 | ||
| Scheme 1 Schematic of the preparation process of CdS/Pt/Mo2C heterostructure. | ||
2. Experimental
2.1 Materials
All the chemicals in the experiments were of analytical reagent grade and used without further purification. Ammonium molybdate ((NH4)6Mo7O24·4H2O), hexamethylenetetramine (C6H12N4), active charcoal powder, cadmium nitrate tetrahydrate (Cd(NO3)2·4H2O), and Na2S, chloroplatinic acid hexahydrate (H2PtCl6·6H2O) were purchased from Sinopharm Chemical Reagent Co., Ltd, China.2.2 Sample synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) were dissolved in 20% NH3·H2O (7 mL). Then, 0.2 g active charcoal powder was added into the solution. The suspension was then further homogenized in an ultrasonic bath for 40 min. The slurry was dried at room temperatures for 24 h. The sample was vacuum-dried for 12 h at 60 °C. For carburization, the solid mixture was annealed in a tube furnace under Ar flow at 100 mL min−1 from ambient to 800 °C at a rate of 2 °C min−1 and then held at 800 °C for 2 h. Ar was kept purging through the reactor while it was cooled to ambient temperature.
:2) were dissolved in 20% NH3·H2O (7 mL). Then, 0.2 g active charcoal powder was added into the solution. The suspension was then further homogenized in an ultrasonic bath for 40 min. The slurry was dried at room temperatures for 24 h. The sample was vacuum-dried for 12 h at 60 °C. For carburization, the solid mixture was annealed in a tube furnace under Ar flow at 100 mL min−1 from ambient to 800 °C at a rate of 2 °C min−1 and then held at 800 °C for 2 h. Ar was kept purging through the reactor while it was cooled to ambient temperature.2.3 Photocatalytic hydrogen evolution
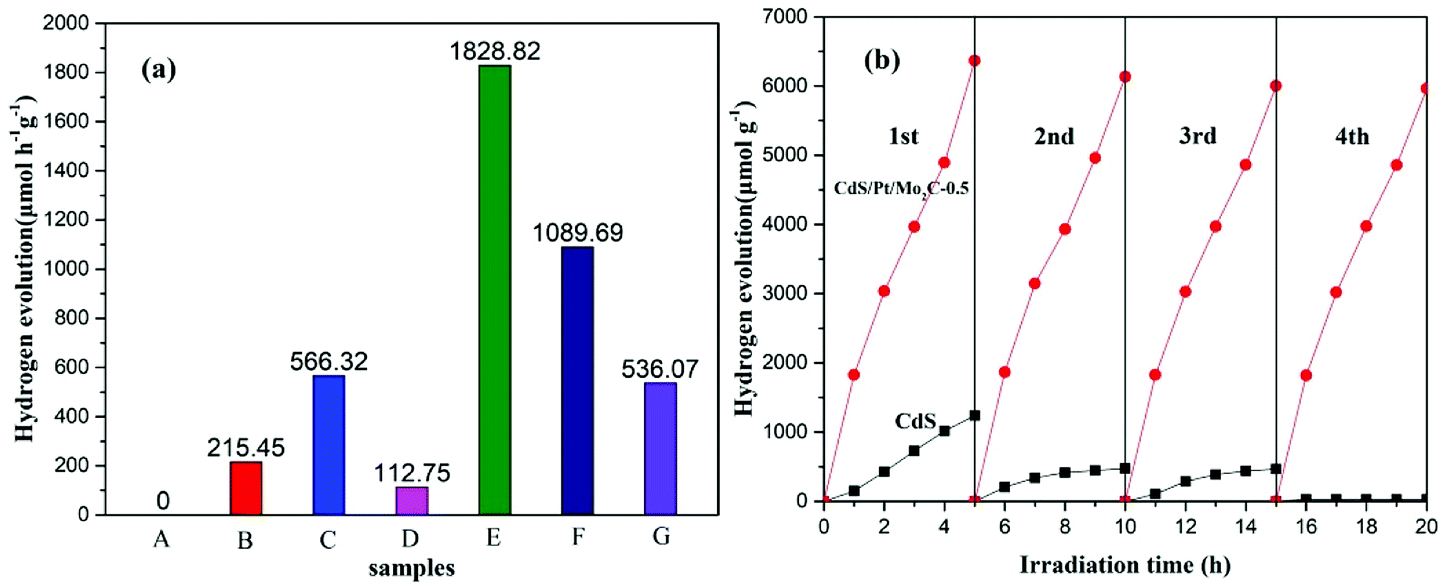
The photocatalytic hydrogen evolution reactions were conducted in a side-irradiation Pyrex reactor which was connected to a glass-closed gas circulation system at ambient temperature. In a typical photocatalytic experiment, 50 mg of catalyst power was suspended in 100 mL aqueous solution containing Na2S (0.1 M) and Na2SO3 (0.1 M) as the sacrificial agent for hole scavenging. Before visible light irradiation, the reactant system was degassed by evacuation to remove air and ensure that the reaction system was under anaerobic conditions. It was then was irradiated by a 300 W Xe lamp with a cutoff filter of 400 nm for H2 evolution under magnetic stirring. The amount of hydrogen evolution was analyzed by an online gas chromatograph (Agilent 7890, TCD, Ar as carrier).The apparent quantum yield (AQY) for photocatalytic H2 production was calculated as follows:
The number of incident photons were measured by a calibrated Si photodiode (SRC-1000-TC-QZ-N, Oriel, USA), and the distance between the calibrated Si photodiode and Xe-lamp with a 400 nm filter was 10 cm.
