
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImparting sensitivity and selectivity to a gold nanoparticle chemiresistor through thiol monolayer functionalization for sensing acetone
Zhenzhen Xiea,
Mandapati V. Ramakrishnam Rajub,
Andrew C. Stewartb,
Michael H. Nantz*b and
Xiao-An Fu *a
*a
aChemical Engineering Department, University of Louisville, Louisville, Kentucky 40292, USA. E-mail: xiaoan.fu@louisville.edu
bDepartment of Chemistry, University of Louisville, Louisville, Kentucky 40292, USA. E-mail: Michael.nantz@louisville.edu
First published on 18th October 2018
Abstract
Chemiresistor-based gas sensors for detection of target volatile organic compounds (VOCs) in air face common challenges of poor sensitivity and selectivity as well as suffering from interference by other constituent gases and/or humidity. This work demonstrates that functionalizing gold nanoparticles (AuNPs) with a designed thiol monolayer improves sensitivity and selectivity of the derived AuNPs gas sensor. We report the synthesis and application of a thiol ligand fitted with both a urea motif and a tert-butyl end group for functionalizing AuNPs. The AuNPs sensor prepared using the urea thiol ligand demonstrated significantly increased acetone sensing in comparison with tested commercially available thiol-functionalized AuNPs. The sensor worked under ambient temperature and high humidity conditions, and demonstrated a linear relationship between the sensor response and the common logarithm of analyte concentration.
Introduction
The analysis of volatile organic compounds (VOCs), either in air or in exhaled breath, has become a highly active research area, due in large part to increasing air pollution concerns as well as growing interest in using breath analysis for detection of diseases. Acetone is one of the most abundant VOCs in environmental air. Acetone is also an endogenous product of human metabolism for which reported concentration levels in exhaled breath of patients with diabetes were much higher than in the breath of healthy subjects.1 As a result, nanostructured metal oxides,2–12 carbon nanotubes,13,14 polymer nanofibers,15,16 and nanoparticles17 all have been explored as sensing materials for detection of acetone.18,19 However, performance issues, such as low sensitivity, low selectivity, lack of reproducibility, interference of humidity, and sensor aging, continue to plague many of these current sensor materials. Thus, there is a need to develop real-time, portable, and high performance sensors to detect VOCs of interest such as acetone in the range of sub-ppm to ppm.Chemiresistors based on thiol ligand-functionalized metal nanoparticles are promising for sensing VOCs due to their controllable selectivity, ambient operating temperature, and their ease of integration with other microelectronic systems.20–22 Since the first thiol ligand-stabilized gold nanoparticles (AuNPs) chemiresistor for detection of toluene was reported in 1998,23 numerous researchers have explored AuNPs-based sensing platforms to detect VOCs24–32 under various conditions, including high humidity.33 However, the development of a thiol-functionalized AuNPs sensor that responds with high sensitivity and selectively to acetone still remains a challenge.
Devising a functional ligand-based chemiresistor to impart high sensitivity and selectivity for sensing target VOCs requires molecular-level control.34 The high level of control afforded by H-bonded ensembles in numerous supramolecular structures has inspired us to harness H-bonding to develop a functional sensing device.35,36 The seminal report by Kaftory et al.37 on the role of electronic interactions between acetone and urea derivatives in determining the crystal structures of adducts has prompted much effort toward developing urea motifs as receptors for the carbonyl functional group. We thus set out to couple the carbonyl selection properties of urea H-bonded networks with the conductive properties of AuNPs to develop a carbonyl-selective sensor.
We previously disclosed the prototype ligand 1-(tert-butyl)-3-((11-mercapto-undecyl)oxy)urea for preparing a AuNPs-based sensor array to detect VOCs in air.38 The current work reports details of the synthesis of this unprecedented thiol-terminated monoalkoxyurea motif through acylation of an aminooxy intermediate and its incorporation onto AuNPs for detecting trace acetone and other VOCs in air. A VOC-sensing comparison study testing the urea thiol ligand and three commercially available thiols is also presented.
