
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe expression and biological function of the PHF2 gene in breast cancer
Lu Zhang†
 a,
Tian-Li Hui†a,
Yu-Xian Weib,
Zhu-Min Caoc,
Fan Fengd,
Guo-Sheng Renab and
Fan Li*b
a,
Tian-Li Hui†a,
Yu-Xian Weib,
Zhu-Min Caoc,
Fan Fengd,
Guo-Sheng Renab and
Fan Li*b
aDepartment of Oncology, Chongqing Key Laboratory of Molecular Oncology and Epigenetics, The First Affiliated Hospital of Chongqing Medical University, China
bDepartment of Endocrine Surgery and Breast Cancer Center, The First Affiliated Hospital of Chongqing Medical University, #1 YouYi Road, YuZhong District, Chongqing 400016, China. E-mail: lf628@163.com
cDepartment of Oncology, The Seventh People’s Hospital of Chongqing, 400016, China
dDepartment of Breast Surgery, Hangzhou Women’s Hospital, Zhejiang 310000, China
First published on 27th November 2018
Abstract
PHD Finger Protein 2 (PHF2), as a protein code and a transcription regulatory gene, is a member of the Jumonji-C domain (JmjC). PHF2 is located at human chromosome 9q22.31 and is frequently decreased in various malignancies. However, the definite role of PHF2 in breast cancer remains unclear. To detect the expression and function of PHF2 in breast cancer, a q-PCR assay was used to detect the mRNA expression of PHF2 in breast cancer cell lines and paired breast cancer tissues, and immunohistochemistry was used to test the protein expression in breast cancer tissues and adjacent tissues. In addition, an adenovirus vector system was utilized to upregulate the expression of PHF2 in breast cancer cells. In our study, we found that PHF2 was down-expressed in breast cancer on both the mRNA and protein levels and the low expression of PHF2 was significantly associated with lymph node metastasis, Ki67 positive rate, ER negative expression and poor prognosis in breast cancer patients. The ectopic expression of PHF2 obviously inhibited the proliferation of breast cancer cell lines and the growth of xenograft tumors. Due to the tumor suppressor signature of PHF2 in breast cancer, we have reasons to believe that it could be a promoting marker and target for the prognosis and therapy of breast cancer.
Introduction
Breast cancer is one of the most common malignancies in women, and it is the leading cause of female deaths worldwide.1 Recently, the incidence and mortality of breast cancer has increased year by year in China. Although the diagnosis and treatment of breast cancer has been greatly improved, the prognosis for patients is still very poor, especially for those who have been diagnosed at an advanced stage.2,3 Therefore, it is an urgent mission for us to thoroughly understand the underlying biological features of breast cancer. Meanwhile, it is crucial for us to explore new targets for the early diagnosis and treatment of breast cancer. Generally, emerging evidence has shown that the inactivation of tumor suppressor genes plays a vital role in the development of malignance.4–6 PHD Finger Protein 2 (PHF2), as a protein code and transcription regulatory gene, is a member of the Jumonji-C domain (JmjC) and contains a zinc finger-like PHD (plant homeodomain).7,8 A large number of studies have shown that PHF2 can mediate the demethylation of dimethylated Lys-9 of histone H3 (H3K9me2), subsequently activating the expression of target genes.9,10 PHF2 is located at the human chromosome 9q22.31, but numerous studies have revealed that the human chromosome 9q22.32-22.33 was frequently deleted in various malignancies, such as basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), as well as bladder, prostate, esophageal, and blood cancer, and head and neck cancer.11,12 Consequently, PHF2 may be a tumor suppressor gene. Until now, the role of PHF2 in breast cancer remains unclear. So, the aim of the present study was to explore the expression and functions of PHF2 in breast cancer.Materials and methods
Cell culture
Breast cancer cell lines (MDA-MB-231, MDA-MB-468, BT549, MCF-7, and SK-BR-3) and normal human mammary epithelial cells (MCF-10A) were obtained from the American Type Culture Collection (Rockville, MD, USA). MCF10A cells were grown in DMEM/F12 containing 5% horse serum (Sigma USA; H0146), EGF 20 ng ml−1, hydrocortisone 0.5 mg ml−1, cholera toxin 100 ng ml−1 and insulin 10 μg ml−1. Other cell lines were cultured in RPMI 1640 (Gibco BRL, Karlsruhe, Germany) containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA) with 100 U ml−1 penicillin and 100 mg ml−1 streptomycin and maintained in a humidified incubator at 37 °C with 5% CO2.Patients and samples