2.4 Electrochemical measurements
Photo-electrochemical measurements were performed on an electrochemical analyzer (Chenhua CHI 660) in a standard three-electrode cell. The working electrodes were prepared by drop-coating homogeneous catalyst suspensions directly onto the precleaned indium tin oxide glass (ITO glass) surfaces (1 × 2 cm). Samples on ITO glass with an active area of ca. 1 × 1 cm (1 mg of photocatalyst) were prepared as the working electrode. Platinum wire was used as the counter electrode, and a saturated calomel electrode (SCE) was used as the reference electrode. The E0 is 0.241 V vs. NHE at 25 °C for a saturated calomel electrode. Na2SO4 (0.5 M) aqueous solution was used as the supporting electrolyte. Electrochemical impedance spectroscopy (EIS) plots were collected at the open circuit potential (0.5 V vs. SCE), with the frequency ranging from 100 kHz to 0.01 Hz and modulation amplitude of 5 mV. The transient photocurrent measurements were recorded at an applied potential of 0.5 V vs. SCE under visible light illumination. A 300 W Xe lamp equipped with an optical cutoff filter of 400 nm was employed for the visible-light excitation.2.5 Characterizations
Powder X-ray diffraction (PXRD) data were collected using a Bruker AXS D8 X-ray diffractometer using Cu Kα radiation (λ = 1.54056 Å). Scanning electron microscope (SEM) images were recorded with a JEOL JSM-6490LV. Transmission electron microscope (TEM) and high-resolution transmission electron microscope (HRTEM) images were obtained by using a FEI Tecnai G2. The photocurrent response curves (PR) and electrochemical impedance spectroscopy (EIS) plots were collected by a CHI 660 electrochemical analyzer. Chemical states of the obtained samples were characterized by an Escalab 250 Xi system (a monochromatic Al Kα X-ray radiation). The binding energies of all elements were calibrated by the C 1s peak at 284.6 eV. UV-vis spectra were collected using a Thermo Fisher spectrometer with BaSO4 as a reference. The photoluminescence (PL) spectra were taken on a FLS 980 fluorescence spectrometer with an excitation wavelength of 318 nm.3. Results and discussion
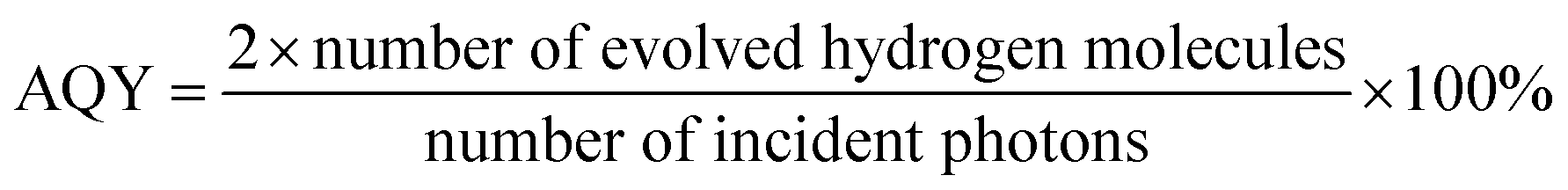