Material and methods
Material
Commercial thiols 1-dodecanethiol, 4-methoxy-α-toluenethiol, and 11-mercaptoundecanoic acid as well as hydrogen tetrachloroaurate (HAuCl4), tetraoctylammonium bromide (TOAB), and Tedlar bags were purchased from Sigma-Aldrich in USA. Sodium borohydride was purchased from Fluka in USA. Deionized water was used for all experimentation. Reagent grade solvents including acetone, ethanol, benzene, and tetrahydrofuran (THF) were used for extractions and for purifications via flash column chromatography. THF was dried by distillation over Na/benzophenone. Dichloromethane (DCM) and dimethylformamide (DMF) were dried by distillation over CaH2.Nuclear magnetic resonance (NMR) spectra were obtained using a Varian/Agilent 400 MHz NMR spectrometer equipped with a 5 mm z-axis gradient AutoX probe operating at the nominal 1H frequency of 400 MHz and 13C frequency of 100 MHz. All spectra are reported in parts per million (ppm) relative to the residual solvent peak in 1H NMR and the deuterated solvent peak in 13C NMR. Mass samples were analyzed on a hybrid linear ion trap (LIT) Fourier transform-ion cyclotron resonance mass spectrometer (LTQ-FT, Thermo Electron, Bremen, Germany) equipped with a TriVersa NanoMate ion source (Advion BioSciences, Ithaca, NY) with an electrospray chip (nozzle inner diameter 5.5 lm). The TriVersa NanoMate was operated in positive ion mode by applying 1.85 kV with 0.1 psi head pressure. Melting points were acquired using Fisher-Johns melting point apparatus.
Synthesis of alkoxyurea thiol 1
N-Alkoxyphthalimide 2 (Scheme 1) was prepared according to a literature procedure.39 1-(tert-Butyl)-3-((11-mercapto-undecyl)oxy)urea (3) was prepared by analogy to a reported method.40 The progress of reactions was monitored by thin-layer chromatography using silica gel 60 Å F-254 plates. The plates were visualized first by UV illumination and then by staining using a p-anisaldehyde stain. Column chromatography was performed using silica gel (230–400 mesh).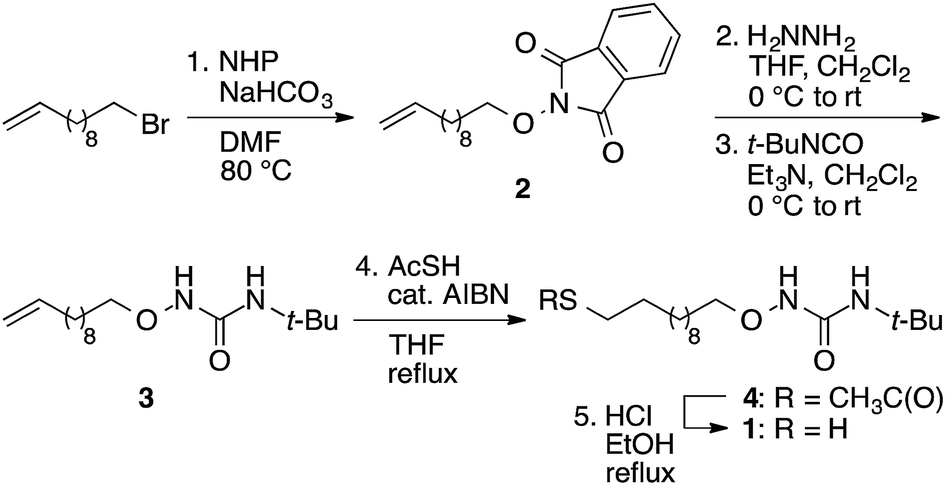 | ||
| Scheme 1 Synthesis of urea thiol 1; NHP = N-hydroxyphthalimide, AIBN = azobis(isobutyronitrile). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 mixture of EtOAc:hexane, to afford 3.51 g (86%) of 2 as a white solid; mp = 35–37 °C (lit. 41 mp = 38–40 °C); TLC, Rf = 0.69 (2.5:7.5, EtOAc:hexane); 1H NMR (CDCl3, 400 MHz) δ 7.84–7.82 (m, 2H), 7.75–7.73 (m, 2H), 5.81–5.79 (m, 1H), 5.00–4.90 (m, 2H), 4.19 (t, J = 5.6 Hz, 2H), 2.04–2.00 (m, 2H), 1.80–1.76 (m, 2H), 1.49–1.47 (m, 2H), 1.36–1.29 (m, 10H) ppm; 13C NMR (CDCl3, 100 MHz) δ 163.7, 139.2, 134.5, 129.1, 123.5, 114.2, 78.7, 33.9, 29.5, 29.4, 29.3, 29.2, 29.0, 28.2, 25.6 ppm; IR 2923, 2851, 1787, 1730, 1464, 1399, 1186 cm−1.
