
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow experimental details matter. The case of a laccase-catalysed oligomerisation reaction†
Keita Kashima ab,
Tomoyuki Fujisakiab,
Sandra Serrano-Luginbühla,
Abbos Khaydarova,
Reinhard Kissnerc,
Aleksandra Janošević Ležaićd,
Danica Bajuk-Bogdanoviće,
Gordana Ćirić-Marjanoviće,
Lukas D. Schulerf and
Peter Walde*a
ab,
Tomoyuki Fujisakiab,
Sandra Serrano-Luginbühla,
Abbos Khaydarova,
Reinhard Kissnerc,
Aleksandra Janošević Ležaićd,
Danica Bajuk-Bogdanoviće,
Gordana Ćirić-Marjanoviće,
Lukas D. Schulerf and
Peter Walde*a
aDepartment of Materials, ETH Zurich, Vladimir-Prelog-Weg 5, 8093 Zürich, Switzerland. E-mail: peter.walde@mat.ethz.ch
bDepartment of Materials Chemistry and Bioengineering, National Institute of Technology, Oyama College, 771 Ohaza-Nakakuki, Oyama, Tochigi 323-0806, Japan
cLaboratory of Inorganic Chemistry, Department of Chemistry and Applied Biosciences, Vladimir-Prelog-Weg 2, 8093 Zürich, Switzerland
dUniversity of Belgrade, Faculty of Pharmacy, Vojvode Stepe Street 450, 11221 Belgrade, Serbia
eUniversity of Belgrade, Faculty of Physical Chemistry, Studentski trg 12-16, 11158 Belgrade, Serbia
fxirrus GmbH, Buchzelgstrasse 36, 8053 Zürich, Switzerland
First published on 26th September 2018
Abstract
The Trametes versicolor laccase (TvL)-catalysed oligomerisation of the aniline dimer p-aminodiphenylamine (PADPA) was investigated in an aqueous medium of pH = 3.5, containing 80–100 nm-sized anionic vesicles formed from AOT, the sodium salt of bis(2-ethylhexyl)sulfosuccinic acid. If run under optimal conditions, the reaction yields oligomeric products which resemble the emeraldine salt form of polyaniline (PANI-ES) in its polaron state, known to be the only oxidation state of linear PANI which is electrically conductive. The vesicles serve as “templates” for obtaining products with the desired PANI-ES-like features. For this complex, heterogeneous, vesicle-assisted, and enzyme-mediated reaction, in which dissolved dioxygen also takes part as a re-oxidant for TvL, small changes in the composition of the reaction mixture can have significant effects. Initial conditions may not only affect the kinetics of the reaction, but also the outcome, i.e., the product distribution once the reaction reaches its equilibrium state. While a change in the reaction temperature from T ≈ 25 to 5 °C mainly influenced the rate of reaction, increase in enzyme concentration and the presence of millimolar concentrations of chloride ions were found to have significant undesired effects on the outcome of the reaction. Chloride ions, which may originate from the preparation of the pH = 3.5 solution, inhibit TvL, such that higher TvL concentrations are required than without chloride to yield the same product distribution for the same reaction runtime as in the absence of chloride. With TvL concentrations much higher than the elaborated value, the products obtained clearly were different and over-oxidised. Thus, a change in the activity of the enzyme was found to have influence not only on kinetics but also led to a change in the final product distribution, molecular structure and electrical properties, which was a surprising find. The complementary analytical methods which we used in this work were in situ UV/vis/NIR, EPR, and Raman spectroscopy measurements, in combination with a detailed ex situ HPLC analysis and molecular dynamics simulations. With the results obtained, we would like to recall the often neglected or ignored fact that it is important to describe and pay attention to the experimental details, since this matters for being able to perform experiments in a reproducible way.
1 Introduction
A well-known prerequisite for carrying out chemical reactions in a controlled and reproducible way is to pay attention to the experimental details.1,2 This is particularly so for complex, heterogeneous chemical transformations, for which it is essential to follow precise protocols to control the outcome of the reactions, for example to obtain a desired product in a desired yield within a desired time. Therefore, thousands of chemical synthesis recipes have been published in the literature. This is very much like in conventional cooking, which is nothing else than the controlled chemical and physical transformation of relatively ill-defined mixtures of chemical compounds.3 Obviously, the more sophisticated a food preparation is, the more detailed the recipe must be; and the more complex a chemical transformation is, the more detailed the protocol must be. Sometimes, one may not be aware of the significance of certain details; it is only when others try to repeat the reaction that one realizes that missing details should have been mentioned since they are essential.4,5Examples of complex chemical reactions are enzyme-catalysed oxidations and oligo- or polymerisations of arylamines,6 in particular of aniline7–18 – or of the linear N–C-para-coupled aniline dimer p-aminodiphenylamine (PADPA)19–23 – in aqueous media in the presence of so-called “templates”. The templates are additives that have a positive influence on the outcome of the reactions in the sense that the transformations are directed by them to obtain maximal yields of desired products.24 Excellent templates for the mentioned reactions are either anionic macromolecules, for example polymers with sulfonated groups,7–12 or anionic polymolecular assemblies, for example, micelles9,13,14,19,25 or vesicles,15–18,20–23 composed of amphiphiles with sulfonated head groups. The products distribution of the reactions not only depends on (i) the type and concentration of template,9 (ii) whether aniline or PADPA is used as monomer,18,20 (iii) the monomer concentration, (iv) the type of enzyme/oxidant system – for example horseradish peroxidase isoenzyme C (HRPC)/H2O2 (ref. 17 and 23) or Trametes versicolor laccase (TvL)/O2 (ref. 18 and 20) – (v) the pH and salt conditions of the aqueous solution which is used for the reaction,16 (vi) the order of adding solutions (or dispersions) of the compounds used for starting the reaction, but also on (vii) the total enzyme concentration20 and (viii) the temperature.16,18 For aniline/HRPC/H2O2 (ref. 17) or PADPA/TvL/O2 (ref. 20) as monomer/enzyme/oxidant systems and vesicles formed from AOT, the sodium salt of bis(2-ethylhexyl)sulfosuccinate, optimal conditions were previously elaborated. These products resemble the conductive polaronic emeraldine salt form of chemically or electrochemically synthesised polyaniline, abbreviated as PANI-ES. Such enzymatic synthesis of PANI-ES is considered as a valuable alternative approach to chemically synthesised PANI-ES, although the advantages and disadvantages of the enzymatic approach, as compared to the purely chemical one, have to be discussed critically, at least in terms of amount and costs of the enzyme used.17,20
The system we investigated in the work presented here consists of (i) an aqueous solution of pH = 3.5 (called “pH = 3.5 solution”),21 prepared from NaH2PO4 and H3PO4 at a total phosphate concentration of 0.1 M, (ii) PADPA as monomer, (iii) TvL/O2 as catalyst/oxidant system, and (iv) 80–100 nm-sized unilamellar AOT vesicles as templates. The AOT concentration was kept constant at 1.5 mM and the concentration of PADPA at the start of the reaction was 1.0 mM. The reaction volume was always 10 mL (containing initially 10 μmol PADPA), and the reaction was carried out without stirring in closed 50 mL glass flasks (Schott bottles). The available amount of O2 content was the sum of the solubility of O2 in the aqueous solution (≈2.6 μmol) and the approximately 40 mL air inside the reaction flask (≈400 μmol), see the ESI† of (Junker et al., 2014).18 Based on stoichiometric considerations, this amount of O2 is sufficient to obtain PANI-ES-type products from 10 μmol PADPA with TvL/O2.20
The main aim of the work was to explore and explain previous observations16 concerning the effect of the amount of TvL on the products obtained. Varying the amount, i.e. concentration, of TvL has a significant influence on the UV/vis/NIR spectrum of the obtained products,16 and not only on the rate of reaction as one may expect at first sight from using an enzyme as catalyst at different concentrations.
In a first series of experiments, the TvL concentration was varied for reactions of PADPA in the presence of AOT vesicles at pH = 3.5 and T ≈ 25 °C (room temperature). In the second part of the investigations, we questioned whether a decrease in reaction temperature from T ≈ 25 °C to T = 5 °C has any significant (and beneficial) effect on the type of reaction products obtained.
2 Materials and methods
2.1 Materials
Laccase from Trametes versicolor (TvL, EC 1.10.3.2; product no. 51639, 13.6 U mg−1, lot no. BCBF7247 V, M ≈ 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1),26–28 bis(2-ethylhexyl)sulfosuccinate sodium salt (AOT, BioUltra ≥99.0%), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt (ABTS2−(NH4+)2, ≥98%), sodium phosphate monobasic (NaH2PO4, ≥99.0%), aqueous orthophosphoric acid (H3PO4, 85 wt%), hydrochloric acid (HCl, ≥37 wt%), acetonitrile (CH3CN, ≥99.5%), aqueous hydrazine (N2H4, 35 wt%), chloroform (CHCl3, ≥99.0%), ethanol (99.99%), and tert-butyl methyl ether (MTBE, ≥99.0%) were purchased from Sigma-Aldrich or Fluka. Aqueous ammonia (NH3, 25 wt%) was purchased from Merck. All these chemicals were used as obtained. N-Phenyl-p-phenylenediamine (p-aminodiphenylamine, PADPA, 98%) was from Sigma-Aldrich or ABCR GmbH and purified by recrystallization from hexane (4–5 times) until white crystals were obtained. Deionized water prepared with a Millipore Synergy system was used for all experiments.
000 g mol−1),26–28 bis(2-ethylhexyl)sulfosuccinate sodium salt (AOT, BioUltra ≥99.0%), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)diammonium salt (ABTS2−(NH4+)2, ≥98%), sodium phosphate monobasic (NaH2PO4, ≥99.0%), aqueous orthophosphoric acid (H3PO4, 85 wt%), hydrochloric acid (HCl, ≥37 wt%), acetonitrile (CH3CN, ≥99.5%), aqueous hydrazine (N2H4, 35 wt%), chloroform (CHCl3, ≥99.0%), ethanol (99.99%), and tert-butyl methyl ether (MTBE, ≥99.0%) were purchased from Sigma-Aldrich or Fluka. Aqueous ammonia (NH3, 25 wt%) was purchased from Merck. All these chemicals were used as obtained. N-Phenyl-p-phenylenediamine (p-aminodiphenylamine, PADPA, 98%) was from Sigma-Aldrich or ABCR GmbH and purified by recrystallization from hexane (4–5 times) until white crystals were obtained. Deionized water prepared with a Millipore Synergy system was used for all experiments.