As showed in Fig. 1a, similar and sharp diffraction peaks (Fig. 1a) of the as-prepared CdS/Pt/Mo2C-0.5, CdS/Pt/Mo2C-1 and CdS/Pt/Mo2C-3 (the names are explained in 2.2 Sample synthesis) indicate excellent crystallinity. Fig. S2† shows the XRD pattern of CdS/Pt/Mo2C-0.5. The five characteristic peaks at 37.76°, 43.69°, 63.39°, 75.72° and 79.86° belonging to (111), (200), (220), (311) and (222) plane can be assigned to molybdenum carbide (JCPDS 77-0720). At the same time, five diffraction peaks at 2θ values of 24.80°, 26.50°, 28.18°, 36.62°, 52.79° correspond to the (100), (002), (101), (102) and (201) planes in CdS (JCPDS no. 77-2306), respectively. It is noteworthy that the diffraction peaks of CdS/Pt/Mo2C-0.5 are weaker than those of the other five samples, suggesting the relatively lower crystallinity. The XRD pattern of Mo2C and CdS correspond well with orthorhombic Mo2C phase (JCPDS no. 77-0720) and hexagonal CdS (JCPDS no. 77-2306), respectively. The XRD patterns of CdS/Pt/Mo2C-0.5 (Fig. S1†) are analogous to that of CdS/Mo2C, but no peaks for Pt can be observed. This is because of the relatively low concentration and relatively small size of Pt nanoparticles,24 which can be confirmed by the full XPS survey spectrum of CdS/Pt/Mo2C-0.5 (Fig. S3a†).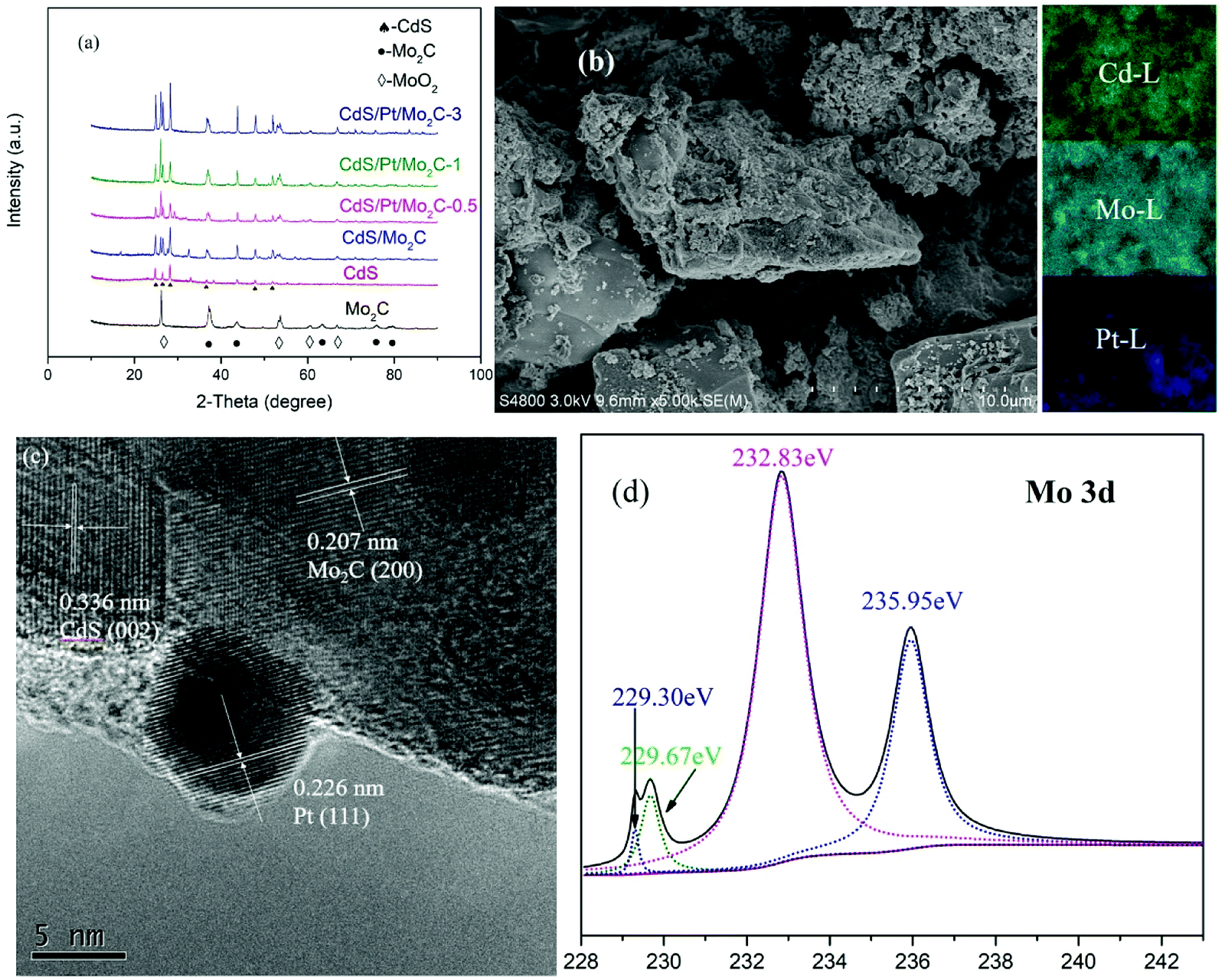 | ||
| Fig. 1 (a) XRD patterns of all samples. (b) SEM image and elemental mapping patterns. (c) HRTEM images of the CdS/Pt/Mo2C-0.5 sample. (d) The XPS peaks of Mo 3d of CdS/Pt/Mo2C-0.5. | ||
To investigate the morphology of the CdS/Pt/Mo2C-0.5 sample, scanning electron microscope (SEM) measurements were performed. From the SEM (Fig. 1b), it can be seen that CdS is closely connected with Mo2C and covers the surface of the active charcoal powder, as confirmed by EDS (Fig. S5†), the elemental mappings and TEM (Fig. S5†). The photodeposited Pt closely interacted with the CdS and Mo2C to form a CdS/Pt/Mo2C triphase structure (Fig. 1c). The lattice spacing with distances of 0.226 nm, 0.336 nm and 0.207 nm originated from Pt (111), CdS (002) and Mo2C (200) planes, respectively and are consistent with the XRD results. TEM and HRTEM images of the CdS/Pt/Mo2C-0.5 sample reveal that rather than a simple physical mixture, Pt and Mo2C, and CdS and Mo2C are in close contact, and form actual interfaces with each other, indicating the successful synthesis of a heterojunction.