:85 mixture of EtOAc:hexane, to afford 3.51 g (86%) of 2 as a white solid; mp = 35–37 °C (lit. 41 mp = 38–40 °C); TLC, Rf = 0.69 (2.5:7.5, EtOAc:hexane); 1H NMR (CDCl3, 400 MHz) δ 7.84–7.82 (m, 2H), 7.75–7.73 (m, 2H), 5.81–5.79 (m, 1H), 5.00–4.90 (m, 2H), 4.19 (t, J = 5.6 Hz, 2H), 2.04–2.00 (m, 2H), 1.80–1.76 (m, 2H), 1.49–1.47 (m, 2H), 1.36–1.29 (m, 10H) ppm; 13C NMR (CDCl3, 100 MHz) δ 163.7, 139.2, 134.5, 129.1, 123.5, 114.2, 78.7, 33.9, 29.5, 29.4, 29.3, 29.2, 29.0, 28.2, 25.6 ppm; IR 2923, 2851, 1787, 1730, 1464, 1399, 1186 cm−1.To a stirred solution of the crude aminooxy intermediate (0.58 g, 3.17 mmol) in dry CH2Cl2 (15 mL) at room temperature under argon was added dry Et3N (0.48 mL, 3.45 mmol) via syringe. The reaction mixture was cooled to 0 °C and then t-butyl isocyanate (0.32 mL, 2.87 mmol) was added dropwise over 5 min. The reaction mixture was slowly allowed to reach room temperature and stirred 10 h before concentrating by rotary evaporation. The residue was purified by SiO2 flash column chromatography, eluting with a 1:4 mixture of EtOAc:hexane, to afford 0.62 g (68%) urea 3 as a colorless liquid; TLC, Rf = 0.51 (2.5:7.5, EtOAc:hexane); 1H NMR (CDCl3, 400 MHz) δ 6.85 (br, NH), 5.84–5.74 (m, 1H), 5.59 (br, NH), 5.00–4.90 (m, 2H), 3.77 (t, J = 6.4 Hz, 2H), 2.05–2.00 (m, 2H), 1.64–1.57 (m, 2H), 1.36–1.27 (m, 21H) ppm; 13C NMR (CDCl3, 100 MHz) δ 159.3, 139.1, 114.2, 76.4, 50.4, 33.8, 29.5, 29.4, 29.18, 29.12, 28.9, 28.1, 26.1 ppm; IR 3422, 3194, 2923, 2855, 1674, 1525, 1457 cm−1.