2.2 Preparation of two “pH = 3.5 solutions”
Two aqueous solutions of pH = 3.5 were prepared, both at [H2PO4−] + [H3PO4] = 0.1 M. Based on in both cases NaH2PO4, the pH of the solution was adjusted to a value of 3.5 either with H3PO4 or with HCl. Therefore, the difference between the two “pH = 3.5 solutions” was that one contained chloride ions, the other did not. For the chloride-free “pH 3.5 solution”, 1.58 mL of 1.0 M H3PO4 in H2O were added to 500 mL of 0.1 M NaH2PO4 in H2O. For the “pH 3.5 solution” containing chloride ions, the pH value of the 0.1 M NaH2PO4 solution was adjusted by adding 1 M HCl (≈1.2 mL per 500 mL, corresponding to [Cl−] = 2.4 mM).2.3 Preparation of AOT vesicle stock suspension
Suspensions of 20 mM AOT consisting of mainly unilamellar vesicles with average diameters of 80–100 nm were prepared with the freezing–thawing/extrusion method.20–22 In a first step, 0.178 g AOT (0.4 mmol) were dissolved in 5 mL chloroform inside a 250 mL round bottom glass flask. After removal of the chloroform by rotary evaporation and successive high vacuum drying overnight, the formed dry AOT film was then hydrated with one of the two “pH = 3.5 solutions”. As discussed below, the chloride-free “pH = 3.5 solution” was used for the majority of experiments. The obtained AOT suspensions were frozen in liquid nitrogen and thawed in a water bath heated to 60 °C. This freezing–thawing process was repeated 10 times. Finally, the obtained AOT suspensions were extruded five times through a Nucleopore polycarbonate membrane of 200 nm pore size and subsequently ten times through a membrane of 100 nm pore size29,30 by using the Extruder from Lipex Biomembranes (Vancouver, Canada).31 The vesicle suspensions were stored at room temperature and used within one month after preparation.2.4 Preparation of PADPA stock solution
A PADPA stock solution of 0.15 M PADPA in ethanol was prepared by dissolving 1.38 mg PADPA in 50 μL ethanol. This solution was stored at T = 5 °C in a refrigerator and used within one day after preparation.2.5 Preparation and activity of TvL stock solution
A TvL stock solution was prepared as follows; 12.92 mg of the laccase powder were dissolved inside a 1.5 mL polypropylene Eppendorf tube in 1 mL deionized water by slow agitation with a Vortex Genie 2 device (from Scientific Industries, Inc.) at the lowest output power level. Afterwards, the solution was centrifuged at 6000 rpm for 2 min in a Eppendorf centrifuge 5415 D. The supernatant was kept as TvL stock solution and the insoluble precipitate was discarded. The molar concentration of TvL in this stock solution was estimated in the same way as described previously,20 and found to be ≈16 μM. This solution was stored at T = 5 °C in a refrigerator. For most of the experiments, the TvL stock solution was first diluted with water to ≈1.6 μM before use. The activity and stability of TvL dissolved in water or in one of the two “pH = 3.5 solutions” were determined spectrophotometrically at pH = 3.5 with 0.25 mM ABTS2− as substrate as described before,20 using quartz cuvettes with pathlengths of 1.0 cm (from Hellma Analytics) and a Cary 1E UV-visible spectrometer (from Varian).2.6 Preparation of reaction mixtures
All reactions with PADPA were carried out in 50 mL Schott Duran® laboratory glass bottles which were kept closed during the reaction with screw caps, see Fig. 1. Different mixtures were prepared and analysed. For the one which we consider our new optimal conditions, the following solutions were mixed in the order given: 9.2 mL of the chloride-free “pH = 3.5 solution”, 0.75 mL of the 20 mM AOT stock suspension, 67 μL of the 0.15 M PADPA stock solution, and finally 16 μL of the diluted TvL stock solution (≈1.6 μM TvL) to yield a total reaction volume of 10 mL. After all components were added individually, the mixture was gently agitated to homogenize sufficiently. The initial concentrations in the reaction mixture were [AOT] = 1.5 mM, [PADPA]0 = 1.0 mM, and [TvL] ≈ 2.6 nM, and the bottle was closed with the screw cap and stored at room temperature (T ≈ 25 °C). For reactions with [TvL] ≈ 320 nM, the volume of the chloride-free “pH = 3.5 solution” was 10.0 mL and 200 μL of the ≈16 μM TvL stock solution were added. For the reaction with the “pH = 3.5 solution” containing chloride ions, the reaction mixtures were prepared in the same way. For other conditions, the volumes of the “pH = 3.5 solution” and of the TvL stock solution were adjusted accordingly. For reactions at T = 5 °C, the glass bottles containing the reaction mixtures were stored in a refrigerator.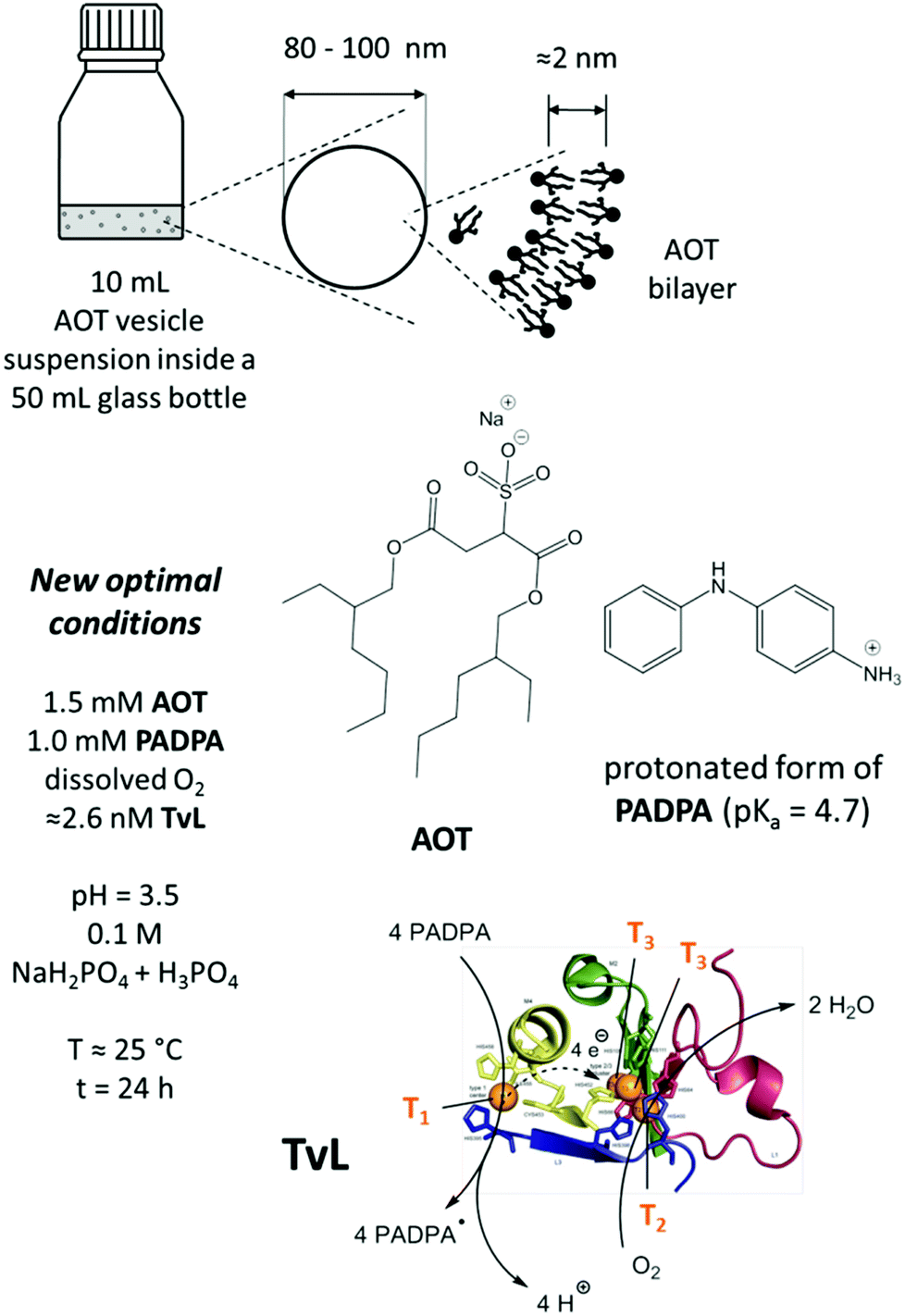 | ||
| Fig. 1 Schematic representation and overview about the main reaction mixture investigated (new optimal conditions), consisting of dispersed unilamellar AOT vesicles with average diameters of 80–100 nm and a membrane thickness of about 2 nm,17 PADPA as monomer, and TvL/O2 as catalyst/oxidant in an aqueous solution of NaH2PO4 and H3PO4 at pH = 3.5. The estimated enzyme concentration for the reaction with 1.0 mM PADPA in the presence of 1.5 mM AOT for obtaining PANI-ES-type products in high yield is ≈2.6 nM for a chloride-free “pH = 3.5 solution”, see text for details. Under these conditions, most of the PADPA molecules are expected to be present in their protonated form (pKa = 4.7).56 TvL is expected to oxidise PADPA in its neutral form at copper site T1 to the PADPA radial (PADPA˙).22 Reoxidation of TvL occurs with dissolved O2 at the trinuclear copper site T2/T3.26,43,57 Non-enzymatic follow-up reactions of PADPA˙ lead to the formation of PANI-ES-type products.22 PDB (TvL): 1GYC, see (Sirim et al., 2011).28 | ||
2.7 Reaction product analysis by in situ UV/vis/NIR spectroscopy measurements
UV/vis/NIR measurements were carried out at room temperature with a V-670 spectrophotometer (from JASCO). Aliquots of 0.3 mL were withdrawn at predetermined times from the reaction mixture, and the absorption spectrum was recorded from 190 nm to 1500 nm using quartz cuvettes with a path length of 0.1 cm (from Hellma Analytics).2.8 Reaction product analysis by in situ EPR spectroscopy measurements
EPR measurements were carried out at room temperature with a Bruker EMX X-band spectrometer equipped with a TM cavity. Aliquots of 0.1 mL of reacting vesicle suspension were introduced into the resonator by means of a 50 mm × 10 mm × 0.2 mm quartz flat cell (Wilmad Glass).2.9 Reaction product analysis by in situ Raman spectroscopy measurements
The Raman spectra were recorded with a DXR Raman microscope (from Thermo Scientific), equipped with a research optical microscope and a CCD detector, using a HeNe gas laser with an excitation wavelength of 633 nm. Aliquots of 5 μL were withdrawn at predetermined times from the reaction mixture and the Raman spectrum was recorded at room temperature from 2000 cm−1 to 300 cm−1. For this, the aliquots were transferred into the sample wells on a sample slide (Gold EZ-Spot Micro Mount sample slide from Thermo Scientific). Each spectrum was measured for a new aliquot, which was taken from the reaction mixture at the specified time and transferred into an empty and clean sample well. The slide with the sample of the reaction mixture was placed on an X–Y motorized sample stage and the laser beam was focused on the sample by using an objective magnification of 10×. The scattered light was analyzed by the spectrograph with a 600 lines mm−1 grating. The laser power on the sample was kept at 4.0 mW. The spectra were recorded using an exposure time of 10 s and 10 exposures per spectrum. All the Raman spectra shown are corrected for fluorescence by the OMNIC software (from Thermo Scientific).2.10 Reaction product analysis by HPLC-DAD and HPLC-MS measurements
The deprotonated and reduced reaction products were separated and analyzed by high performance liquid chromatography with a reverse-phase EC 150/4.6 Nucleodur C18 Isis column (from Machery-Nagel), with either a spectrophotometric diode array detector (HPLC-DAD) or a mass spectrometer as detector (HPLC-MS). The elution started with 20 vol% CH3CN and 80 vol% 10 mM NH4HCO3 (pH = 10) with a gradual change to 95 vol% CH3CN over a period of 75 min with a flow rate of 0.7 mL min−1.The sample preparation was similar to the one reported previously,22 as described here: (1) aliquots of 0.5 mL were withdrawn at predetermined times from the reaction mixture and transferred into 1.5 mL Eppendorf tubes. (2) Immediately afterwards, 1 mL MTBE and 0.1 mL of an aqueous NH3 solution (25 wt%) were added to extract remaining PADPA and the reaction products into MTBE. (3) The tubes were closed and shaken from time to time and then left to stand overnight at room temperature. Complete extraction of the deprotonated products into the organic phase was assumed to have occurred if the remaining aqueous phase was colorless. (4) The colored organic phase was withdrawn and collected in a 15 mL glass bottle. (5) Then, 1 mL CH3CN were added and the organic solvent mixture was removed in vacuo at 70 mbar for 10 min at room temperature. (6) Afterwards, 0.4 mL aqueous H4N2 (35 wt%) were added to reduce the products under vigorous stirring for 10 min. (7) Finally, 1 mL CH3CN and 10 mL 10 mM NH4HCO3 buffer (pH = 10, prepared by adjusting the pH value with 25 wt% aqueous NH3) were added. For the HPLC-DAD analysis, a Dionex UltiMate 3000 UHPLC+ focused system and a Dionex UltimMate 3000 rapid separation diode array detector (DAD-3000RS) were used (injection volume: 5 μL). The chromatograms were elaborated for absorption at λ = 310 nm. For the HPLC-MS analysis, an Agilent 1200 HPLC instrument and a Bruker Daltonics maXis ESI-QTOF MS detector with a capillary voltage of +4.5 kV were applied (injection volume: 5 μL). The covered mass range for the HPLC-MS analysis was 100–1200 Da.