The triphase heterojunction structure leads to a change of valence state and electron density of Cd, Mo and Pt. In order to further ascertain the details of the surface chemical composition and valence state of the CdS/Pt/Mo2C system, the XPS spectrum of the CdS/Pt/Mo2C-0.5 was measured (Fig. S3†). The full XPS spectrum of CdS/Pt/Mo2C-0.5 (Fig. S3a†) shows S, Mo, C, O, and Cd with sharp peaks. These results correspond to the EDS (Fig. S5†). At the same time, the weak peak of Pt is found at a binding energy of 71.78 eV (Fig. S3c†), suggesting that Pt exists in the oxidation state, Pt0. The XPS peaks of Cd 3d (Fig. S3e†) and Mo 3d (Fig. 1d) match well with those in previous studies. The peak fitting suggests oxidation states of Cd2+ for Cd and Mo2+ and Mo4+ for Mo. The binding energies of the Mo 3d5/2 and Mo 3d3/2 peaks were 228.2 and 231.4 eV, respectively and are typical values for Mo2+ in Mo2C. The high resolution XPS data of S 2p exhibits two peaks at 162.24 eV and 163.37 eV (Fig. S3b†), which can be assigned to S 2p3/2 and S 2p1/2, respectively. In addition, there is a distinct peak of 169.63 eV in the high resolution XPS of S 2p, which can be distributed to SO42− that may be created during the process of calcination. The XPS results confirm the existence of CdS, Mo2C and Pt.
For photocatalysts, the most important question is to lower the recombination of photogenerated electron–hole pairs. Photoluminescence (PL) spectroscopy was performed to confirm the capability of the samples in separating the photogenerated electron–hole pairs (Fig. 2a). The PL spectra of different samples (Fig. 2b) over a wavelength range from 410 to 800 nm showed a strong peak at nearly 465 nm while the PL intensity decreased as: CdS > CdS/Mo2C > Mo2C > CdS/Pt/Mo2C-3 > CdS/Pt/Mo2C-1 > CdS/Pt/Mo2C-0.5, as confirmed by EIS analysis (Fig. S2†). The PL intensity of the CdS/Mo2C is much lower than that of CdS, indicating efficient electron transfer in the CdS/Mo2C system from CdS to Mo2C, leading to the spatial separation of electrons and holes. This phenomenon can be attributed to the inspiring separation process of electron–hole pairs connected with the metal character of Mo2C. Furthermore, compared with CdS/Mo2C, the CdS/Pt/Mo2C system results in lower quenching of the PL intensity. The outcome of PL spectroscopy signifies that the photo-excited electrons transfer from CdS to Mo2C, and then migrate to Pt under visible light irradiation.24,25 The much higher transient photocurrent response also confirms the more efficient electron–hole separation for CdS/Pt/Mo2C-0.5 in relation to the pure CdS and Mo2C (Fig. 2c).26 The EIS results of CdS and CdS/Pt/Mo2C-0.5 (Fig. 2d) also prove that the conductivity of CdS/Pt/Mo2C-0.5 is much better than that of CdS.