:85 mixture of EtOAc:hexane, to afford 0.48 g (75%) of 4 as a pale yellow liquid; TLC, Rf = 0.42 (2.5:7.5 EtOAc:hexane); 1H NMR (CDCl3, 400 MHz) δ 6.91 (br, NH), 5.59 (br, NH), 3.76 (t, J = 6.8 Hz, 2H), 2.84 (t, J = 6.8 Hz, 2H), 2.30 (s, 3H), 1.59–1.50 (m, 4H), 1.35–1.25 (m, 23H), ppm; 13C NMR (CDCl3, 100 MHz) δ 196.1, 159.2, 76.4, 50.5, 30.7, 30.6, 29.5, 29.2, 28.8, 28.2, 26.1 ppm; IR 3427, 2925, 2854, 1689, 1673 cm−1; FT-ICR-MS for C18H37N2O3S [M + H]+ calcd 361.2519, found 361.2500.:7.5 EtOAc:hexane); 1H NMR (CDCl3, 400 MHz) δ 6.76 (br s, 1H, NH), 5.60 (br s, 1H, NH), 3.77 (t, J = 6.8 Hz, 2H), 2.51 (q, J = 7.4 Hz, 2H), 1.61–1.55 (m, 4H), 1.36–1.26 (m, 23H, t-butyl + aliphatic chain) ppm; 13C NMR (CDCl3, 100 MHz) δ 159.2, 76.4, 50.4, 34.0, 29.5, 29.4, 29.2, 29.1, 28.3, 28.2, 26.1, 24.6 ppm; IR 3426, 2924, 2853, 1689, 1672, 1525 cm−1; FT-ICR-MS for C16H35N2O2S+ [M + H]+ calcd 319.2414, found 319.2419.Synthesis of thiol-functionalized gold nanoparticles (AuNPs)
AuNPs were synthesized using a two-phase reduction approach as reported by Brust et al.42 Two solutions were prepared as follows: (1) HAuCl4 (0.05 g) was dissolved in deionized water (4 mL); (2) TOAB (0.08 g) was dissolved in toluene (20 mL). The aqueous HAuCl4 solution was added to the toluene TOAB solution with vigorous stirring until all the tetrachloroaurate had transferred into the organic layer. An amount of 1-dodecanethiol, 4-methoxy-α-toluenethiol (4-MTT), 11-mercaptoundecanoic acid (11-MUA) or the urea thiol 1 (Table 1) to give a thiol:Au 1:1 molar ratio was then added to the toluene mixture. An aqueous solution of NaBH4, freshly prepared by dissolving NaBH4 (0.056 g) in deionized water (4 mL), was slowly added with vigorous stirring to the toluene mixture. A rapid color change occurred as the NaBH4 solution was added. After stirring for at least 3 h at room temperature, the organic phase was separated and the AuNPs were precipitated by dropwise addition of the organic phase into ethanol (400 mL) with rapid stirring. After settling overnight, the clear supernatant was decanted and the settled AuNPs were filtered and washed with ethanol. The AuNPs then were dispersed in toluene (10 mL) and precipitated again by dropwise addition into ethanol (200 mL). After standing overnight, the suspension was centrifuged and the collected particles were washed with ethanol and dried at 40 °C in an oven to yield AuNPs (ca. 30 mg) suitable for sensor formation.
| Sensor | Thiol | Thiol structure |
|---|---|---|
| a | 1-Dodecanethiol | 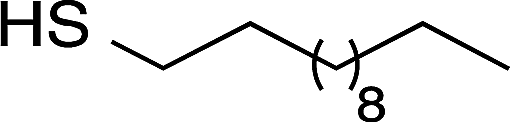 |
| b | 4-Methoxy-α-toluenethiol (4-MTT) | 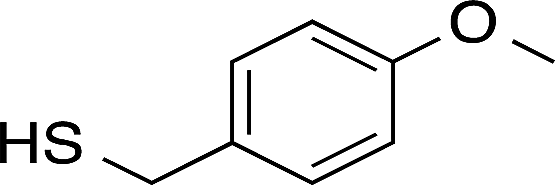 |
| c | 11-Mercaptoundecanoic acid (11-MUA) |  |
| d | Urea thiol 1 | 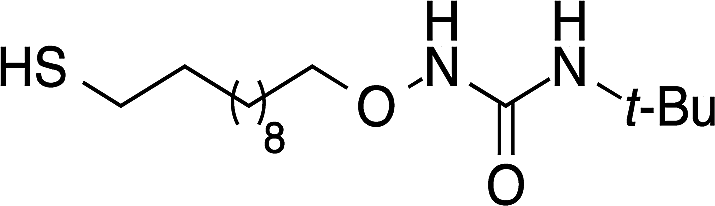 |
Fabrication of interdigitated electrodes and AuNP film deposition
Interdigitated electrodes (IDEs) were designed using the microelectromechanical systems (MEMS) software L-Edit to fit onto a 4-inch silicon wafer with a thin insulating layer of 1 μm of silicon dioxide. The IDEs then were fabricated using the MEMS microfabrication process, beginning with a sacrificial layer of photoresist coated on the silicon substrate. After photoresist was patterned by photolithography, a 10 nm thick Ti adhesion layer and a Pt metal layer (200 nm thick) were deposited. Finally, the sacrificial layer was removed by dissolving the photoresist in a solvent to finish a lift-off process. In this way, the IDEs and contact pads remained on the wafer.The thiol-coated AuNPs in toluene (0.2%) were sonicated at room temperature for 20 min and then cast onto the IDE area by dropwise addition. The toluene was evaporated at room temperature to leave a flat, roughly circular film of gold nanoparticles. The AuNPs-coated sensors were then stabilized overnight at 40 °C in a vacuum oven. After drying, the 1-dodecanethiol-, 4-MTT-, 11-MUA- and urea thiol 1-modified AuNPs sensors were used without further manipulation.