2.11 MD simulations of the AOT bilayer
The setup of our MD simulation of the AOT bilayer including buffer species was already done for our earlier analysis at 25 °C.20,22 The system consisted of 512 negatively charged AOT molecules (64 of each of the eight different stereoisomers), 641 sodium counter ions, 129 di-hydrogen phosphate, 6 phosphoric acid and 62337 water molecules. In the simulation protocol, a simulated annealing procedure to cool down from 25 °C to 5 °C was implemented such that no big shift of the thermodynamic equilibrium was expected. The simulation was started with the equilibrated structure after 10 ns at 25 °C and simulated each cooling step from 25 °C to 5 °C with a decrease of only 1 degree for another 2 ns. Therefore 21 times 2 ns were necessary to reach the lower temperature, which resulted in a time step of 52 ns for the structure at 5 °C. At 5 °C, we simulated the structure for another 30 ns in equilibrium, so that we could be sure the statistical analysis was not biased by the cooling procedure applied beforehand. We did not simulate for longer times, since this AOT- system contains more than 200000 atoms and many ions. For the analysis, we calculated radial distribution functions g(r) between the sulfur head groups, the angular orientation against the transversal (normal) z-axis by taking the average vector of the sulfur head atom to the mean position between the methyl tails of the AOT molecules, and the diffusion coefficient of the sulfur head groups by the mean square displacement.
3 Results and discussion
3.1 Optimal conditions for obtaining PANI-ES-type products at T ≈ 25 °C from 1.0 mM PADPA with 2.6 nM TvL
A few years ago, we wondered whether it is possible to obtain polyaniline in its emeraldine salt form (PANI-ES) from the aniline dimer PADPA as monomer – instead of aniline – with TvL/O2 and AOT vesicles as templates.20 We were particularly interested in obtaining PANI-ES products in their conductive forms, i.e., with a high fraction of polaron states. Polaron transitions originate from radical cations (which give rise to an EPR response)32–35 and show high intensity in the NIR region of the absorption spectrum, at λ ≈ 1000 nm or above.36–39 Indeed, complementary in situ UV/vis/NIR, in situ EPR, in situ Raman spectroscopy, and ex situ HPLC measurements indicated that PANI-ES-type products can be obtained from PADPA with TvL/O2 and AOT vesicles; and that the products formed under the elaborated optimal conditions are not true polymers, but mainly oligomers.21,22 The emeraldine salt of tetraaniline – i.e., the linear PADPA dimer – in its polaron state was identified as the main product after complete conversion of PADPA, at a reaction runtime t ≈ 24 h and T ≈ 25 °C.21,22 The initial concentration of PADPA was 1.0 mM, the concentration of AOT was 1.5 mM, the pH value was 3.5 ([H2PO4−] + [H3PO4] = 0.1 M), and the concentration of TvL was ≈32 nM; the reaction runtime was t ≈ 1 d.20 With ≈64 nM TvL, the outcome of the reaction was about the same.20 At that time, the “pH = 3.5 solution” was prepared from NaH2PO4 by using HCl for adjusting the pH to a value of 3.5.20 To our initial surprise, we recently realised that the reaction proceeds much faster if the “pH = 3.5 solution” is prepared from NaH2PO4 and H3PO4 (instead of HCl), probably because of the competitive inhibitory effect of chloride ions on TvL,40–43 which we had overlooked, see below. Based on literature data, it seems that the chloride ions suppress the electron transfer by blocking the access of the T1 site of the laccase (see Fig. 1), as recently discussed by (Di Bari et al., 2017).43 The influence of the seemingly minor difference in the way the “pH = 3.5 solution” was prepared, is a clear example for how experimental details may matter. We explored the inhibitory effect of chloride ions on the reaction and determined how much we could reduce the TvL for the PADPA reaction to complete within t ≈ 1 d. The results obtained are outlined in the following.If the “pH = 3.5 solution” is prepared from NaH2PO4 and H3PO4 (no chloride ions), the AOT vesicle-guided transformation of PADPA (1.0 mM) into PANI-ES-type products, with high absorption around 1000 nm, proceeds well in the presence of 1.5 mM AOT by using 2.6 nM TvL instead of 32 or 64 nM, see Fig. 2. Therefore, our new optimal conditions for this reaction are 1.0 mM PADPA, 1.5 mM AOT, 2.6 nM TvL, aqueous “pH = 3.5 solution” (0.1 M NaH2PO4/H3PO4), T ≈ 25 °C. Under these conditions, a rather stable UV/vis/NIR spectrum is obtained after t = 1 d, with λmax ≈ 1050, 420, and 298 nm, see Fig. 2A.
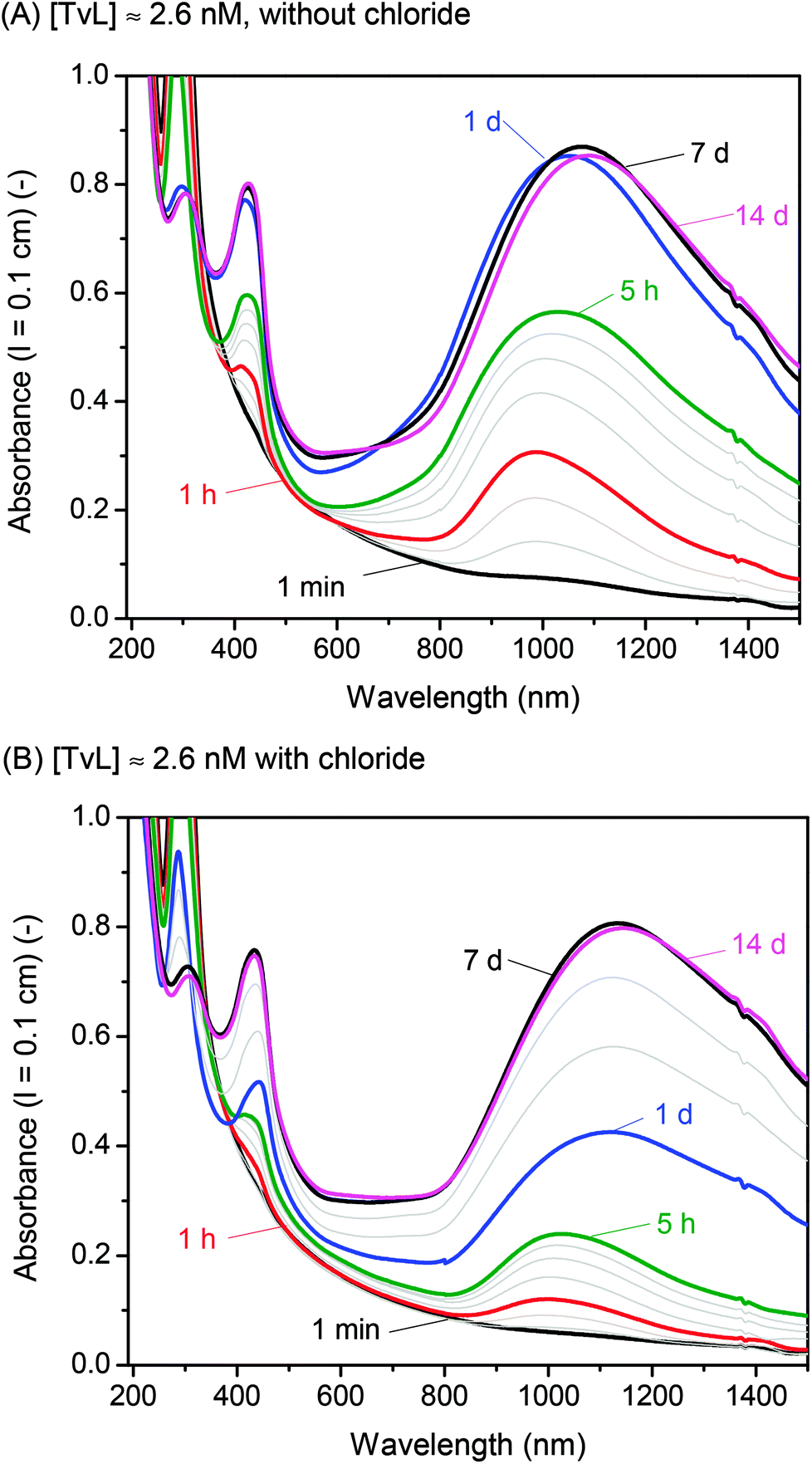 | ||
| Fig. 2 Effect of chloride ions. Comparison of the UV/vis/NIR absorption spectra of two reaction mixtures recorded during the reaction with [TvL] ≈ 2.6 nM, [PADPA]0 = 1.0 mM, [AOT] = 1.5 mM, [H2PO4−] + [H3PO4] = 0.1 M, pH = 3.5, and T ≈ 25 °C. (A) Use of the chloride-free “pH = 3.5 solution” (new optimal conditions). The grey lines are the spectra recorded for t = 10 min, 30 min, 2 h, 3 h, and 4 h. (B) Use of the “pH = 3.5 solution” containing chloride ions (≈2.4 mM). The grey lines are the spectra recorded for t = 10 min, 30 min, 2 h, 3 h, 4 h, 2 d, 3 d, and 4 d. | ||
If a “pH = 3.5 solution” is used which was prepared from NaH2PO4 and small amounts of HCl, the reaction with 2.6 nM TvL is much slower and results in a stable UV/vis/NIR spectrum only after t = 7 d, with λmax ≈ 1100, 433, and 304 nm, see Fig. 2B. A direct comparison of the two kinetic measurements is shown in Fig. S-1.† With 32 nM TvL, the reaction with the “pH = 3.5 solution” prepared from NaH2PO4 and HCl is very similar to the one measured previously for the same conditions,20 with λmax = 1014, 421, and 304 nm after t = 1 day, while for [TvL] = 32 nM and the chloride-free solution, the reaction was much faster than with [TvL] = 2.6 nM, see Fig. S-2.† For both conditions with 32 nM TvL, continuous spectral changes occur well beyond t = 1 d, as shown in Fig. S-2:† between t = 1 d and t = 14 d, the absorbance at λ = 1000 nm (A1000) decreases and A>1100 increases.
The activity and stability of TvL in the two “pH = 3.5 solutions” were measured at T ≈ 25 °C with ABTS2− as substrate for [TvL] = 2.6 nM and [TvL] = 32 nM, see Fig. S-3.† For the same given TvL concentration, the rate of ABTS2− oxidation in the presence of the chloride ions is about half the one in the chloride-free solution; in both solutions, the enzyme activity decreased with time within one day to about 50% or less of the initial value; dissolved in pure water, TvL is much more stable than in the two “pH = 3.5 solutions” (Fig. S-3†).
Overall, the data confirm at least qualitatively, the inhibitory effect of chloride ions on the activity of TvL.40–43 This enzyme inhibition by chloride ions explains why with a chloride-free “pH = 3.5 solution” much less TvL (about one tenth) can be used to obtain the same spectroscopic product characteristics as in the case of the chloride-containing “pH = 3.5 solution” (Fig. 2).