 | ||
| Fig. 2 (a) PL spectra of the pure CdS nanoparticles and CdS/Pt/Mo2C-0.5. (b) PL spectra of different samples. (c) Transient photocurrent response of the pure CdS nanoparticles, Mo2C and CdS/Pt/Mo2C-0.5. (d) EIS Nyquist plots of CdS and CdS/Pt/Mo2C-0.5 samples. (e) DRS spectra of Mo2C, CdS and CdS/Pt/Mo2C-0.5. (f) Schematic illustration of band structures of the different samples vs. NHE, (A) CdS, (B) CdS/Mo2C, (C) CdS/Pt/Mo2C-0.5, (D) CdS/Pt/Mo2C-1 and (E) CdS/Pt/Mo2C-3. | ||
The optical properties of different samples were investigated by UV-vis diffuse reflection spectroscopy (DRS). An obvious enhancement, compared with that of CdS, in the absorption of wavelengths longer than 530 nm is seen in CdS/Mo2C based on the UV-vis spectrum (Fig. 2e). The corresponding main band structures of the different samples (Fig. S4a†) in visible light region are calculated by the transformed Kubelka–Munk function αhν = A(hν − Eg)2. The valence band potentials (Fig. S4b†) are 1.25, 0.99, 1.8, 1.05, 1.49 eV for CdS, CdS/Mo2C, CdS/Pt/Mo2C-0.5, CdS/Pt/Mo2C-1 and CdS/Pt/Mo2C-3, respectively. More clearly, the band positions of ECB and EVB for the different samples are illustrated in Fig. 2f. It can be found that the valence band and conduction band positions of CdS/Pt/Mo2C-0.5 shift from 1.25 eV to 1.8 eV and from −0.63 eV to −0.07 eV, respectively, compared with those of CdS, which can be due to the SPR effect of Pt nanoparticles.27 The results of optical test indicate the heterostructure system can efficiently improve the reductive capacity of the photo-generated electrons and significantly increase the photocatalytic activity of CdS.
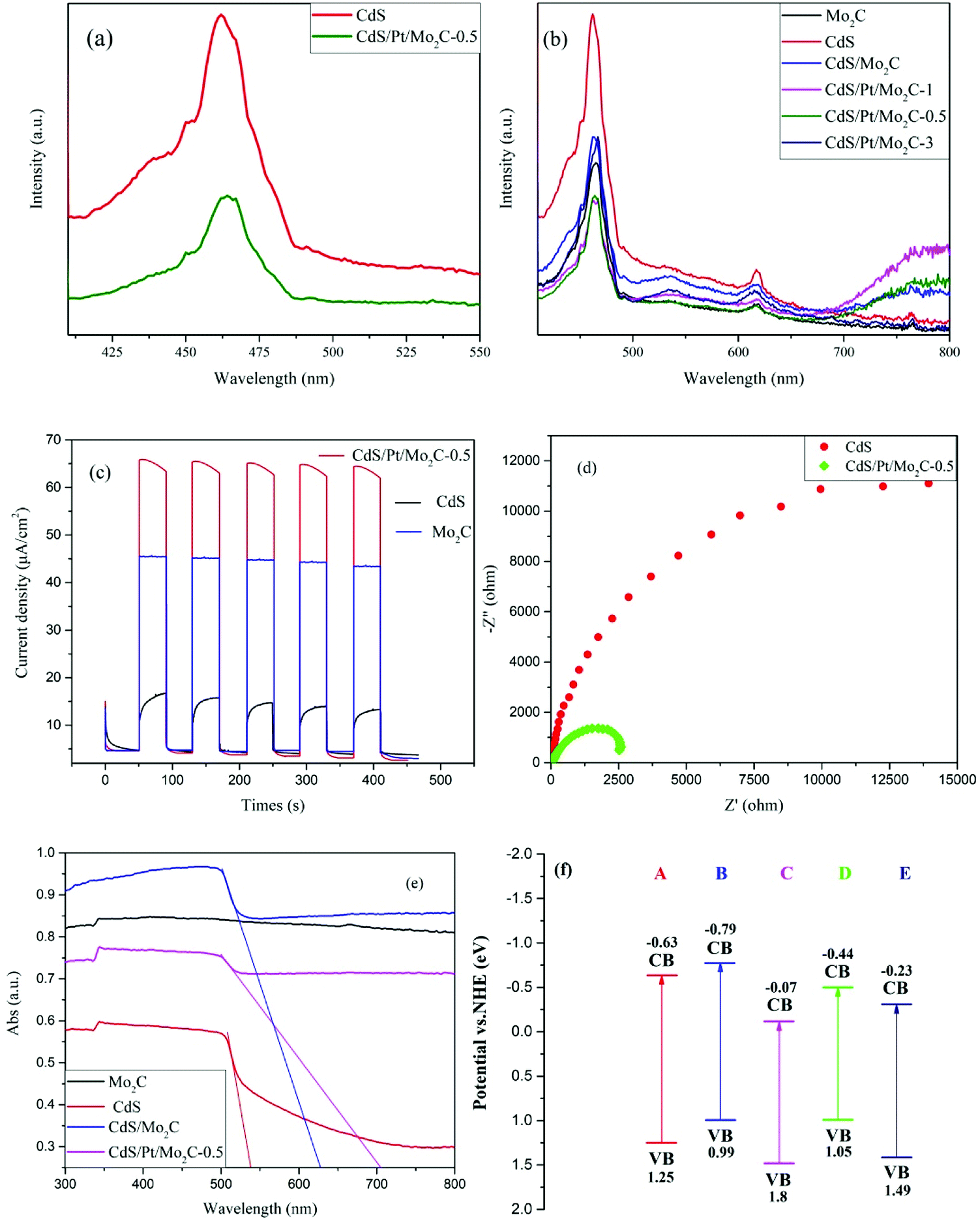
The photocatalytic H2 evolution in response to visible-light were examined for all samples in the presence of Na2S (0.1 M) and Na2SO3 (0.1 M) as the sacrificial agent of the hole and the results are illustrated in Fig. 3a. The CdS/Pt/Mo2C-0.5 shows the best H2 evolution performance with a rate of 1828.82 μmol h−1 g−1, which is 8.5 times and 16.2 times higher than that of pure CdS and CdS/Mo2C, respectively. Compared with other samples, CdS/Pt/Mo2C-0.5 