The paired breast cancer tissues (n = 28) were obtained from the First Affiliated Hospital of Chongqing Medical University (2014/08-2014/12). Paraffin embedded specimens including breast cancer tissue (n = 80), and normal breast tissue (n = 40) which have the total clinical parameters (Table 2) were obtained from the Clinic Pathology Test Center of the First Affiliated Hospital of Chongqing Medical University. All patients who were involved in this study signed an informed consent. The study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication no. 85-23 Rev. 1985) and was approved by the Institutional Ethics Committee of The First Affiliated Hospital of Chongqing Medical University (Chongqing, China).Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from the tissues and cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and complementary DNA was synthesized by 1 μg of total RNA to a final volume of 20 μl, according to M-MLVE reverse transcription (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s instructions. The semi-quantitative PCR conditions were as follows: initial denaturation at 95 °C for 2 min, denaturation at 94 °C for 30 s with 32 cycles, annealing at 55 °C for 30 s, and extension at 72 °C for 30 s, with extension at 72 °C for 3 min in the final step. The RNA and c-DNA solution was stored at 80 °C. The quantitative polymerase chain reaction (qPCR) conditions were as follows: initial denaturation at 95 °C for 30 s, followed by 40 cycles with denaturation at 95 °C for 5 s, annealing and extension at 60 °C for 34 s. The reaction was performed on ABI 7500 Primes (Applied Biosystems, Foster City, CA, USA) using a SYBR Green PCR Master Mix/Rox II (Takara Biotechnology Co., Ltd.). Each sample with 10 μl of the mixture (containing 2 μl cDNA, 1.8 μl of ddH2O (RNase Free), 5 μl of One Step SYBR Premix Buffer (2×), 0.2 μl of ROX Reference Dye II (50×), 0.4 μl of PCR forward primer (10 μM) and 0.4 μl of PCR reverse primer (10 μM)) was run in triplicate, and the average value was analysed. The relative expression of target gene ΔΔCt was normalized with the GAPDH. The primers for the reaction were: PHF2-F: GCCTCTAACCACAGCGAGAT, PHF2-R: GTAGATCCAGCCTGAGGGGA; and GAPDH-F: GGAGTCAACGGATTTGGT, and GAPDH-R: GTGATGGGATTTCCATTGAT.Immunohistochemistry
Immunohistochemistry was performed by a two-step method. In short, the sections were maintained in a 60 °C incubator for 2 h. After this, the sections were deparaffinized in dimethylbenzene and hydrated in gradient ethanol, then underwent sodium citrate (pH 6.0) antigen retrieval in a microwave at a low heat for 20 min. Sections were incubated with 3% hydrogen peroxide to block endogenous peroxidase activity. Next, the sections were incubated with the primary antibody PHF2 (HPA010831, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :250), which was produced in rabbits and was purchased from Sigma (USA), overnight at 4 °C. The sections were washed three times with PBS for 5 min each and were subsequently immerged with goat anti-rabbit secondary antibody (SSA004, 1:1000), which was purchased from Beijing Yiqiao (China), at 37 °C for 20 min and stained by DAB for 3–5 min under a microscope. Finally, the slides were counterstained with hematoxylin, dehydrated, made transparent and sealed. The results were scored by two pathologists without knowing the information. Firstly, they were scored according to the staining percentage of the tumor cell: 0 = positive staining ≤5%, 1 = positive staining 6–25%, 2 = positive staining 26–50%, 3 = positive staining 51–75%, and 4 = positive staining >75%; and secondly, the intensity of the staining: 0 = negative, 1 = buff, 2 = yellow, and 3 = brown. The total score was determined by the multiplication of the intensity of the staining and the percentage of the positive cells: scores of ≤4 were negative, and scores of 5–12 were positive. Negative staining means a low expression of PHF2, and positive means a high expression of PHF2.
:250), which was produced in rabbits and was purchased from Sigma (USA), overnight at 4 °C. The sections were washed three times with PBS for 5 min each and were subsequently immerged with goat anti-rabbit secondary antibody (SSA004, 1:1000), which was purchased from Beijing Yiqiao (China), at 37 °C for 20 min and stained by DAB for 3–5 min under a microscope. Finally, the slides were counterstained with hematoxylin, dehydrated, made transparent and sealed. The results were scored by two pathologists without knowing the information. Firstly, they were scored according to the staining percentage of the tumor cell: 0 = positive staining ≤5%, 1 = positive staining 6–25%, 2 = positive staining 26–50%, 3 = positive staining 51–75%, and 4 = positive staining >75%; and secondly, the intensity of the staining: 0 = negative, 1 = buff, 2 = yellow, and 3 = brown. The total score was determined by the multiplication of the intensity of the staining and the percentage of the positive cells: scores of ≤4 were negative, and scores of 5–12 were positive. Negative staining means a low expression of PHF2, and positive means a high expression of PHF2.
Survival analysis
An online data base (http://kmplot.com/analysis/) was used to analyze the prognosis of breast cancer patients with different expression levels of PHF2.The transfection of adenovirus Ad-PHF2 and Ad-RFP