Sensor measurements
AuNPs sensors individually functionalized with the thiol panel listed in Table 1 were placed inside a homemade small stainless steel test chamber with a volume of about 500 mL. The chamber was first evacuated and then a pulse of either synthetic air or a VOC sample was introduced from a sample bag attached to the test chamber and the pressure of the chamber reached atmospheric pressure in a few seconds. After a testing time, the chamber was evacuated again. The VOC samples were prepared using Tedlar bags that had been flushed with synthetic air three times. A known amount of analyte, for example, 3 μL of acetone, was injected into a Tedlar bag containing 1 L synthetic air. As the acetone vaporized within the bag, a gaseous concentration of 1000 ppm was achieved. Then, 10 mL of the above gas sample was collected by an air-tight glass syringe and injected into a new 1 L Tedlar bag followed by a dilution process using 990 mL synthetic air, resulting in an acetone concentration of 10 ppm. For lower concentrations of analytes, a similar dilution process was carried out again.The resistance of an individual sensor was monitored at a fixed voltage of 5 V by a Keithley 2400 I–V meter and recorded as a function of time using the Labview program. The output voltage was set and a pulse current of the sensor was sent to the sensor every second. The resistance was calculated based on Ohm's law. In a typical experiment, the sensor response was measured over 5 min under vacuum (28 in Hg below atmospheric pressure), followed by synthetic air or VOC sample exposure for 5 min at atmospheric pressure, and then again for 5 min under vacuum. The measurements of synthetic air and VOC in air samples were done at atmospheric pressure. The cycles were repeated at least three times to test reproducibility. Each sensor was examined at different concentrations of acetone, ethanol, ethylbenzene and benzene in air. All experiments were performed at room temperature.
Results and discussion
Fig. 1a shows a micrograph of scanning electron microscopy of the fabricated IDEs at the core of the sensor. All IDEs were designed to fit into a 400 μm × 400 μm area with both the width of the electrodes and spaces between electrodes of 10 μm. The two contact pads were 3 × 3 mm3 for convenient connection to an I–V meter. The overall size of the sensor is rather small in comparison to a one-cent coin (Fig. 1b). Table 1 shows the molecular structures of the synthesized urea thiol and three commercial thiols used to modify AuNPs. | ||
| Fig. 1 (a): SEM micrograph of the microfabricated IDE electrodes; (b): size comparison between the sensor and a US one-cent coin. | ||
To probe the VOC-tracing capability, the urea thiol 1-functionalized AuNPs sensor was exposed to VOCs in air with varying concentrations in the test chamber. Alternating cycles of sample introduction into the chamber and evacuation were performed while the sensor resistance was monitored and recorded as a function of time. The sensor resistance in the presence of added acetone decreased in comparison with that in synthetic air because of the interactions between acetone and urea thiol 1 on the surface of the AuNPs. The sensor response is defined by the following equation for resistance decrease from air to analyte:2,3
| Response = (Rair − Rgas)/Rgas |
Fig. 2 shows the responses of the urea thiol-AuNPs sensor for sensing acetone in air at a concentration range of 10 ppb to 10 ppm. As the concentration of acetone increases, the sensor resistance decreases and thus the response increases. The urea thiol-AuNPs sensor is sensitive toward acetone at a concentration as low as 10 ppb, a level of detection more sensitive than most current metal oxide-based acetone sensing devices and thiol functionalized AuNPs sensors.2–12,30–32 Furthermore, the response increased with increasing acetone concentration up to 10 ppm, thus demonstrating a wide dynamic range of measurements.