possesses apparent quantum yield of 9.39% (AQY = 9.39%) under visible-light irradiation.25,28–33 Interestingly, upon further increasing the percent of Pt nanoparticles, the rate for photocatalytic hydrogen generation decreases, which is probably due to the extra percent of Pt nanoparticles in the CdS/Pt/Mo2C system. The excessive Pt may act as the center of recombination, which has been proven by previous research.20 The photocatalytic activity of the noble-metal-based CdS/Pt photocatalyst was also explored and an optimal H2 evolution rate of 566.32 μmol h−1 g−1 was obtained (Fig. S7†). Taking the H2 evolution rate of CdS/Pt/Mo2C-0.5 is 1828.82 μmol h−1 g−1 into consideration, which can prove that Mo2C can be used to boost the photo-activity. The role of CdS in the CdS/Pt/Mo2C system, as proven by the photocatalytic activity tests, was that of a photocatalyst. At the same time Pt nanoparticles were acting as the photosensitizer and activity sites.
 | ||
| Fig. 3 (a) Hydrogen evolution rate of different samples in 0.1 M Na2S and 0.1 M Na2SO3 aqueous solution under visible light irradiation (λ ≥ 400 nm); (A) Mo2C, (B) CdS, (C) Pt/Mo2C, (D) CdS/Mo2C, (E) CdS/Pt/Mo2C-0.5, (F) CdS/Pt/Mo2C-1, (G) CdS/Pt/Mo2C-3. (b) Recyclability testing and comparison of H2 evolution over CdS and CdS/Pt/Mo2C-0.5 in 0.1 M Na2S and 0.1 M Na2SO3 aqueous solution as sacrificial reagents. | ||
In order to verify the stability of the photocatalyst, an experiment to determine the recyclability of CdS and CdS/Pt/Mo2C-0.5 was carried out (Fig. 3b). From Fig. 3b, it can be observed that CdS/Pt/Mo2C-0.5 shows excellent stable performance under visible-light irradiation, and there is almost no significant decrease in the amount of photocatalytic H2 evolution for CdS/Pt/Mo2C-0.5, compared with that of CdS. The results confirm that CdS/Pt/Mo2C-0.5 is a robust photocatalyst. In addition, XRD and SEM analyses of fresh and used samples (Fig. S8 and S9†) reveal that there is no obvious difference between the two samples regarding composition and microstructure, which indicates that the CdS/Pt/Mo2C-0.5 is a stable catalyst under the designed reaction conditions.