We purchased the adenovirus of the over-expressed PHF2 gene (Ad-PHF2/PHF2) and the negative control which lacked the PHF2 gene and expressed enhanced red fluorescence (Ad-RFP/vector) from the Hanbio Shanghai company. Breast cancer cell lines were seeded in six well plates at a density of 1 × 105 per well and cultured overnight. Then the cells were transfected with the Ad-PHF2 and Ad-RFP (the multiplicity of infection is 60) in serum-free medium for 2 h, then replaced by the complete medium.The colon formation assay of cells
After 72 h post-transfection, cells were collected and seeded in six well plates at a density of 600 per well following ectopic expression of PHF2 by Ad-PHF2 and the control (Ad-RFP/vector), selected for 2 weeks. Surviving colonies (≥50 cells per colony) were counted after staining with gentian violet. Finally, photos were taken.Cell proliferation assay
Breast cancer cells were cultured in 96-well plates at a density of 2000 cells per well and allowed to grow overnight. At 0 h, 24 h, 36 h, 48 h and 72 h, Cell Counting Kit-8 (CCK-8, DojindoMolecular Technologies, Inc., Kumamoto, Japan) was added, according to the manufacturer’s protocol, then incubated for 2 h at 37 °C in the dark. Finally, cell proliferation was measured at 450 nm by a microplate reader.Analysis of cell cycle distribution
Breast cancer cells were seeded in six well plates (2 × 105 per well) and grown overnight. Cells were harvested and centrifuged at 800 × g for 5 min after transfection for 48 h. Cells were subsequently washed twice with PBS, resuspended and fixed in ice-cold 70% ethanol for 24 h at 4 °C. Finally, 100 μg ml−1 of RNase and 50 μg ml−1 of PI were added and incubated for 30 min. Then the cell cycles were determined by fluorescence-activated cell sorting (BD Influx, Bec-tone Dickinson, San Jose, CA, USA) analysis.Nude-mouse transplanted tumor model assay
BALB/c nude female mice (n = 8) that were 4 weeks old (18–20 g) were purchased from Beijing HFK Bioscience, and maintained under specific pathogen free (SPF) conditions in the Experimental Animal Department of the Chongqing Medical University. MDA-MB-231 cells (2 × 106) in 200 μl of serum-free medium were subcutaneously injected in the right dorsal flank of the mice. When the volume of the tumor was about 100 mm3, the mice were randomized and grouped with 4 in each group (the PHF2 group and the vector group), then the mice received an intratumoral injection with Ad-PHF2 and Ad-RFP respectively (∼109 PFU (plaque-forming units)) divided into three parts, in a total volume of 45 μl. The injection was repeated every week. After 4 weeks from the last viral injection, the mice were sacrificed with their tumors harvested for further research. The in vivo study was performed in strict accordance with the NIH guidelines for the care and use of laboratory animals (NIH Publication no. 85-23 Rev. 1985) and was approved by the Institutional Animal Care and Use Committee of The First Affiliated Hospital of Chongqing Medical University (Chongqing, China).Statistical analysis
SPSS version 19.0 (IBM, USA) was used for all of the statistical analysis. The χ2 test was used to analyse expression status and clinicopathological parameters. Student’s t-test and analysis of variance were used to compare the value with the groups. For all of the tests, according to a = 0.05 standard, P < 0.05 was considered to be statistically significant.Results
The expression of PHF2mRNA in tissues and cells
Firstly, we systematically obtained PHF2 mRNA expression levels between breast cancer tissues and normal breast samples from the publicly available database Oncomine. According to the online database analysis, we recognized that PHF2 was significantly down-regulated in breast cancer samples compared to normal tissues (Fig. 1A).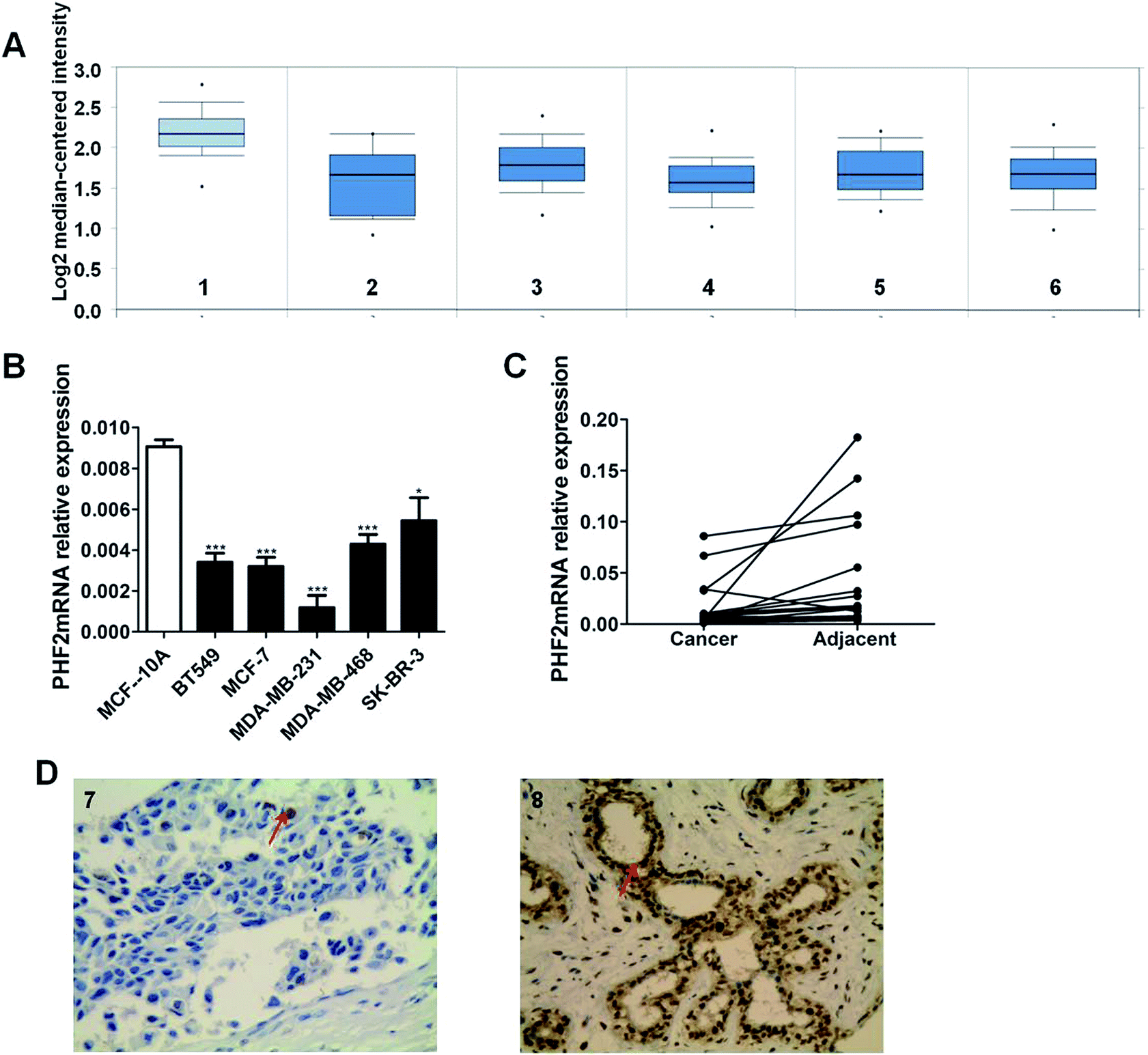 | ||
| Fig. 1 Down-expression of PHF2 in breast cancer cells and tissues. (A) The expression profile of PHF2mRN from the online database. (1) Breast (n = 144), (2) ductal breast cancer in situ (n = 10), (3) tubular breast cancer (n = 67), (4) medullary breast cancer (n = 32), (5) mucinous breast cancer (n = 46), and (6) invasive ductal and lobular breast cancer (n = 90). (B) The expression of PHF2 mRNA in breast cancer cell lines (n = 5) was detected by RT-qPCR. (C) The expression of PHF2 mRNA in paired breast cancer tissues (n = 28) was detected by RT-qPCR. (D) The expression of PHF2 protein in breast cancer tissues (n = 80) and normal breast tissues (n = 40): (7) low expression in breast cancer tissues (original magnification, ×400). (8) High expression in normal breast tissues (original magnification, ×400). *P < 0.05 and ***P < 0.001, vs. vector. | ||
To further validate the expression profile of PHF2, we detected PHF2 expression in tissues and cells. We found that the expression of PHF2 mRNA in breast cancer cells was lower than the normal breast epithelial cells (MCF-10A) (P < 0.05) (Fig. 1B). The expression of PHF2 mRNA in paired breast cancer tissues and cells was detected using q-PCR. Consistent with other studies, the expression of PHF2 mRNA was significantly down-regulated in 85% of breast cancer tissues compared to the paired adjacent tumor tissues (P < 0.05) (Fig. 1C).