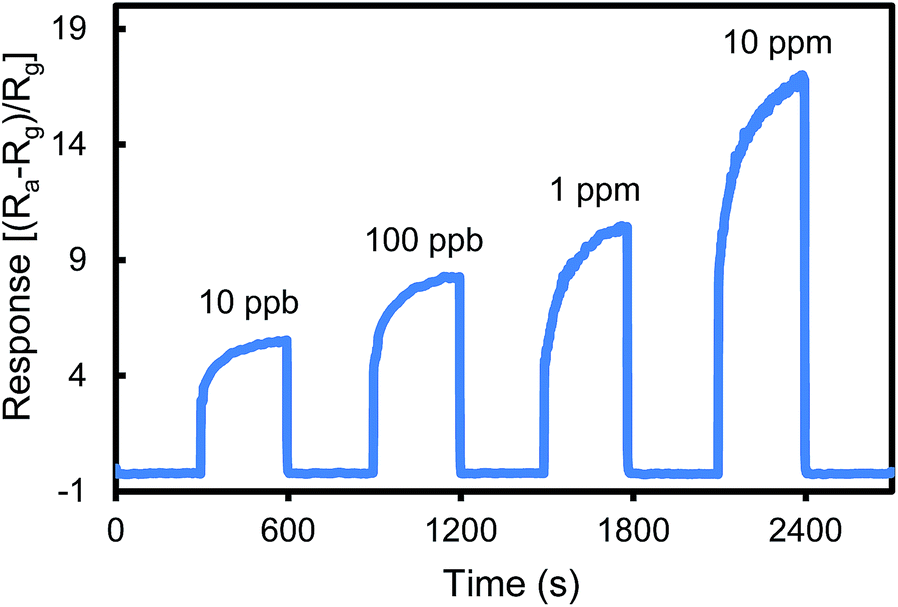 | ||
| Fig. 2 Response of the urea thiol 1 AuNPs sensor to acetone exposure at 10 ppb to 10 ppm. | ||
Given this high sensitivity toward acetone, we set out to test the carbonyl-sensing selectivity of the urea thiol-AuNPs sensor by measuring the sensor responses to non-carbonyl VOCs. Fig. 3 shows linear relationships between the sensor response and the common logarithm of the concentrations of acetone, ethanol, benzene, ethylbenzene and water, ranging from 10 ppb to 10 ppm. As the slope of the linear regression curve is a direct measure of sensitivity, the relatively flat slopes obtained for the non-carbonyl analytes (all slopes below 1.6) imply that the sensor discriminates in its interactions with VOCs. The considerably stronger response to acetone (slope = 4.6) suggests the hydrogen bonding design concept for acetone is operative and imparts both sensitivity and selectivity. The sensor is not sensitive to ethanol and water moisture in the tested concentration range, but is relatively sensitive to benzene. The large slope difference between the sensor response curves of acetone and benzene shown in Fig. 3 may provide a way to distinguish these two compounds by a series of simple dilutions of the samples to obtain the slopes of the response curves. For water and ethanol concentration even at a high concentration of 10 ppm, the responses of the sensor are smaller than that for sensing 10 ppb acetone. Therefore, the interference of water and ethanol can be negligible up to 10 ppm for sensing acetone in air.