3.2 Effect of increasing [TvL] from 2.6 nM to 320 nM
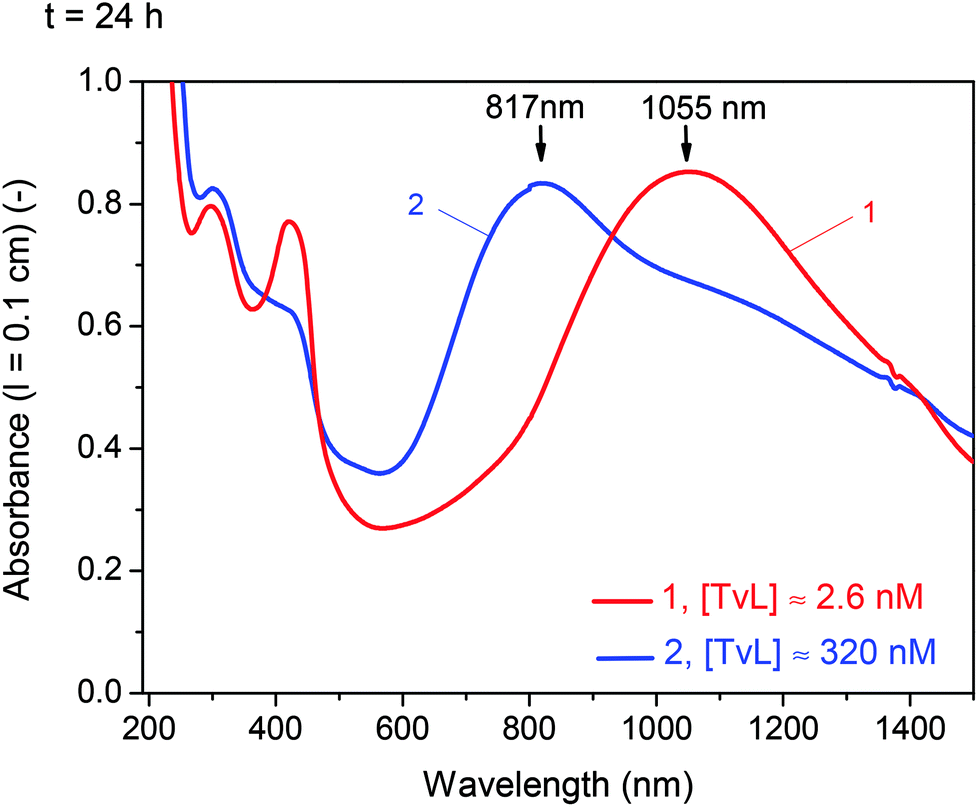 | ||
| Fig. 3 Effect of laccase concentration. Comparison of the UV/vis/NIR absorption spectra of two reaction mixtures recorded after t = 24 h for [PADPA]0 = 1.0 mM, [AOT] = 1.5 mM, [H2PO4−] + [H3PO4] = 0.1 M (chloride ion-free), pH = 3.5, T ≈ 25 °C, and [TvL] ≈ 2.6 nM (1, new optimal conditions) or [TvL] ≈ 320 nM (2). | ||
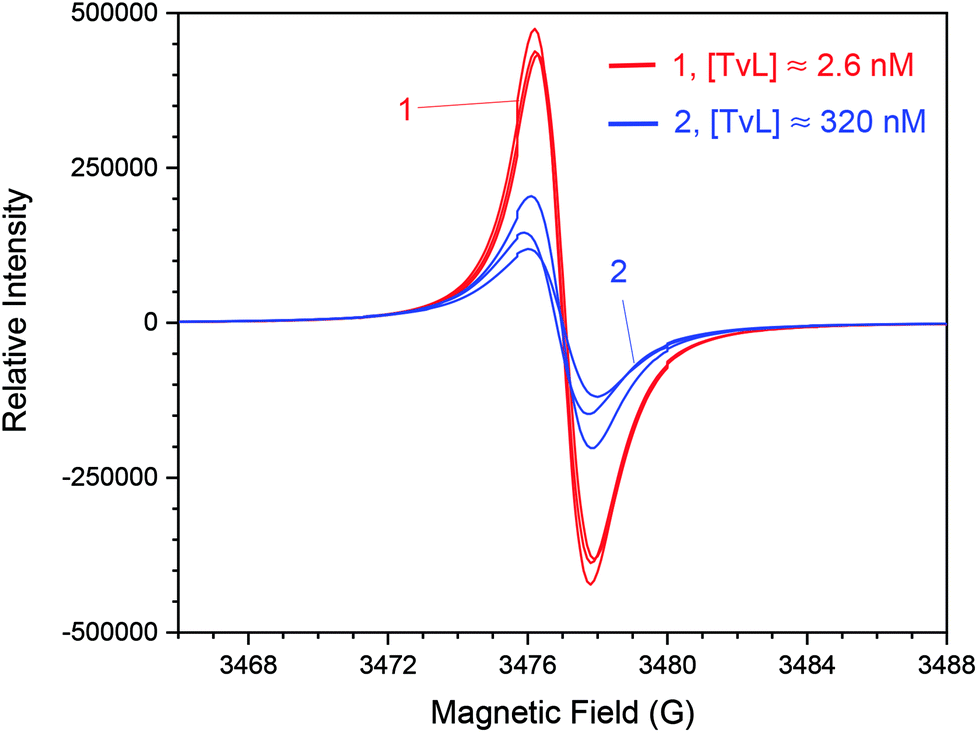 | ||
| Fig. 4 Effect of laccase concentration. Comparison of the EPR spectra of the two reaction mixtures recorded after t = 24 h for [PADPA]0 = 1.0 mM, [AOT] = 1.5 mM, [H2PO4−] + [H3PO4] = 0.1 M (chloride ion-free), pH = 3.5, T ≈ 25 °C, and [TvL] ≈ 2.6 nM (red, new optimal conditions) or [TvL] ≈ 320 nM (blue); ν = 9.766 GHz, g = 2.0068 ± 0.0002 (2 s confidential interval) for both products. The spectra shown represent three reaction runs for each TvL concentration, under otherwise identical conditions, and indicate good reproducibility of radical yield for [TvL] ≈ 2.6 nM. With [TvL] ≈ 320 nM, the radical yields are less than half of the value obtained with [TvL] ≈ 2.6 nM and vary almost by a factor of 2. | ||
For [TvL] ≈ 2.6 nM a stable radical content is achieved after t = 24 h, for [TvL] ≈ 320 nM, conversion processes last much longer (t ≈ 4 d) (Fig. S-7†) with a radical yield lower than obtained with [TvL] ≈ 2.6 nM. This observation correlates with the UV/vis/NIR spectra which do not change significantly after t = 24 for [TvL] ≈ 2.6 nM (Fig. 2A), but still change for [TvL] ≈ 320 nM (Fig. S-4†).
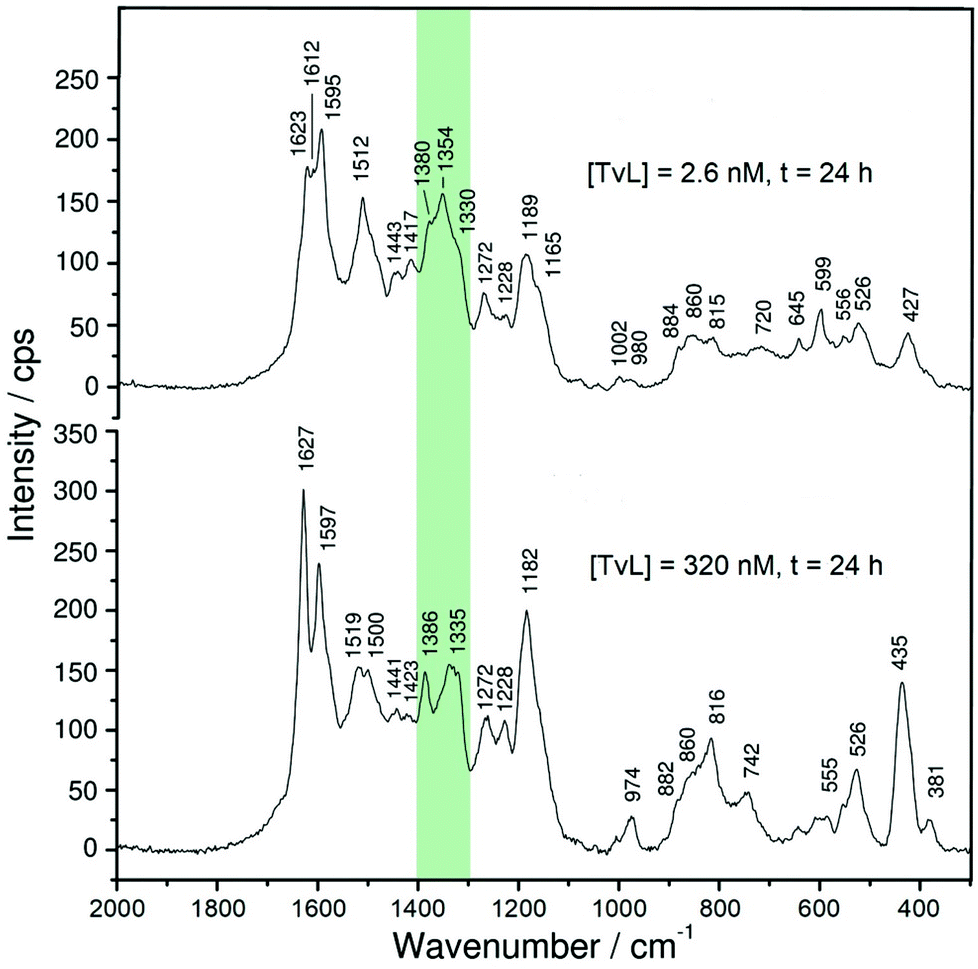 | ||
| Fig. 5 Effect of laccase concentration. Comparison of the Raman spectra of the two reaction mixtures recorded after t = 24 h for [PADPA]0 = 1.0 mM, [AOT] = 1.5 mM, [H2PO4−] + [H3PO4] = 0.1 M (chloride ion-free), pH = 3.5, T ≈ 25 °C, and [TvL] ≈ 2.6 nM (top, new optimal conditions) or [TvL] ≈ 320 nM (bottom). Excitation wavelength: 633 nm. The characteristic polaron bands are highlighted in green. | ||
Although there are great similarities in the Raman spectra of the two reaction mixtures, there are also clear differences. One important difference is related to intensity changes of the ‘polaron band’ during the reaction, attributed to C∼N˙+ stretching vibrations of polaronic (semiquinonoid) structures, ν(C∼N˙+)SQ. These vibrations are observed in the region of ν ≈ 1320–1380 cm−1. Two peaks due to ν(C∼N˙+)SQ vibration bands are present at ν ≈ 1330 cm−1 and ν ≈ 1350 cm−1, for both TvL concentrations. The symbol ′′∼′′ denotes a bond intermediate between a single and a double bond. A third peak is present at ν ≈ 1370–1380 cm−1, whose origin is still debatable, but can be attributed to ν(C∼N˙+)SQ vibrations in localized polaron sites and/or to ν(C∼N+) vibrations in N-phenylphenazine units.43
For [TvL] ≈ 320 nM, the relative intensity of the ‘polaron band’ in the spectrum recorded at t = 24 h (with a peak at ν ≈ 1335 cm−1 and a shoulder at ν ≈ 1355 cm−1) is significantly reduced with respect to the relative intensity of the ‘polaron band’ in the spectrum recorded at t = 1 h (with a peak at ν = 1354 cm−1 and a shoulder at ν = 1332 cm−1) (Fig. S-8 and S-9†). These changes are in accordance with the changes observed in the corresponding UV/vis/NIR spectra for [TvL] ≈ 320 nM (Fig. 3 and S-4†): the absorption band indicative for polarons appeared at λ ≈ 900 nm for t = 1 h, while for t = 24 h a blue-shift to λ = 817 nm was observed, accompanied with a decrease in band intensity.