PHF2 expression and the clinical parameters
Next, we used immunohistochemistry to measure the expression of PHF2 protein in tissues. Our results showed that PHF2 was mainly located in the nucleus and the PHF2 protein expression level in breast cancer tissues was significantly lower than that in normal breast tissue (P < 0.05) (Fig. 1D) (Table 1). Then we further analyzed the relationship between the PHF2 expression level and the clinical parameters in 80 patients. The results suggested that the low expression of PHF2 was obviously associated with the clinical parameters of lymph node metastasis, and the Ki67 positive and ER negative rate (P < 0.05) (Table 2). Meanwhile, we further investigated the prognostic significance of PHF2 in breast cancer by an online data base (http://kmplot.com/analysis/). Prognostic analysis showed a high PHF2 expression with a good OS (overall survival) and RFS (recurrence free survival) in breast cancer patients, compared to the low-expressed PHF2 group (Fig. 2). When stratified by different molecular subtypes, high PHF2 expression of basal-like, luminal A and HER2+ breast cancer have a better OS and RFS, but only the RFS showed a similar result in luminal B breast cancer. PHF2 results indicated that the down-expression of PHF2 was associated with a poor prognosis of breast cancer.| Clinical parameter | N = 80 | PHF2 | % | P-value | |
|---|---|---|---|---|---|
| + | − | ||||
| a Non-significant (P > 0.05) other data were not listed.b P < 0.05. | |||||
| Age (year) | |||||
| ≤60 | 20 | 8 | 12 | 40.0 | 1.000 |
| >60 | 60 | 26 | 34 | 43.3 | |
|
|||||
| Lymph node metastasis | |||||
| + | 23 | 2 | 21 | 8.7 | 0.014b |
| − | 57 | 21 | 36 | 36.8 | |
|
|||||
| ER | |||||
| + | 46 | 22 | 24 | 47.8 | 0.036b |
| − | 34 | 8 | 26 | 23.5 | |
|
|||||
| PR | |||||
| + | 44 | 17 | 27 | 38.6 | 0.648 |
| − | 36 | 12 | 24 | 33.3 | |
|
|||||
| Ki67 (%) | |||||
| ≤14 | 35 | 22 | 13 | 62.9 | 0.024b |
| >14 | 45 | 16 | 29 | 35.6 | |
 | ||
| Fig. 2 A low expression of PHF2 indicates a poor prognosis of breast cancer patients. Data taken from the Kmplot online database http://kmplot.com/analysis/. PHF2 expression and the over survival and the recurrence-free survival (RFS) in patients with different types of breast cancer. | ||
PHF2 inhibition of the proliferation and potential molecular mechanism
Accumulative research has uncovered that PHF2 is down-regulated in most tumors, and it may act as a tumor suppressor gene.11–14 To investigate the role of PHF2 in breast cancer, we used Ad-PHF2 to over-express PHF2, with Ad-RFP as a control, and then the exogenous expression of PHF2 was tested by RT-qPCR (P < 0.001) (Fig. 3A). The CCK8 and colony assays were performed to detect the role of PHF2 on cell proliferation. Up-regulation of PHF2 significantly inhibited the growth of MDA-MB-231 and MCF-7 