3.1 Photocatalytic mechanism of the CdS/Pt/Mo2C system
On the basis of the above experimental results and discussions, a possible mechanism for H2 evolution under visible-light in the CdS/Pt/Mo2C system is shown in Scheme 2. The enhanced photocatalytic activity H2 evolution of CdS/Pt/Mo2C under visible-light condition could be attributed to the introduction of Pt nanoparticles and Mo2C, which created narrow bandgap energy and excellent separation efficiency of photo-generated electrons–holes. This consequently improved the absorption region of photocatalyst and improved charge carrier separation. Owing to the close connection of heterostructures and excellent separation of photo-generated electrons and holes caused by the metal character of Mo2C, the CdS/Pt/Mo2C heterostructure showed enhanced photocatalytic activity for hydrogen production under visible-light irradiation, compared with CdS and Pt/Mo2C. When irradiated by visible-light, CdS generates electron–hole pairs and the electrons are exited into the conduction band and then injected into Mo2C. The photogenerated electrons can be efficiently transferred to Pt nanoparticles where hydrions are reduced in the presence of the photogenerated electrons to produce H2. The hole left on the valence band of CdS reacts with the electron donor (Na2S/Na2SO3).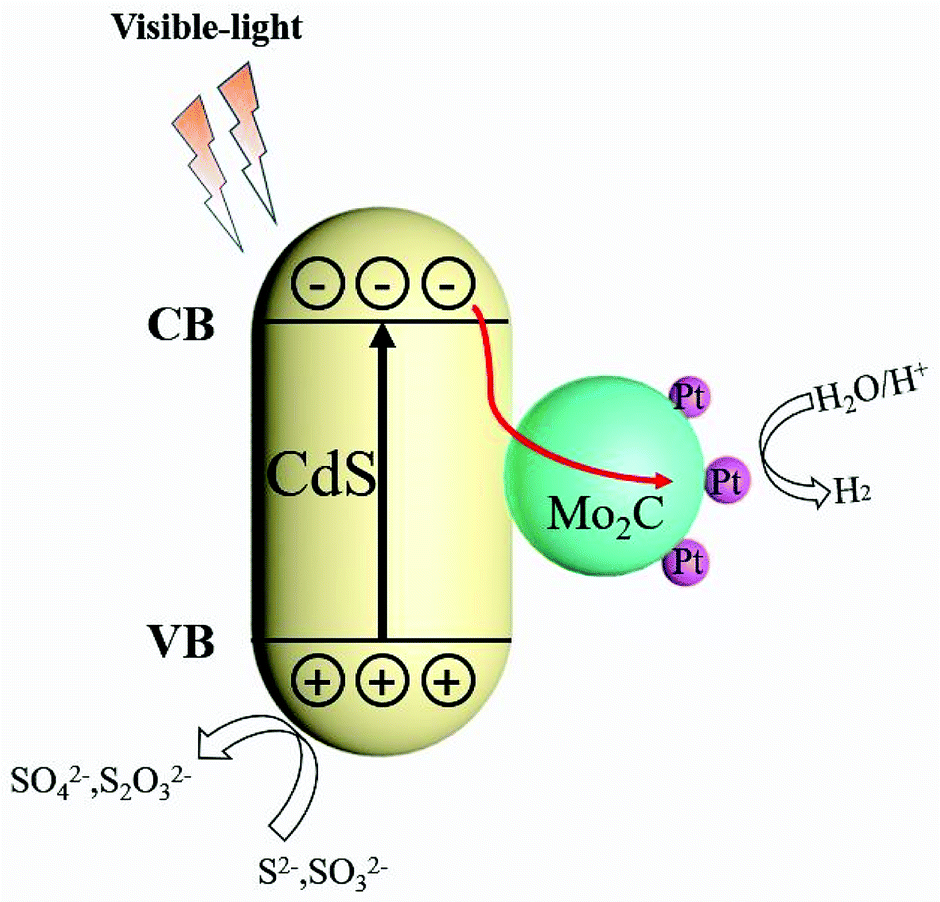 | ||
| Scheme 2 Photocatalytic mechanism of CdS/Pt/Mo2C system under visible-light irradiation (λ > 400 nm). | ||
4. Conclusion
In summary, triphase CdS/Pt/Mo2C heterostructure is a highly efficient and wide-spectrum-responsive photocatalyst. The investigation of the visible-light photocatalytic properties of the CdS/Pt/Mo2C composites shows a high photoactivity with H2 evolution rate of 1828.82 μmol h−1 g−1 and AQY of 9.39%. The aim of decreasing the amount of Pt used in the photocatalyst and increasing the activity of the photocatalyst was achieved, as proven by the optimal H2 evolution rate of 566.32 μmol h−1 g−1 in Pt/CdS. The superior performance can be due to the excellent separation of photo-generated electrons and holes and the electron transfer efficiency caused by the metal character of Mo2C which can not only inhibit the recombination of the photo-generated electron–hole pairs but also increase the visible-light absorption in CdS. Mo2C may provide some insight into the construction of novel and significantly efficient heterostructure photocatalysts for practical utilization in solar energy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (grant No. 21267020).References
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y.-W. Li, C. Shi, X.-D. Wen and D. Ma, Nature, 2017 DOI:10.1038/nature21672.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- H. Maleki and N. Hüsing, Appl. Catal., B, 2018, 221, 530–555 CAS.
- X. Sheng, Z. Liu, R. Zeng, L. Chen, X. Feng and L. Jiang, J. Am. Chem. Soc., 2017, 139, 12402–12405 CrossRef CAS PubMed.
- M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227–6240 CAS.
- J. Ferreira de Brito, F. Tavella, C. Genovese, C. Ampelli, M. V. B. Zanoni, G. Centi and S. Perathoner, Appl. Catal., B, 2018, 224, 136–145 CrossRef CAS.
- H. Mou, C. Song, Y. Zhou, B. Zhang and D. Wang, Appl. Catal., B, 2018, 221, 565–573 CrossRef CAS.