A different behaviour was observed in the case of [TvL] ≈ 2.6 nM, for which the relative intensity of the Raman band characteristic for polarons, with maxima at ν ≈ 1354 cm−1 and 1330 cm−1, increased with increasing reaction runtime from t = 1 h to t = 24 h, and the peak at ν = 1354 cm−1 became particularly strengthened for t = 24 h (Fig. S-8 and S-9†). This is again consistent with the change observed in the corresponding UV/vis/NIR spectra (Fig. 2A): the intensity of the ‘polaron band’ at λ ≈ 1100 nm for t = 24 h increased compared to its intensity for t = 1 h.
A comparison of the Raman and UV/vis/NIR spectra recorded at t = 24 h for [TvL] ≈ 2.6 nM and [TvL] ≈ 320 nM indicates a higher electrical conductivity of the products obtained with [TvL] ≈ 2.6 nM as compared to the products obtained with [TvL] ≈ 320 nM; this conclusion is drawn on the basis of the following features: (i) in the two Raman spectra recorded at t = 24 h, the characteristic ‘polaron’ band is stronger for the mixture with [TvL] ≈ 2.6 nM (ν ≈ 1355 cm−1 with a shoulder at ν ≈ 1330 cm−1) than the corresponding band for the mixture with [TvL] ≈ 320 nM (ν ≈ 1335 cm−1 with a shoulder at ν ≈ 1355 cm−1), see Fig. 5 and S-9;† (ii) the ‘polaron band’ in the UV/vis/NIR spectrum of the mixture with [TvL] ≈ 2.6 nM has a maximum at λ ≈ 1055 nm (Fig. 3), as expected for highly conducting PANI-ES-like materials,36–39 while the ‘polaron band’ in the spectrum of the mixture with [TvL] ≈ 320 nM is observed at λ ≈ 817 nm indicating lower electron delocalization and conductivity.
There are also other interesting changes in the features in the Raman spectra recorded during the reaction for the two TvL concentrations (Fig. S-8 and S-9†). For example, besides the bands characteristic for PANI-ES at ν ≈ 1625 cm−1 (C∼C stretching vibrations of benzenoid (B) rings, ν(C∼C)B)21,23,44 and ν = 1595 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C∼C stretching vibrations of quinonoid (Q) and semiquinonoid (SQ) rings, ν(CC)Q and ν(C∼C)SQ)21,23,44 that are observed during the reaction for both TvL concentrations, the spectra of the reaction mixture with [TvL] = 2.6 nM have an additional distinct peak at ν ≈ 1610 cm−1 (Fig. S-8A†), also attributable to ν(C∼C)B vibrations, which is not seen in the spectra of the reaction mixture with [TvL] ≈ 320 nM (Fig. S-8B†). The reason for this may be the strong band intensity at ν ≈ 1625 cm−1 for the reaction with [TvL] ≈ 320 nM.
C and C∼C stretching vibrations of quinonoid (Q) and semiquinonoid (SQ) rings, ν(CC)Q and ν(C∼C)SQ)21,23,44 that are observed during the reaction for both TvL concentrations, the spectra of the reaction mixture with [TvL] = 2.6 nM have an additional distinct peak at ν ≈ 1610 cm−1 (Fig. S-8A†), also attributable to ν(C∼C)B vibrations, which is not seen in the spectra of the reaction mixture with [TvL] ≈ 320 nM (Fig. S-8B†). The reason for this may be the strong band intensity at ν ≈ 1625 cm−1 for the reaction with [TvL] ≈ 320 nM.
Spectral differences between systems with [TvL] ≈ 2.6 nM and 320 nM also exist regarding the bands which are not typical for PANI-ES-like structures. For both TvL concentrations such bands are seen at ν ≈ 1410 cm−1 (attributed to phenazine-type units), ν = 1450 cm−1 (attributable to ring CC stretching vibrations, possibly in short chains/short branches, or in substituted phenazine- and N-phenylphenazine-type oligomers), and ν = 1570 cm−1 (attributable to phenazine-, phenoxazine, or N-phenylphenazine-type structures).23 They all indicate the formation of branched and substituted phenazine unit-containing oligomers. The intensity ratio of the ‘polaron band’ (ν ≈ 1355 cm−1 or 1330 cm−1) to the bands at ν = 1410 cm−1 and 1450 cm−1 is higher in the final spectrum (at t = 24 h) for the reaction mixture with [TvL] = 2.6 nM than in the corresponding spectrum for [TvL] = 320 nM (Fig. 5 and S-9†). This feature indicates the presence of more regular structures and higher conductivity of the products obtained after t = 24 h with [TvL] ≈ 2.6 nM as compared to [TvL] ≈ 320 nM. It is interesting to note that in the spectra recorded at t = 1 h (and before) the relative intensity of the bands at ν ≈ 1410 cm−1 and 1450 cm−1 (e.g. in relation to the intensity of the ‘polaron band’) is higher for the system with [TvL] ≈ 2.6 nM than for the system with [TvL] ≈ 320 nM, but the situation becomes reverse at t = 24 h, when the relative intensity of the bands at ν ≈ 1410 cm−1 and 1450 cm−1 (in relation to the ‘polaron band’) is lower for the system with [TvL] = 2.6 nM (Fig. 5 and S-9†). This feature, together with the previously described Raman and UV/vis/NIR characteristics, suggests that a longer reaction runtime (t ≈ 24 h) favors the formation of delocalized polarons in the case of the mixture with [TvL] ≈ 2.6 nM, while this is not the case for the mixture with [TvL] ≈ 320 nM. The formation of overoxidised structural units containing CO group in the products formed with high TvL concentration is indicated by the shoulder at ≈1660 cm−1, attributed to the CO stretching (Fig. 5)45 Such a shoulder is not observed in the spectrum for the case of low TvL concentration (Fig. 5).
Overall, all the mentioned Raman spectral features are in good agreement with the UV/vis/NIR and EPR spectra and indicate a strong influence of the TvL concentration on the reaction mechanism and on the molecular structure and electrical properties of the final oligomeric products.
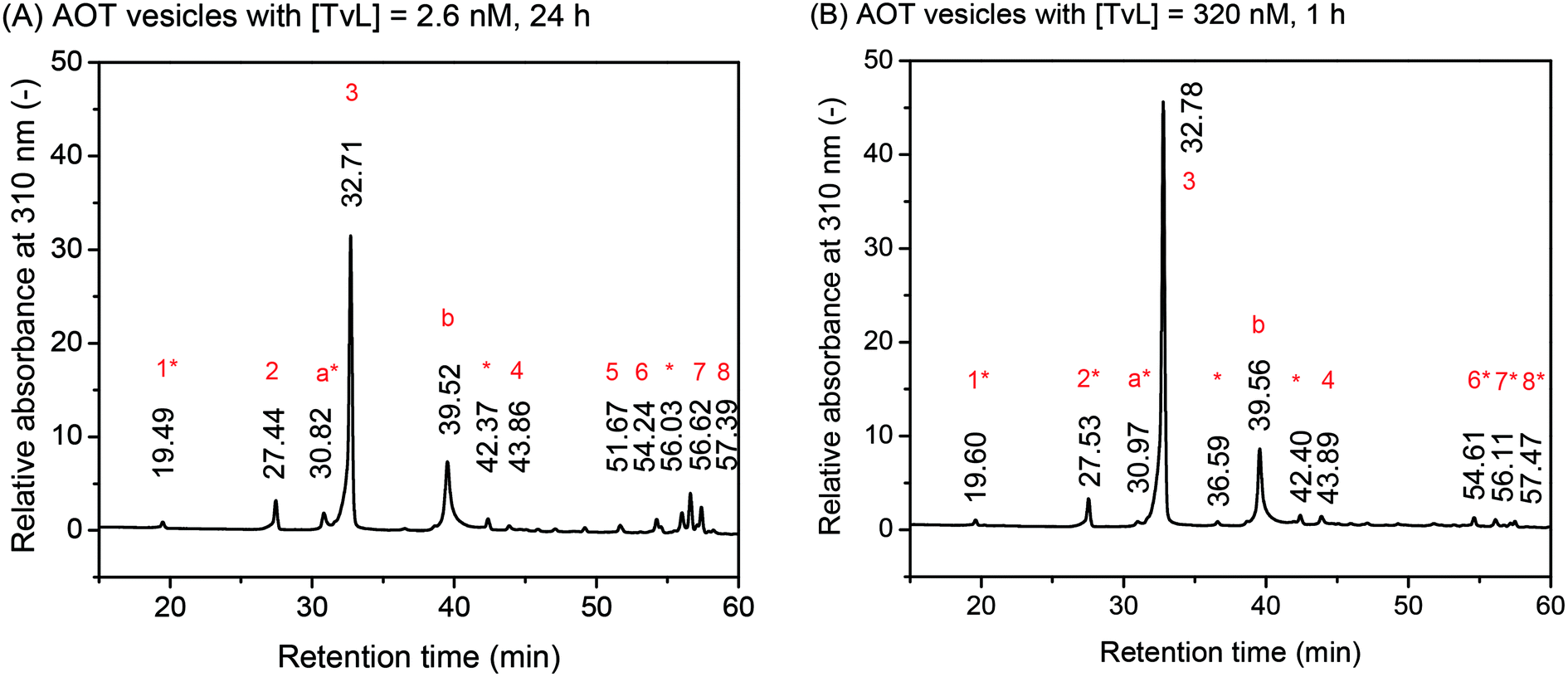 | ||
| Fig. 6 Effect of laccase concentration. Comparison of the chromatograms for the HPLC-DAD analysis of the deprotonated and reduced reaction products, as obtained from [PADPA]0 = 1.0 mM, [AOT] = 1.5 mM, [H2PO4−] + [H3PO4] = 0.1 M (chloride ion-free), pH = 3.5, T ≈ 25 °C, and [TvL] ≈ 2.6 nM after t = 24 h (A, new optimal conditions) or [TvL] ≈ 320 nM after t = 1 h (B). The relative absorbance at λ = 310 nm is plotted against the retention time. For the assignment of the different peaks, see Tables 1 and 2. | ||
The chromatograms shown in Fig. 6 are very similar with a main peak at retention time rt ≈ 32.7 min, a second most intense peak at rt ≈ 39.5 min, and small peaks at rt ≈ 19.5, 27.4, 30.8, 42.3, and 43.8 min. Furthermore, there is a group of peaks at rt ≈ 54–57 min. This peak pattern is very similar to the one obtained previously for the same reaction, but run with [TvL] = 62 nM, see Fig. 2A in Luginbühl et al. (2016).22 The most significant differences between the chromatogram for the reaction with [TvL] ≈ 2.6 nM, t = 24 h (Fig. 6A, new optimal conditions) and the reaction with [TvL] = 320 nM, t = 1 h (Fig. 6B) are (i) the lower intensity of the peak at rt ≈ 32.7 min, and (ii) the higher intensity for the group of peaks at rt ≈ 54–57 min for the reaction run with 2.6 nM (t = 24 h), as compared to the reaction with 320 nM TvL (t = 1 h).