cells: about a 51% reduction in the colony number was observed compared to the control group (P < 0.001) (Fig. 3B and C). Meanwhile, CCK8 results showed that the MDA-MB-231 and MCF-7 viability was dramatically decreased at 24 h and 48 h, respectively (P < 0.05) (Fig. 3D and E). At the same time, we used fluorescence-activated cell sorting (FACS) to detect the cell cycle distribution. This revealed the over-expression of PHF2 induced G1 and G2/M arrest in MDA-MB-231 compared to the vector. Together, these data suggested that PHF2 inhibited cell proliferation, likely by inducing cell cycle arrest in the G1 and G2/M phases (P < 0.05) (Fig. 3F and G). | ||
| Fig. 3 PHF2 represses the proliferation of breast cancer cells. (A) PHF2 expression levels in MDA-MB-231 and MCF-7 after infection by Ad-RFP (vector) and Ad-PHF2 (PHF2) detected by RT-qPCR. (B) Over-expression of PHF2 markedly diminished the ability of colony formation in MDA-MB-231 and MCF-7. (C) The statistics of the colonies; the number of colonies in the vector group was set to 100%. (D and E) Ectopic expression of PHF2 obviously inhibited the proliferation of MDA-MB-231 and MCF-7 by CCK-8, respectively. (F) PHF2 arrested the cell cycle of MDA-MB-231 mainly in the G1, and G2/M phases. (G) The statistics of the cell cycles. *P < 0.05,**P < 0.01 and ***P < 0.001, vs. vector. | ||
PHF2 inhibition of the xenograft tumor growth in vivo
Next, in order to probe the effects of PHF2 on breast cancer in vivo, a nude-mouse transplanted tumor model of MDA-MB-231 (n = 8) was constructed. We observed that the average tumor volume in the PHF2 group (n = 4) was much smaller than the vector group (n = 4), after 2 weeks adenovirus injection (6 weeks after cell injection) (Fig. 4A and B). Similarly, the average weight of the PHF2 treated tumors was markedly lower than the vector group (P < 0.05) (Fig. 4C). Our results showed that over-expressed PHF2 in MDA-MB-231 xenografts significantly inhibited the tumor growth, and PHF2 may be a tumor suppressor gene in breast cancer.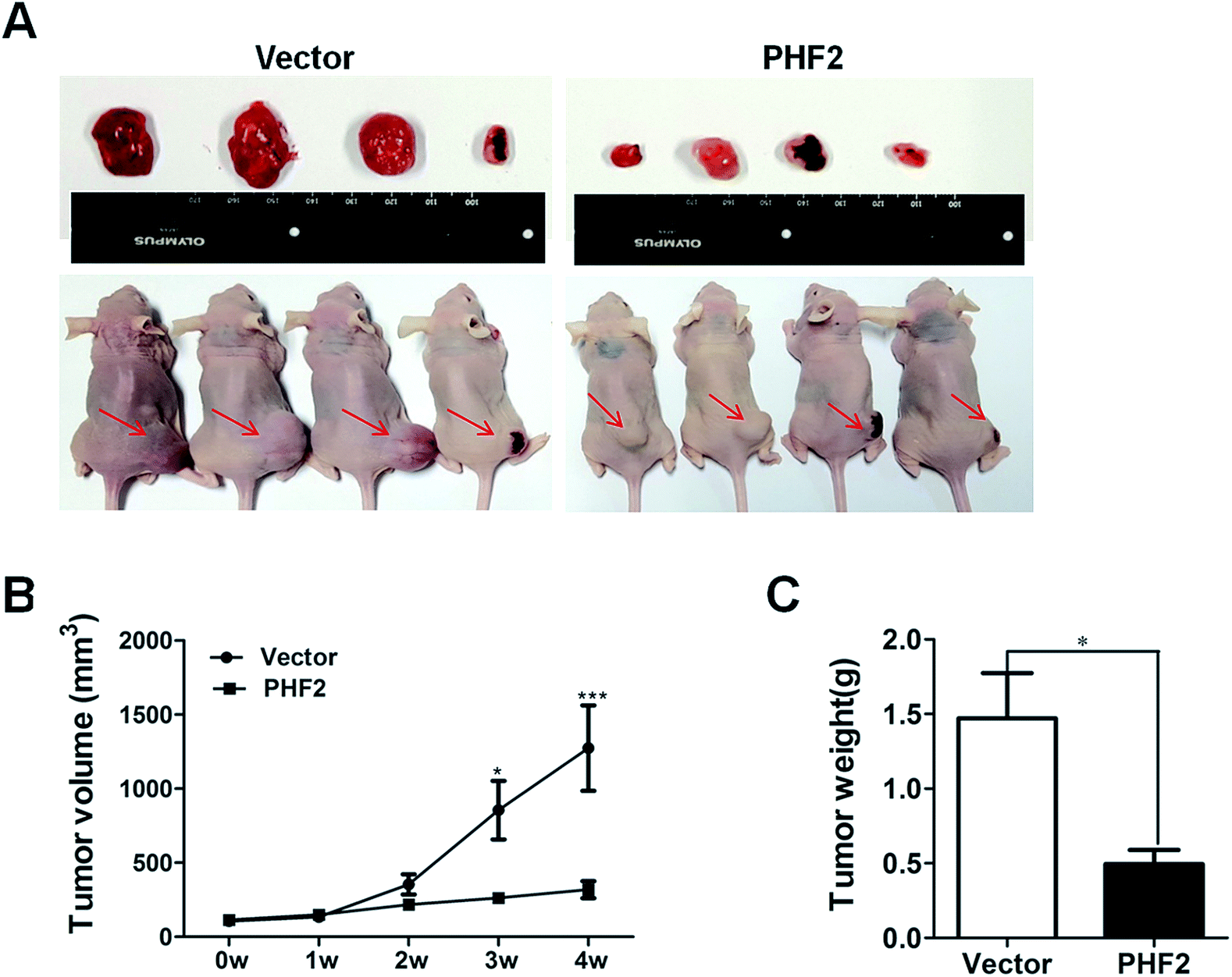 | ||
| Fig. 4 PHF2 inhibits the tumor growth in vivo. (A) Xenograft tumor size in both groups (n = 4 per group). (B) Growth curve of the tumors. The growth was significantly inhibited after 2 weeks of the injected Ad-PHF2 in the tumor. (C) Final weight of the xenograft tumors in both groups. *P < 0.05, ***P < 0.001, vs. vector. | ||
Lastly, to further explore the underlying molecular mechanism of PHF2 involved in regulating gene expression and tumor suppression, we analysed the PPI (protein interaction) network for PHF2 by searching using the online tool on the STRING database. As we expected, PHF2 showed complex interactions with other proteins which were associated with histone demethylase, DNA transcription, and the regulation of the cell-cycle (Fig. 5).