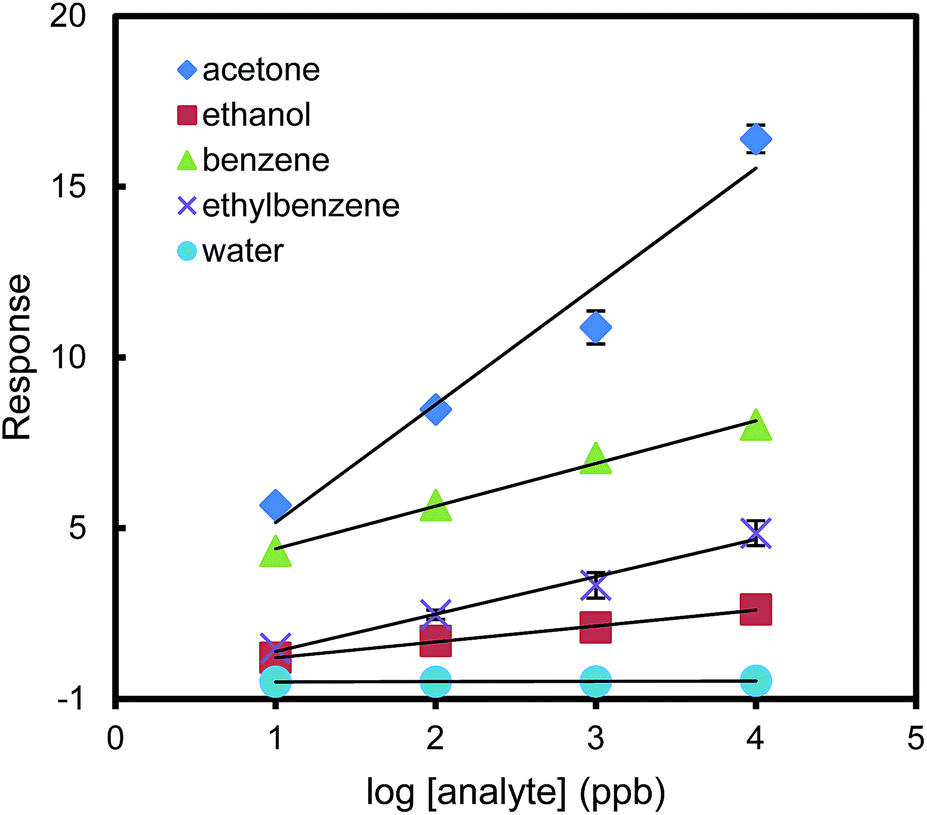 | ||
| Fig. 3 Sensor response versus log[analyte (ppb)] in detection of acetone, ethanol, benzene, ethylbenzene and water vapor. The responses are nearly linear (R2 > 0.97). Error bars indicate the variability among three different sensors. | ||
Fig. 4 shows a schematic illustration of a proposed model of the urea thiol molecular self-assembly and interaction with acetone on the surfaces of AuNPs. The urea motifs form hydrogen bonds among adjacent chains on the surfaces of AuNPs as well as hydrogen bonds with incoming acetone. It is reasonable to expect that hydrogen bonding between adjacent ureas is weakened when bulky tert-butyl groups are placed on nitrogen of the urea functionality,43 thus accommodating more acetone as its concentration increases.
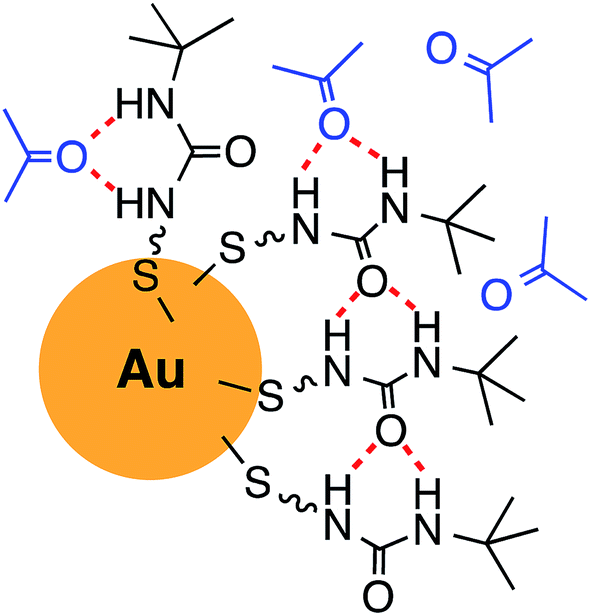 | ||
| Fig. 4 Schematic illustration of the urea thiol self-assembly on the surface of a AuNP and interaction with acetone. | ||
We surmise that the urea thiol is sensitive to acetone and insensitive to ethanol and water due to the strong molecular interactions of the carbonyl moieties with the urea motifs, whereas ethanol and water may have weaker interactions with the urea motif due to stronger intermolecular interactions with other ethanol or water molecules. The higher responses to benzene and ethyl benzene in comparison with ethanol and water suggest that benzene and ethyl benzene may nest within the hydrophobic chain domains of the urea thiol network in the absence of any significant intermolecular interactions among these analytes.