The peak assignments listed in Tables 1 and 2 are based on HPLC-MS measurements and our previous analysis.22 The UV/vis spectra of the different chromatographic peaks are shown in Fig. S-12† (for the peaks of the chromatogram in Fig. 6A) and Fig. S-13† (for the peaks of the chromatogram in Fig. 6B). With these assignments, the key findings from this comparative HPLC analysis can be summarized as follows: (i) in both cases, the linear PADPA dimer, i.e., the N–C-para coupled tetraaniline is the main product (rt ≈ 32.7 min); (ii) in both cases, hexaaniline (rt ≈ 39.5 min) and products with phenazine units (rt ≈ 54–57 min) also form; (iii) for the reaction with [TvL] ≈ 2.6 nM (t = 24 h), the amount of tetraaniline (rt ≈ 32.7 min) is lower and the amount of products with phenazine units (rt ≈ 54–57 min) is higher than in the case of the reaction with [TvL] ≈ 320 nM (t = 1 h). Products with phenazine-units seem to form at later stages of the reaction, see Fig. S-10.† This is in qualitative agreement with the increase in the absorbance around λ ≈ 500 nm with reaction runtime, see Fig. 2A ([TvL] ≈ 2.6 nM) and compare the spectra recorded for t = 1 d and t = 7 d, or see Fig. S-4† ([TvL] ≈ 320 nM) and compare the spectra recorded for t = 1 h and t = 1 d.
| HPLC peak number | HPLC-DAD, rt (min) | Spectrum ESI Fig. S-12 | Absorption maximum, λmax (nm) | Measured molecular mass (HPLC-MS)a (Da) | Calculated molecular mass (Da) | Molecular formula | Structure,b see ESI and Luginbühl et al. (2016)22 |
|---|---|---|---|---|---|---|---|
| a The measured molecular mass is the molar mass corresponding to the most intense m/z signal of the MS spectrum of the HPLC peak number (separate HPLC-MS analysis).b The superscripts I and II are used to distinguish between isomers,22 (PADPA)1.5 = linear aniline trimer; (PADPA)I2 = linear aniline tetramer; (PADPA)2.5 = aniline pentamer; (PADPA)I3 = aniline hexamer (with a phenazine unit); (PADPA)4 = aniline octamer (with a phenazine unit); (PADPA)II2–NH2 is an aniline tetramer in which one amino group is missing, i.e., replaced by a hydrogen atom; the presence of oxidised forms indicates that the reduction with hydrazine was not complete. | |||||||
| 1 | 19.49 | A | 286 | — | — | — | (1) PADPA |
| 2 | 27.44 | B | 300 | 274.1338 | 274.1339 | C18H16N3+ | (2) (PADPA)1.5 |
| a* | 30.82 | C | 298, 560 | — | — | — | ? |
| 3 | 32.71 | D | 312 | 365.1762 | 365.1761 | C24H21N4+ | (3) (PADPA)I2 oxidised |
| 367.1911 | 367.1917 | C24H23N4+ | (4) (PADPA)I2 | ||||
| b | 39.52 | E | 318, 590 | 548.2655 | 548.2677 | C36H31N62+ | (5) (PADPA)I3 oxidised |
| * | 42.37 | F | 312 | — | — | — | ? |
| 4 | 43.86 | G | 320, 610 | 352.1803 | 352.1808 | C24H22N3+ | (6) (PADPA)II2–NH2 |
| 5 | 51.67 | H | 298, 322, 438 | 456.2175 | 456.2183 | C30H26N5+ | (7) (PADPA)2.5 |
| 6 | 54.24 | I | 290, 352, 490 | 547.2600 | 547.2605 | C36H31N6+ | (8) (PADPA)II3 |
| * | 56.03 | J | 302, 510 | — | — | — | ? |
| 7 | 56.62 | K | 302, 510 | 729.3438 | 729.3449 | C48H41N8+ | (9) (PADPA)4 |
| 8 | 57.39 | L | 314, 528 | 729.344 | 729.3449 | C48H41N8+ | |
| HPLC peak number | HPLC-DAD, rt (min) | Spectrum ESI Fig. S-13 | Absorption maximum, λmax (nm) | Measured molecular mass (HPLC-MS)a (Da) | Calculated molecular mass (Da) | Molecular formula | Structure,b see ESI and Luginbühl et al. (2016)22 |
|---|---|---|---|---|---|---|---|
| a See Table 1.b See Table 1. | |||||||
| 1* | 19.60 | AA | 286 | — | — | — | (1) PADPA |
| 2* | 27.53 | BB | 308 | — | — | — | ? |
| a* | 30.97 | — | — | — | — | — | ? |
| 3 | 32.78 | CC | 312 | 365.1762 | 365.1761 | C24H21N4+ | (3) (PADPA)I2 oxidised |
| 367.1917 | 367.1917 | C24H23N4+ | (4) (PADPA)I2 | ||||
| * | 36.59 | DD | 312 | — | — | — | ? |
| b | 39.56 | EE | 318 | 548.2669 | 548.2677 | C36H31N62+ | (5) (PADPA)I3 oxidised |
| * | 42.40 | FF | 314 | — | — | — | ? |
| 4 | 43.89 | GG | 316, 600 | 352.1809 | 352.1808 | C24H22N3+ | (6) (PADPA)II2–NH2 |
| 6* | 54.61 | HH | 314, 484 | — | — | — | (8) (PADPA)II3 |
| 7* | 56.11 | II | 300, 498 | — | — | — | ? |
| 8* | 57.47 | JJ | 316, 528 | — | — | — | ? |
3.3 Effect of decreasing the reaction temperature from T ≈ 25 °C to T = 5 °C
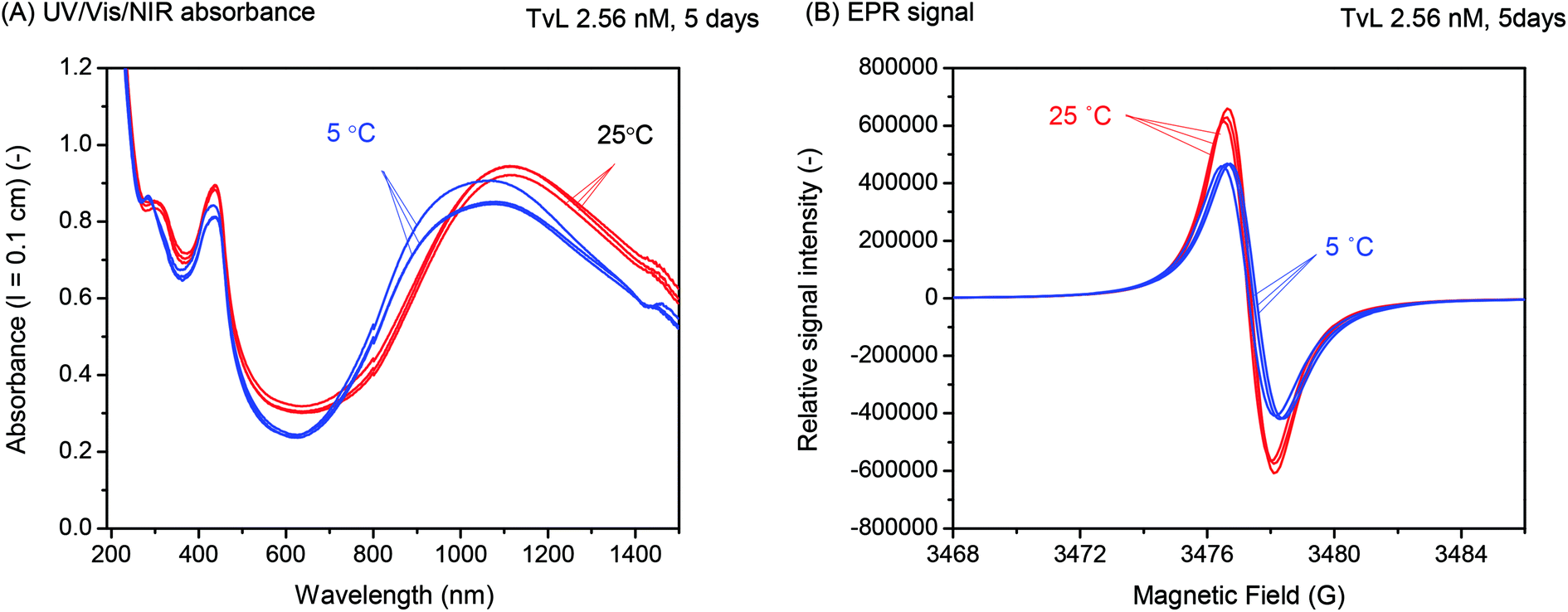 | ||
| Fig. 7 Effect of temperature. UV/vis/NIR (A) and EPR spectra (B) of reaction mixtures, as obtained for reactions carried out at either T ≈ 25 °C or T = 5 °C. [PADPA]0 = 1.0 mM, [AOT] = 1.5 mM, [H2PO4−] + [H3PO4] = 0.1 M (chloride ion-free), pH = 3.5, [TvL] ≈ 2.6 nM and t = 5 d. | ||
Overall, there was no indication of a significant improvement of the products distribution by running the reaction at T = 5 °C instead of room temperature. This finding is in contrast to what we had observed previously for the oxidation and polymerisation of aniline in the presence of AOT vesicles with TvL/O2 at pH = 3.5 (ref. 18) or HRPC/H2O2 at pH = 4.3,16 where a considerable increase in the ratio of A730 to A500 (for the reaction with TvL/O2)18 or A1000 to A500 (for the reaction with HRPC/H2O2)16 resulted when the reaction was carried out at T = 7 or 8 °C as compared to T = 25 °C. This indicated a lower extent of branching at the lower reaction temperature, i.e., less ortho-coupling.18 Furthermore, it is known that in the case of the chemical oxidation and polymerisation of aniline to PANI-ES with ammonium peroxydisulfate in 0.2 or 1.2 M aqueous HCl, a decrease in the reaction temperature is advantageous since it leads to products with higher average molar masses.38,47,48
For the complex reaction mixtures which we are investigating, a change in reaction temperature may have different effects, and it is difficult to predict whether and how they might influence each other. A decrease in temperature leads (i) to a decrease in the TvL-catalysed rate of PADPA oxidation, as observed for other monomers,49 (ii) possibly to an increase in TvL stability, (iii) to an increase in O2 solubility;50 (iv) to a change in the pH of the “pH = 3.5 solution” (prepared at pH ≈ 25 °C) due to changes in the pKa values of H3PO4; and (v) a decrease in the fluidity of the AOT membrane. With respect to (iv), we measured the pH value of the “pH = 3.5 solution” at T = 5 °C and did not find a substantial deviation from the value at 25 °C. This is in agreement with the small temperature dependency of the two relevant pKa-values of H3PO4: dpKa1/dT = +0.0044/°C (pKa1 = 2.15) and dpKa2/dT = −0.0028/°C (pKa2 = 7.21).51 With respect to (v), the temperature dependence of the AOT bilayer fluidity was determined by MD simulations as outlined in the following section.