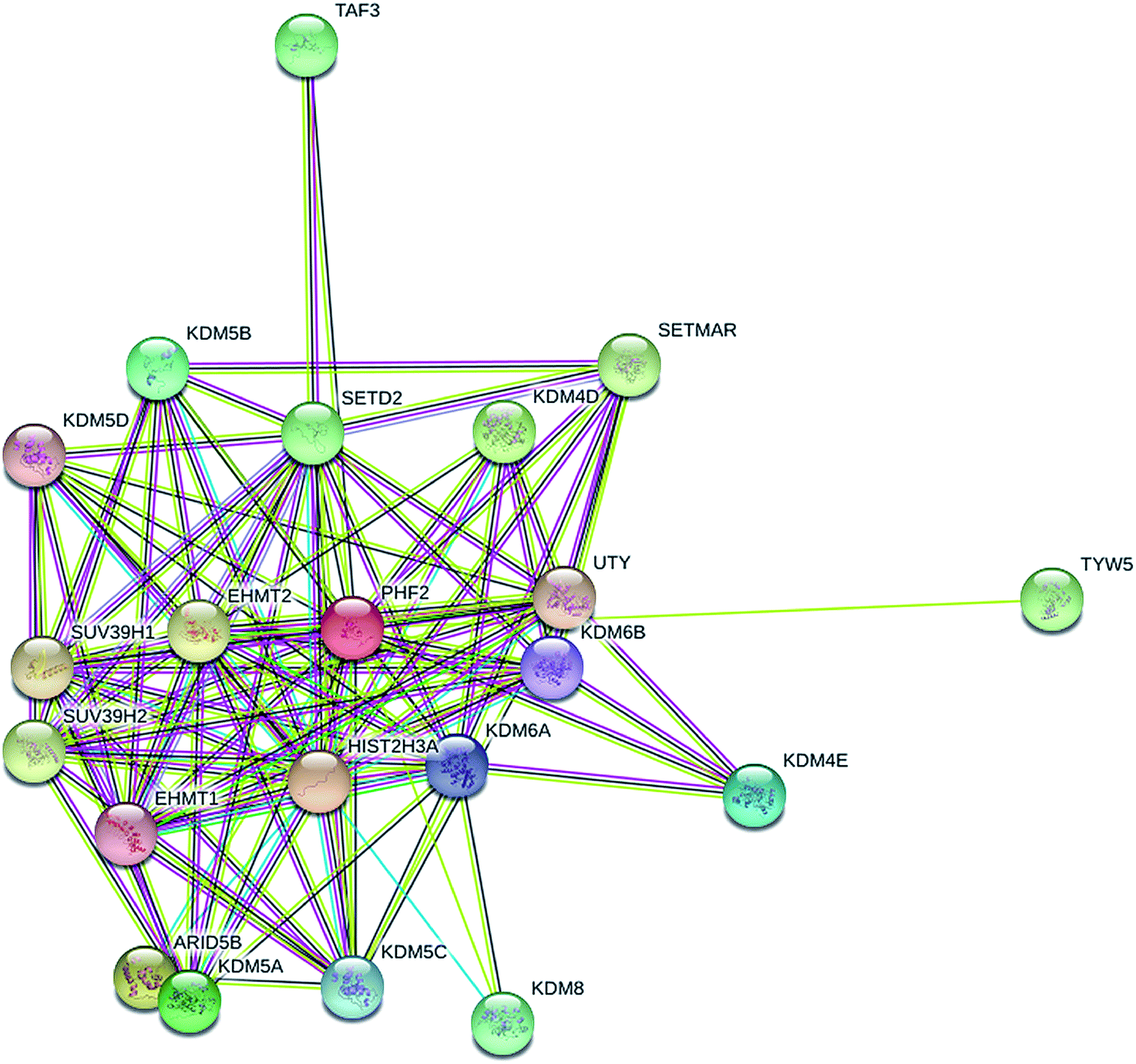 | ||
| Fig. 5 Molecular functional pathways and process of PHF2. | ||
Discussion
PHF2, located in 9q22.31, belongs to the KDM7 family (KDM7 is a subfamily of JmjC) containing a zinc finger-like PHD (plant homeodomain). The KDM7 family contains KDM7A (KIAA1718), KDM7B (PHF8), and KDM7C (PHF2).15 The expression of KDM7B and KIAA1718 is associated with the development of the brain, and these also regulate gene transcription by demethylating H3K9Me2, H3K27Me2 and H4K20Me.16,17 A large number of molecular function studies showed that PHF2 can mediate the demethylation of dimethylated Lys-9 of histone H3 (H3K9me2), subsequently activating the transcription of the target gene. PHF2 can also bind to the trimethylated Lys-4 of histone H3 (H3K4me3) at rDNA promoters and promotes the expression of rDNA.18,19 In the beginning, PHF2 was found as a candidate gene for hereditary sensory neuropathy type-I.20 Subsequent studies showed that 9q22.31 included some potential tumor suppressor genes such as PHF2, FANCC, PTCH1, XDP and it was often deleted in basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) as well as bladder, prostate, esophageal, and blood cancer, and head and neck cancer. Consequently, PHF2 may be regarded as a tumor suppressor gene.11,12,14In the present study we validated that PHF2 expression was frequently and significantly down-regulated in breast cancer tissue and cell lines, which was in line with the analysis outcome of an online database and previous studies.12 Epigenetic inactivation includes promoter methylation and histone modification, which further regulates gene expression.21,22 Some studies showed that the deletion of PHF2 was associated with PHF2 methylation in head and neck tumors.11 It is unclear whether the promoter methylation contributes to PHF2 silencing, and further investigation on this problem is needed.