- H. Shi, Y. Yu, Y. Zhang, X. Feng, X. Zhao, H. Tan, S. U. Khan, Y. Li and E. Wang, Appl. Catal., B, 2018, 221, 280–289 CAS.
- C. Xue, S. Hu, Q. Chang, Y. Li, X. Liu and J. Yang, RSC Adv., 2017, 7, 49759–49768 RSC.
- Z. Fang, S. Weng, X. Ye, W. Feng, Z. Zheng, M. Lu, S. Lin, X. Fu and P. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 13915–13924 CAS.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- A. Celzard, J. F. Marêché, G. Furdin, V. Fierro, C. Sayag and J. Pielaszek, Green Chem., 2005, 7, 784 RSC.
- N. M. Schweitzer, J. A. Schaidle, O. K. Ezekoye, X. Pan, S. Linic and L. T. Thompson, J. Am. Chem. Soc., 2011, 133, 2378–2381 CrossRef CAS PubMed.
- J.-S. Li, Y. Wang, C.-H. Liu, S.-L. Li, Y.-G. Wang, L.-Z. Dong, Z.-H. Dai, Y.-F. Li and Y.-Q. Lan, Nat. Commun., 2016, 7, 11204 CrossRef CAS PubMed.
- R. Ma, Y. Zhou, Y. Chen, P. Li, Q. Liu and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 14723–14727 CrossRef CAS PubMed.
- H. B. Wu, B. Y. Xia, L. Yu, X.-Y. Yu and X. W. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed.
- W.-F. Chen, S. Iyer, S. Iyer, K. Sasaki, C.-H. Wang, Y. Zhu, J. T. Muckerman and E. Fujita, Energy Environ. Sci., 2013, 6, 1818–1826 RSC.
- B. Zhu, L. Zhang, B. Cheng and J. Yu, Appl. Catal., B, 2018, 224, 983–999 CrossRef CAS.
- Y.-N. Liu, C.-C. Shen, N. Jiang, Z.-W. Zhao, X. Zhou, S.-J. Zhao and A.-W. Xu, ACS Catal., 2017, 7(12), 8228–8234 CrossRef CAS.
- B. Ma, H. Xu, K. Lin, J. Li, H. Zhan, W. Liu and C. Li, ACS Sustainable Chem. Eng., 2016, 9, 820–824 CAS.
- A. A. Ismail and D. W. Bahnemann, Sol. Energy Mater. Sol. Cells, 2014, 128, 85–101 CrossRef CAS.
- Z. Xing, J. Zhang, J. Cui, J. Yin, T. Zhao, J. Kuang, Z. Xiu, N. Wan and W. Zhou, Appl. Catal., B, 2018, 225, 452–467 CrossRef CAS.
- Z. Zhao, G. Ge and D. Zhang, ChemCatChem, 2018, 10, 62–123 CrossRef CAS.
- W. Zhao, L. Xie, M. Zhang, Z. Ai, H. Xi, Y. Li, Q. Shi and J. Chen, Int. J. Hydrogen Energy, 2016, 41, 6277–6287 CrossRef CAS.
- D. Jiang, X. Chen, Z. Zhang, L. Zhang, Y. Wang, Z. Sun, R. M. Irfan and P. Du, J. Catal., 2017, 357, 147–153 CrossRef CAS.
- P. Xiao, J. Lou, H. Zhang, W. Song, X.-L. Wu, H. Lin, J. Chen, S. Liu and X. Wang, Catal.: Sci. Technol., 2018, 8, 201–209 RSC.
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS PubMed.
- Z. B. Yu, Y. P. Xie, G. Liu, G. Q. Lu, X. L. Ma and H.-M. Cheng, J. Mater. Chem. A, 2013, 1, 2773–2776 RSC.
- J. L. DiMeglio and B. M. Bartlett, Chem. Mater., 2017, 29, 7579–7586 CrossRef CAS.
- X. Guo, Y. Chen, Z. Qin, J. Su and L. Guo, ChemCatChem, 2018, 10, 153–158 CrossRef CAS.
- P. Wang, Y. Sheng, F. Wang and H. Yu, Appl. Catal., B, 2018, 220, 561–569 CrossRef CAS.
- F. Wang, Y. Wang, Y. Feng, Y. Zeng, Z. Xie, Q. Zhang, Y. Su, P. Chen, Y. Liu, K. Yao, W. Lv and G. Liu, Appl. Catal., B, 2018, 221, 510–520 CrossRef CAS.
- Y. Zhang, Z. Peng, S. Guan and X. Fu, Appl. Catal., B, 2018, 224, 1000–1008 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05473h |
| This journal is © The Royal Society of Chemistry 2018 |