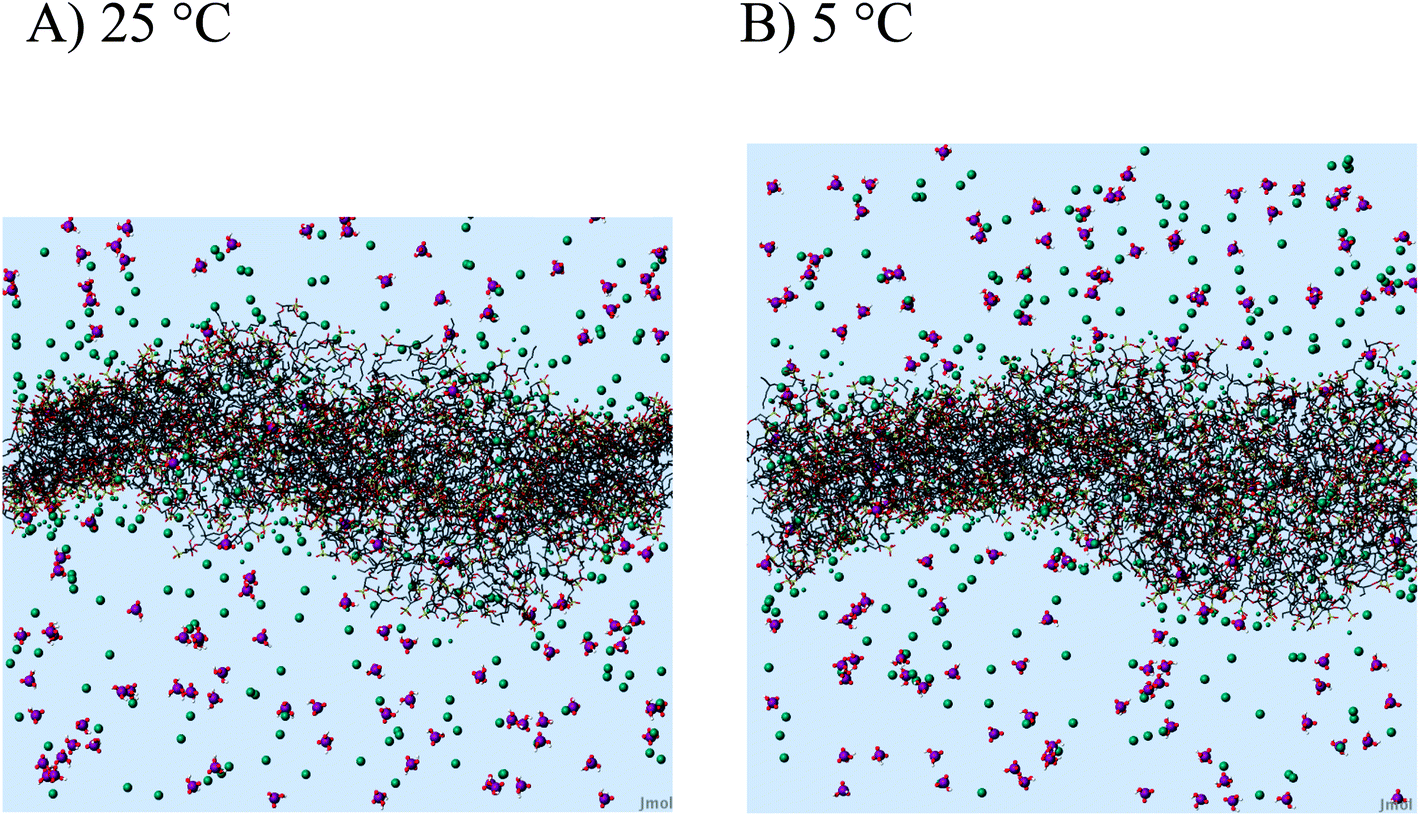 | ||
| Fig. 8 Effect of temperature. Side view of the “bilayer” membrane (wireframe) in water (not shown) including H2PO4 ions, H3PO4 species (purple/red) and sodium counter-ions (blue-green, smaller when close to the surface). The overall bumpy and noisy surface at T = 25 °C does not fully calm down when cooled to T = 5 °C. We still observe undulations, pores and a tendency to micellar bumps. | ||
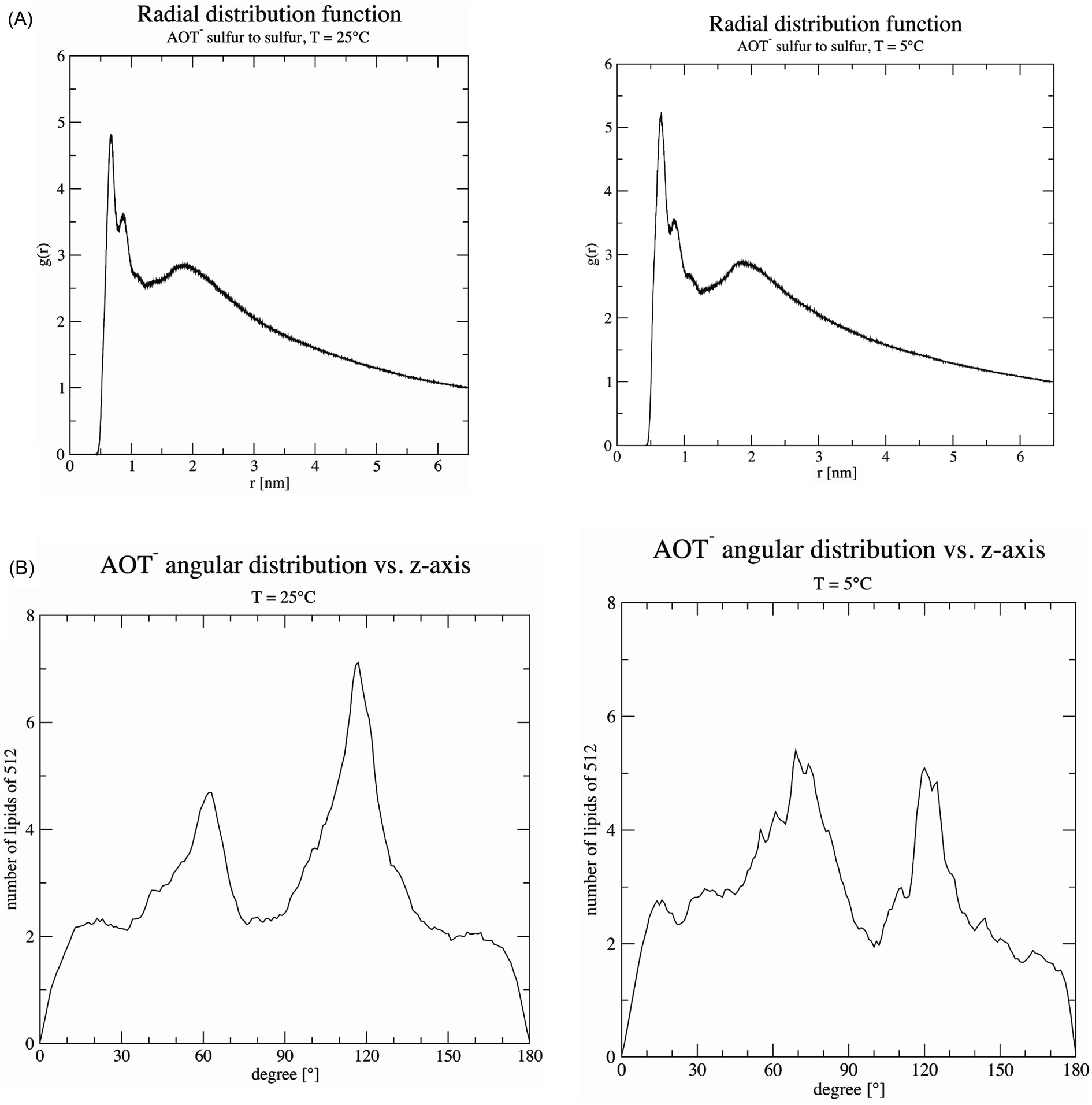 | ||
| Fig. 9 Effect of temperature. (A) The radial distribution function of sulfur atoms reveals the average distances between the AOT head groups. For AOT membranes at T = 25 °C, the distances are similar with a value of 0.664 nm compared to 0.662 nm at T = 5 °C. The peak at T = 5 °C (g(r) > 5) is higher than at T = 25 °C (g(r) < 5), indicating that at T = 5 °C more than 5 AOT molecules surround each AOT at the similar distance (average g(r) = 5.20). At T = 25 °C temperature, the peak height is “softer”; and there are below 5 surrounding AOT in the vicinity of each AOT (average g(r) = 4.81). (B) The preferred orientation of AOT compared to the z-axis, for both temperatures, is more pronounced at 60 and 120°, than around 90° (flat) against the z-axis. The drop at exactly the z-axis must be ignored, its origin are numerical instabilities of the calculation due to divisions close to zero. At T = 5 °C, it seems that the angular distribution (peak height) has become a bit more equal than at T = 25 °C. | ||
4 Concluding remarks
Using peroxidases or laccases for the polymerisation of aniline or PADPA in the presence of sulfonated polystyrene,7–12 micelles from sodium dodecylbenzenesulfonate,9,19,25 AOT vesicles,16–18,20–23 or any other type of additive that acts as “template”, is considered as an environmentally friendly approach for obtaining PANI-ES or PANI-ES-like products. Similarly, the use of glucose oxidase and D-glucose for initiating the polymerisation of aniline in a template-free system is another environmentally friendly approach for the preparation of PANI.52–54 In this latter case, D-glucose is oxidised to yield H2O2, which in turn oxidises aniline. One drawback of using enzymes, however, may be the large amounts that are required for achieving high conversions. This was shown to be the case for aniline as monomer, AOT vesicles as “templates”, and either HRPC/H2O2 (at pH = 4.3),17 soybean peroxidase/H2O2 (at pH = 4.3)55 or TvL/O2 (at pH = 3.5)18 as enzyme/oxidant systems. With PADPA instead of aniline – and again AOT vesicles as “templates” – the situation is very different. Much lower amounts of enzyme are required, as shown for TvL/O2 (ref. 20–22) as well as for HRPC/H2O2.23 In both cases, however, the majority of the products obtained are not true polymers but only oligomers, mainly tetraaniline, obtained as ES in its polaron state. Furthermore, with HRPC/H2O2 and PADPA, the formation of substantial amounts of products containing undesired phenazine-type units could not be avoided.23 Recently, it was shown that this phenazine-type units formation can be reduced significantly by running the reaction instead of PADPA alone with a mixture of PADPA and aniline.46By using TvL/O2 and PADPA in the presence of AOT vesicles, the amount of TvL required to reach high conversion of PADPA into the desired oligomeric PANI-ES-like products, one apparently minor detail is important to be taken in account. It is the way how the aqueous “pH = 3.5 solution” is prepared from a solution of NaH2PO4. If the pH value is adjusted with H3PO4 instead of HCl, the TvL concentration can be reduced by one order of magnitude (from ≈32 or 64 nM in the presence of 2.4 mM chloride ions, to 2.6 nM without chloride). This resulted in our new optimal conditions for this reaction: 1.0 mM PADPA, 1.5 mM AOT, 2.6 nM TvL, aqueous “pH = 3.5 solution” (0.1 M NaH2PO4/H3PO4), T ≈ 25 °C, t = 24 h, see Section 3.1. This reaction is very reproducible.
At first sight one may think that increasing the TvL concentration – but keeping all other conditions the same – would simply result in an increase of reaction rate, as one expects for a “standard” enzyme-catalysed reaction. The situation here is, however, different due to non-enzymatic follow-up reactions that lead to the formation of the final products. Furthermore, the enzyme is inactivated during the reaction, which means that the amount of active enzyme present at one time point during the reaction depends on the initially applied enzyme concentration. As a result, if the TvL concentration is much higher than the optimal concentration (≈320 nM instead of 2.6 nM), the obtained products are overoxidised. This was studied in detail by using in situ UV/vis/NIR (Fig. 3), EPR (Fig. 4) and Raman spectroscopy measurements (Fig. 5), as well as with ex situ HPLC-DAD and HPLC-MS analyses (Fig. 6).
Decreasing the reaction temperature from T = 25 °C to 5 °C did not have a distinct effect on the products distribution of the PADPA reaction (Fig. 7). This differs from the previous studies with HRPC/H2O2 and aniline16 or TvL/O2 and aniline,18 both with AOT vesicles as templates. This indicates that a possible variation of the AOT bilayer fluidity apparently does not affect the reaction product composition within the chosen reaction temperature range. MD simulations showed, that there is no substantial change in AOT membrane fluidity in the range of T = 5 °C and 25 °C (Fig. 9). Therefore, for our future investigations of this system with TvL/O2 and PADPA we will keep the reaction temperature at T ≈ 25 °C. The aim of the next step is to explore whether the type of “template” used for the same reaction matters. More precisely, we will compare the effect of vesicles, micelles and polyelectrolytes, all with sulfonate groups, on one and the same reaction, the TvL/O2-catalysed oxidation of PADPA. The results obtained in this study will be reported elsewhere.