Moreover, the down-expression of PHF2 was associated with the ER negative rate, the Ki67 positive rate and lymph node metastasis. Numerous studies showed that ER negative breast cancer patients frequently possess characteristic resistance to endocrinetherapy.23 As we all know, Ki67 is a good marker of cell proliferation in a variety of tumors and lymph node metastasis indicates high invasion and migration of tumors, which can be used for prognostic assessments of patients with cancer.24–29 Further prognostic analysis of the Kmplot online database confirmed this point which showed that the breast cancer patients with low PHF2 expression frequently have shorter OS and RFS compared to the high expression group. In further studies, we will assess the prognosis of different histological types of breast cancer with a large sample and a longer follow-up period. Based on these results, it was indicated that PHF2 down-regulation may impede the suppressor role in breast cancer, which leads to malignant progression of tumors.11,12
Next, we further proved the role of ectopic expression of PHF2 in breast cancer cell lines. The results showed that the over-expression of PHF2 significantly inhibited the proliferation of breast cancer cells by inducing the cell cycle arrest in G1 and G2/M phases, which was consistent with the result of clinical pathological analysis. Meanwhile, we observed a similar phenomenon, that PHF2 also significantly inhibited the xenograft tumor growth through intratumoral injection of PHF2. Although the number of mice involved in our study was limited, this is only a preliminary exploration and the difference in the two groups has statistical significance. More nude mice will be involved in the next study which will enhance the statistical and persuasive power.
In our study PHF2 inhibited the proliferation and vitality of breast cancer cells by inducing the cell cycle arrest in the G1 and G2/M phases. Few studies reported its related mechanism in tumorigenesis. A previous study confirmed that PHF2, as a histone demethylase, activated P21, which is a downstream target gene of P53, by relieving histone modification silencing (H3K9 methylation), then exerted its tumor suppressing role with the P53 signal in colon cancer.14 P21, as a crucial cell cycle regulator protein, mediated the proliferation of tumors by arresting the cell cycle and regulating the DNA reproduction.30–32 In our study, we confirmed that the ectopic expression of PHF2 contributes to the cell cycle arresting in breast cancer cells. Considering previous studies and our study, we suppose that PHF2 inhibits cell proliferation by up-regulating P21, which is a downstream gene of P53, signaling by derepressing H3K9me silencing in breast cancer. It is also possible that PHF2 works with other components which play a key role in suppressing tumor proliferation. Further investigations focusing on these questions are warranted in future studies.
Alternatively, PHF2 down-regulation was associated with lymph node metastasis in breast cancer patients, which indicated that PHF2 down-regulation may contribute to the metastasis of tumors. Metastasis is a major cause of poor prognosis in malignancy. Numerous studies revealed that epithelial-mesenchymal transition (EMT) played a crucial role in tumor metastasis and treatment resistance in breast cancer.33–36 However, mesenchymal-epithelial-transition (MET) which is the reverse of EMT can increase cell–cell adhesion and reduce the mobility and invasion of cells by enhancing epithelial marker expressions such as E-cadherin and occluding, then enhances the sensitivity of the cell to a variety of drugs with potentially important therapeutic implications.13 Emerging evidence revealed that PHF2, with PKA-induced activation, promoted mesenchymal-to-epithelial transition (MET) through relieving the H3K9-mediated silencing of epithelial genes.13 This may be the reason why PHF2 inhibits the lymph node metastasis of breast cancer. Further intensive research focusing on the related biological functions and molecular mechanisms are needed in future studies.
Combined previous reports and the PPI network of PHF2 showed that PHF2, a histone-modifier enzyme, may derepress genes, silencing by repressing histone methyltrans-ferases (SUV39H1).10 Histone methyltransferase that counteracts the PHF2 function may also provide a direction for molecular therapeutic targets in cancer.
In conclusion, we confirm that PHF2 may serve as a tumor suppressor, but is frequently down-regulated in breast cancer. We have reasons to believe that PHF2 could be a novel molecular target for personalized therapy and prognosis estimation of breast cancer.
Conflicts of interest
There are no conflicts to declare.Abbreviations
| PHF2 | PHD Finger Protein 2 |
| JmjC | Jumonji-C domain; |
References
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef PubMed.
- T. Fujii, F. Le Du, L. Xiao, T. Kogawa, C. H. Barcenas, R. H. Alvarez, V. Valero, Y. Shen and N. T. Ueno, JAMA Oncol., 2015, 1, 1311–1318 CrossRef PubMed.
- R. C. M Clarke, S. Darby, C. Davies, V. Evans, J. Godwin, R. Gray, P. McGale, R. Peto and Y. Wang, Lancet, 2005, 365, 1687–1717 CrossRef.
- J. Lv, P. Zhu, X. Zhang, L. Zhang, X. Chen, F. Lu, Z. Yu and S. Liu, Molecular medicine reports, 2017 Search PubMed.
- J. Chai, L. Du, J. Ju, C. Ma, Z. Shen, X. Yang, L. Liang, Q. Ni and M. Sun, Molecular medicine reports, 2017 Search PubMed.
- Z. H. Liang Chang, Z. Zhou and H. Zhang, RSC Adv., 2018, 8, 8 Search PubMed.
- K. Hasenpusch-Theil, B. P. Chadwick, T. Theil, S. K. Heath, D. G. Wilkinson and A. M. Frischauf, Mamm. Genome, 1999, 5, 294–298 CrossRef.
- A. Yokoyama, Y. Okuno, T. Chikanishi, W. Hashiba, H. Sekine, R. Fujiki and S. Kato, Genes Cells, 2010, 15, 867–873 CAS.
- A. Baba, F. Ohtake, Y. Okuno, K. Yokota, M. Okada, Y. Imai, M. Ni, C. A. Meyer, K. Igarashi, J. Kanno, M. Brown and S. Kato, Nat. Cell Biol., 2011, 13, 668–675 CrossRef PubMed.
- G. Shi, M. Wu, L. Fang, F. Yu, S. Cheng, J. Li, J. X. Du and J. Wong, J. Biol. Chem., 2014, 289, 29691–29700 CrossRef CAS PubMed.
- A. Ghosh, S. Ghosh, G. P. Maiti, S. Mukherjee, N. Mukherjee, J. Chakraborty, A. Roy, S. Roychoudhury and C. K. Panda, Ann. Surg. Oncol., 2012, 19(suppl. 3), S528–S538 CrossRef PubMed.
- S. Sinha, R. K. Singh, N. Alam, A. Roy, S. Roychoudhury and C. Panda, Mol. Cancer, 2008, 7, 84 CrossRef PubMed.
- D. R. Pattabiraman, B. Bierie, K. I. Kober, P. Thiru, J. A. Krall, C. Zill, F. Reinhardt, W. L. Tam and R. A. Weinberg, Science, 2016, 351, aad3680 CrossRef PubMed.
- K. H. Lee, J. W. Park, H. S. Sung, Y. J. Choi, W. H. Kim, H. S. Lee, H. J. Chung, H. W. Shin, C. H. Cho, T. Y. Kim, S. H. Li, H. D. Youn, S. J. Kim and Y. S. Chun, Oncogene, 2015, 34, 2897–2909 CrossRef CAS PubMed.
- M. Bjorkman, P. Ostling, V. Harma, J. Virtanen, J. P. Mpindi, J. Rantala, T. Mirtti, T. Vesterinen, M. Lundin, A. Sankila, A. Rannikko, E. Kaivanto, P. Kohonen, O. Kallioniemi and M. Nees, Oncogene, 2012, 31, 3444–3456 CrossRef CAS PubMed.
- W. Feng, M. Yonezawa, J. Ye, T. Jenuwein and I. Grummt, Nat. Struct. Mol. Biol., 2010, 17, 445–450 CrossRef CAS PubMed.
- J. D. Stender, G. Pascual, W. Liu, M. U. Kaikkonen, K. Do, N. J. Spann, M. Boutros, N. Perrimon, M. G. Rosenfeld and C. K. Glass, Mol. Cell, 2012, 48, 28–38 CrossRef CAS PubMed.
- K. Fortschegger and R. Shiekhattar, Epigenetics, 2011, 6, 4–8 CrossRef CAS PubMed.
- H. Wen, J. Li, T. Song, M. Lu, P. Y. Kan, M. G. Lee, B. Sha and X. Shi, J. Biol. Chem., 2010, 285, 9322–9326 CrossRef CAS PubMed.
- G. A. Nicholson, J. L. Dawkins, I. P. Blair, M. L. Kennerson, M. J. Gordon, A. K. Cherryson, J. Nash and T. Bananis, Nat. Genet., 1996, 13, 101–104 CrossRef CAS PubMed.
- Y. Okuno, F. Ohtake, K. Igarashi, J. Kanno, T. Matsumoto, I. Takada, S. Kato and Y. Imai, Diabetes, 2013, 62, 1426–1434 CrossRef CAS PubMed.
- J. L. Schussel, L. P. Kalinke, L. M. Sassi, B. V. de Oliveira, P. A. Pedruzzi, M. Olandoski, L. E. Alvares, G. P. Garlet and P. C. Trevilatto, Cancer Biomarkers, 2015, 15, 11–17 CAS.
- B. S. Abdulkarim, J. Cuartero, J. Hanson, J. Deschenes, D. Lesniak and S. Sabri, J. Clin. Oncol., 2011, 29, 2852–2858 CrossRef PubMed.
- A. Bleckmann, L. C. Conradi, K. Menck, N. A. Schmick, A. Schubert, E. Rietkotter, J. Arackal, P. Middel, A. Schambony, T. Liersch, K. Homayounfar, T. Beissbarth, F. Klemm, C. Binder and T. Pukrop, Clin. Exp. Metastasis, 2016, 33, 309–323 CrossRef CAS PubMed.
- J. Konsti, M. Lundin, H. Joensuu, T. Lehtimäki, H. Sihto, K. Holli, T. Turpeenniemi-Hujanen, V. Kataja, L. Sailas, J. Isola and J. Lundin, BMC Clin. Pathol., 2011, 11 DOI:10.1186/1472-6890-11-3.
- B. Belev, World J. Gastroenterol., 2013, 19, 523 CrossRef PubMed.
- N. U. Lin, A. Vanderplas, M. E. Hughes, R. L. Theriault, S. B. Edge, Y. N. Wong, D. W. Blayney, J. C. Niland, E. P. Winer and J. C. Weeks, Cancer, 2012, 118, 5463–5472 CrossRef PubMed.
- D. Jiang, Z. Gao, Z. Cai, M. Wang and J. He, BMC Cancer, 2015, 15, 727 CrossRef PubMed.
- H. Mahmood, M. Faheem, S. Mahmood, M. Sadiq and J. Irfan, Asian Pac. J. Cancer Prev., 2015, 16, 1019–1024 CrossRef PubMed.
- N. Zhao, G. Zhang, M. He, H. Huang, L. Cao, A. Yin, P. Wang and L. Wang, J. Cancer, 2017, 8, 2132–2141 CrossRef PubMed.
- W. S. El-Deiry, Cancer Res., 2016, 76, 5189–5191 CrossRef CAS PubMed.
- J. W. Harper, G. R. Adami, N. Wei, K. Keyomarsi and S. J. Elledge, Cell, 1993, 75, 805–816 CrossRef CAS PubMed.
- I. Roxanis, J. Clin. Pathol., 2013, 66, 517–521 CrossRef CAS PubMed.
- Y. Wang and B. P. Zhou, Cancer Hallmarks, 2013, 1, 38–49 CrossRef PubMed.
- P. Mallini, T. Lennard, J. Kirby and A. Meeson, Cancer Treat. Rev., 2014, 40, 341–348 CrossRef CAS PubMed.
- L. Gammon and I. C. Mackenzie, J. Oral Pathol. Med., 2016, 45, 77–82 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2018 |