With our detailed investigations of a complex reaction we like to draw attention to experimental details which are important to be considered and to be reported to ensure experimenting in a reproducible way.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the Swiss National Science Foundation projects 200020_150254, IZ73Z0_152457 (P. W., S. S.-L., G. Ć.-M., D. B.-B., and A. J. L.) and IZK0Z2_170360 (A. K.), the Ministry of Education, Science and Technological Development of Serbia project OI172043 (G. Ć.-M., D. B.-B. and A. J. L) and through the generous fellowships given to K. K (National Institute of Technology, Faculty Research Abroad Program, Japan) and T. F. (TOBITATE! Young ambassador program from The Japan Public-Private Partnership Student Study Abroad Program) for their stay at ETH. The authors like to thank Louis Bertschi and Daniel Wirz from the Mass Spectrometry Service Facility of the Department of Chemistry and Applied Biosciences of the ETH for the HPLC-MS measurements, and the two Materials Science bachelor students Tizian Keller and Pascal Studer for their preliminary investigations of the temperature dependence of the reaction.Notes and references
- A textbook example is the reaction of but-3-en-2-one with NaCN/HCN to yield either cyanohydrin (direct addition of the nucleophilic cyanide to the carbonyl carbon atom) if the reaction is peformed at 5–10 °C, or 4-oxopentanenitrile (Michael-type addition of the cyanide) if the reaction is carried out at 80 °C: J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, 2nd edn, 2012, p. 504 Search PubMed.
- Since molecular oxygen has an influence on the course of many chemical polymerisation reactions, it usually must be eliminated carefully in order to obtain desired polymeric products: D. Braun, H. Cherdron, M. Rehan, H. Ritter and B. Voit, Polymer Synthesis: Theory and Practice, Springer-Verlag, Berlin Heidelberg, 4th edn, 2005, p. 63 Search PubMed.
- H. McGee, On Food and Cooking. The Science and Lore of the Kitchen, Unwin Hyman Ltd., London, 1984 Search PubMed.
- M. Baker and D. Penny, Nature, 2016, 533, 452–454 CrossRef CAS PubMed.
- R. G. Bergman and R. L. Danheiser, Angew. Chem., Int. Ed., 2016, 55, 12548–12549 (Angew. Chem., 2016, 128, 12736–12737) CrossRef CAS PubMed.
- G. Ćirić-Marjanović, M. Milojević-Rakić, A. Janoševic Ležaić, S. Luginbühl and P. Walde, Chem. Pap., 2017, 71, 199–242 CrossRef PubMed.
- L. A. Samuelson, A. Anagnostopoulos, K. S. Alva, J. Kumar and S. K. Tripathy, Macromolecules, 1998, 31, 4376–4378 CrossRef CAS.
- W. Liu, J. Kumar, S. Tripathy, K. J. Senecal and L. Samuelson, J. Am. Chem. Soc., 1999, 121, 71–78 CrossRef CAS.
- W. Liu, A. L. Cholli, R. Nagarajan, J. Kumar, S. Tripathy, F. F. Bruno and L. Samuelson, J. Am. Chem. Soc., 1999, 121, 11345–11355 CrossRef CAS.
- I. Y. Sakharov, A. C. Vorobiev and J. J. Castillo Leon, Enzyme Microb. Technol., 2003, 33, 661–667 CrossRef CAS.
- A. V. Karamyshev, S. V. Shleev, O. V. Koroleva, A. I. Yaropolov and I. Y. Sakharov, Enzyme Microb. Technol., 2003, 33, 556–564 CrossRef CAS.
- A. V. Caramyshev, E. G. Evtushenko, V. F. Ivanov, A. Ros Barceló, M. G. Roig, V. L. Shnyrov, R. B. van Huystee, I. N. Kurochkin, A. K. Vorobiev and I. Y. Sakharov, Biomacromolecules, 2005, 6, 1360–1366 CrossRef CAS PubMed.
- V. Rumbau, J. A. Pomposo, J. A. Alduncin, H. Grande, D. Mecerreyes and E. Ochoteco, Enzyme Microb. Technol., 2007, 40, 1412–1421 CrossRef CAS.
- A. V. Caramyshev, V. M. Lobachov, D. V. Selivanov, E. V. Sheval, A. K. Vorobiev, O. N. Katasova, V. Y. Polyakov, A. A. Makarov and I. Y. Sakharov, Biomacromolecules, 2007, 8, 2549–2555 CrossRef CAS PubMed.
- Z. Guo, H. Rüegger, R. Kissner, T. Ishikawa, M. Willeke and P. Walde, Langmuir, 2009, 25, 11390–11405 CrossRef CAS PubMed.
- Z. Guo, N. Hauser, A. Moreno, T. Ishikawa and P. Walde, Soft Matter, 2011, 7, 180–193 RSC.
- K. Junker, G. Zandomeneghi, Z. Guo, R. Kissner, T. Ishikawa, J. Kohlbrecher and P. Walde, RSC Adv., 2012, 2, 6478–6495 RSC.
- K. Junker, R. Kissner, B. Rakvin, Z. Guo, M. Willeke, S. Busato, T. Weber and P. Walde, Enzyme Microb. Technol., 2014, 55, 72–84 CrossRef CAS PubMed.
- G. Shumakovich, A. Streltsov, E. Gorshina, T. Rusinova, V. Kurova, I. Vasil'eva, G. Otrokhov, O. Morozova and A. Yaropolov, J. Mol. Catal. B: Enzym., 2011, 69, 83–88 CrossRef CAS.
- K. Junker, S. Luginbühl, M. Schüttel, L. Bertschi, R. Kissner, L. D. Schuler, B. Rakvin and P. Walde, ACS Catal., 2014, 4, 3421–3434 CrossRef CAS.
- A. Janoševic Ležaić, S. Luginbühl, D. Bajuk-Bogdanović, I. Pašti, R. Kissner, B. Rakvin, P. Walde and G. Ćirić-Marjanović, Sci. Rep., 2016, 6, 30724 CrossRef PubMed.
- S. Luginbühl, L. Bertschi, M. Willeke, L. D. Schuler and P. Walde, Langmuir, 2016, 32, 9765–9779 CrossRef PubMed.
- S. Luginbühl, M. Milojević-Rakić, K. Junker, D. Bajuk-Bogdanović, I. Pašti, R. Kissner, G. Ćirić-Marjanović and P. Walde, Synth. Met., 2017, 226, 89–103 CrossRef.
- P. Walde and Z. Guo, Soft Matter, 2011, 7, 316–331 RSC.
- W. Liu, J. Kumar, S. Tripathy and L. A. Samuelson, Langmuir, 2002, 18, 9696–9704 CrossRef CAS.
- K. Piontek, M. Antorini and T. Choinowski, J. Biol. Chem., 2002, 277, 37663–37669 CrossRef CAS PubMed.
- H. Kellner, N. Jehmlich, D. Benndorf, R. Hoffmann, M. Rühl, P. J. Hoegger, A. Majcherczyk, U. Kües, M. von Bergen and F. Buscot, Enzyme Microb. Technol., 2007, 41, 694–701 CrossRef CAS.
- D. Sirim, F. Wagner, L. Wang, R. D. Schmid and J. Pleiss, Database, 2011, bar006 Search PubMed.
- F. Olson, C. A. Hunt, F. C. Szoka, W. J. Vail and D. Papahadjopoulos, Biochim. Biophys. Acta, 1979, 557, 9–23 CrossRef CAS.
- L. D. Mayer, M. J. Hope and P. R. Cullis, Biochim. Biophys. Acta, 1986, 858, 161–168 CrossRef CAS.
- M. J. Hope, R. Nayar, L. D. Mayer and P. R. Cullis, in Liposome Technology, ed. G. Gegoriadis, CRC Press, Boca Raton, 2nd edn, 1993, vol. I, pp. 123–139 Search PubMed.
- A. V. Kulikov, V. R. Bogatyrenko, O. V. Belonogova, L. S. Fokeeva, A. V. Lebedev, T. A. Echmaeva and I. G. Shunia, Russ. Chem. Bull., 2002, 51, 2216–2223 CrossRef CAS.
- V. I. Krinichnyi, H.-K. Roth, M. Schrödner and B. Wessling, Polymer, 2006, 47, 7460–7468 CrossRef CAS.
- L. Dennany, P. C. Imnis, S. T. McGovern, G. G. Wallace and R. J. Forster, Phys. Chem. Chem. Phys., 2011, 13, 3303–3310 RSC.
- D. Carić, B. Rakvin, M. Kveder, K. Junker, P. Walde and E. Reijerse, Curr. Appl. Phys., 2015, 15, 1516–1520 CrossRef.
- Y. Xia, J. M. Wiesinger, A. G. MacDiarmid and A. J. Epstein, Chem. Mater., 1995, 7, 443–445 CrossRef CAS.
- G. M. do Nascimento and M. A. de Souza, in Nanostructured Conductive Polymers, ed. A. Eftekhari, John Wiley & Sons, Chichester, 2015, ch. 8, pp. 341–373 Search PubMed.
- J. Tarver and Y.-L. Loo, in Conjugated Polymers: A Practical Guide to Synthesis, ed. K. Müllen, J. R. Reynolds and T. Masuda, The Royal Society of Chemistry, 2014, ch. 12, pp. 248–264 Search PubMed.
- J. Stejskal, M. Trchová, P. Bober, P. Humpolíček, V. Kašpárková, I. Sapurina, M. A. Shishov and M. Varga, in Encyclopedia of Polymer Science and Technology, John Wiley & Sons, 2015 Search PubMed.
- F. Xu, Biochemistry, 1996, 35, 7608–7614 CrossRef CAS PubMed.
- E. Enauda, M. Trovaslet, F. Naveau, A. Decristoforo, S. Bizet, S. Vanhulle and C. Jolivalt, Enzyme Microb. Technol., 2011, 49, 517–525 CrossRef PubMed.
- N. Raseda, S. Hong, O. Y. Kwon and K. Ryu, J. Microbiol. Biotechnol., 2014, 24, 1673–1678 CrossRef CAS PubMed.
- C. Di Bari, N. Mano, S. Shleev, M. Pita and A. L. De Lacey, J. Biol. Inorg Chem., 2017, 22, 1179–1186 CrossRef CAS PubMed.
- G. Ćirić-Marjanović, M. Trchová and J. Stejskal, J. Raman Spectrosc., 2008, 39, 1375–1387 CrossRef and references cited therein.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Elsevier, 1991, pp. 117–154 Search PubMed.
- Y. Zhang, S. Serrano-Luginbühl, R. Kissner, M. Milojević-Rakić, D. Bajuk-Bogdanović, G. Ćirić-Marjanović, Q. Wang and P. Walde, Langmuir, 2018, 34, 9153–9166 CrossRef CAS PubMed.
- J. Stejskal, A. Riede, D. Hlavatá, J. Prokeš, M. Helmstedt and P. Holler, Synth. Met., 1998, 96, 55–61 CrossRef CAS.
- L. H. C. Mattoso, A. G. MacDiarmid and A. J. Epstein, Synth. Met., 1994, 68, 1–11 CrossRef CAS.
- M.-J. Han, H.-T. Choi and H.-G. Song, J. Microbiol., 2005, 43, 555–560 CAS.
- F. J. Millero, F. Huang and A. F. Laferiere, Geochim. Cosmochim. Acta, 2002, 66, 2349–2359 CrossRef CAS.
- R. J. Beyon and J. S. Easterby, Buffer Solutions, Oxford University Press, 1996, p. 79 Search PubMed.
- A. Kausaite, A. Ramanaviciene and A. Ramanavicius, Polymer, 2009, 50, 1846–1851 CrossRef CAS.
- A. Kausaite-Minkstimiene, V. Mazeiko, A. Ramanaviciene and A. Ramanavicius, Sens. Actuators, B, 2011, 158, 278–285 CrossRef CAS.
- N. German, A. Popov, A. Ramanaviciene and A. Ramanavicius, Polymer, 2017, 115, 211–216 CrossRef CAS.
- K. Junker, I. Gitsov, N. Quade and P. Walde, Chem. Pap., 2013, 67, 1028–1047 CAS.
- J. P. Malval, J. P. Morand, R. Lapouyade, W. Rettig, G. Jonusauskas, A. Oberlé, C. Trieflinger and J. Daub, Photochem. Photobiol. Sci., 2004, 3, 939–948 RSC.
- S. M. Jones and E. I. Salomon, Cell. Mol. Life Sci., 2015, 72, 869–883 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra05731a |
| This journal is © The Royal Society of Chemistry 2018 |