To further escalate the role of the H-bonding motif of the urea thiol in recognition of carbonyl moieties, the urea thiol-AuNPs sensor was compared with AuNPs sensors derived from different thiols, namely 1-dodecanethiol, 4-MTT, and 11-MUA shown in Table 1. Fig. 5 shows that the commercial thiol-derived AuNPs sensors respond to acetone vapor with considerably lower sensitivities than the urea thiol-AuNPs sensor. Indeed, at a general concentration of acetone of 1 ppm, the response of the urea thiol AuNPs sensor was ∼10, 12 and 23 times higher than the 1-dodecanethiol-, 4-MTT and 11-MUA-functionalized AuNPs sensors, respectively. These results strongly support that incorporation of terminal functionality on a thiol monolayer surrounding AuNPs can be used to confer sensitivity and selectivity in analyte detection. Along these lines, the sensor derived from 4-MTT responded selectively to ethanol, albeit at a substantially lower response relative to the urea thiol-derived sensor in its detection of acetone.38 Fig. 6 shows the responses of the sensors derived from AuNPs coated with the four thiols in detection of a mixture of 1 ppb benzene, 1 ppb ethanol and acetone varied from 1 ppb to 100 ppb. 1 ppb of interfering VOCs was chosen because normally the concentrations of these VOCs in environmental air are lower than 1 ppb. The urea thiol AuNPs sensor responded with increases in acetone concentration, as shown in Fig. 3. Responses to VOC mixtures were higher than that of only acetone spiked into synthetic air. The contribution of benzene to the sensor response for a mixture of 1 ppb each of benzene, ethanol and acetone likely is larger than acetone. However, in comparison, the other thiol-derived sensors showed little sensitivity to VOC mixtures with increasing acetone concentrations.
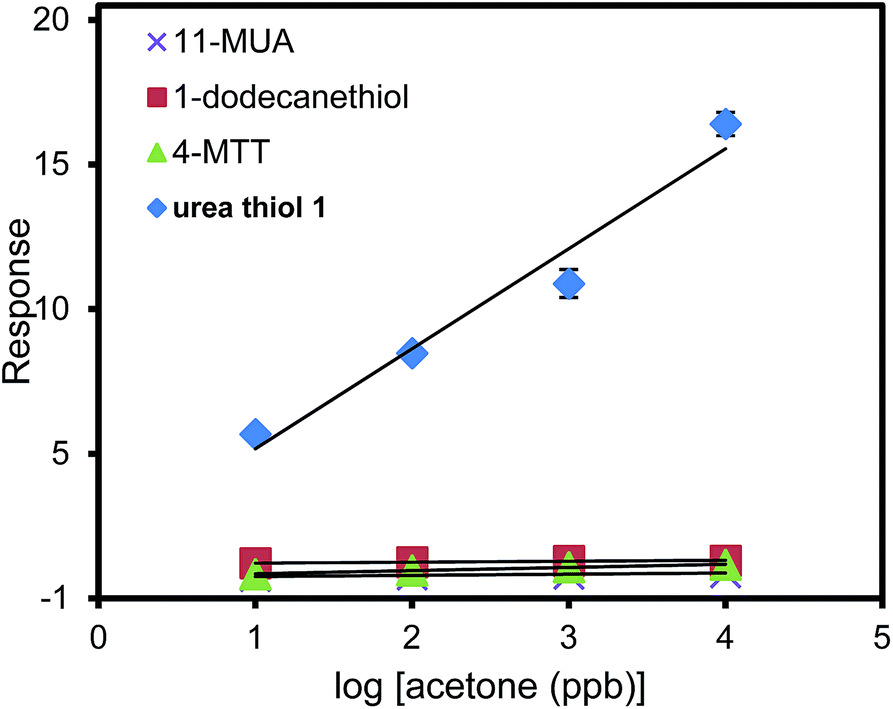 | ||
| Fig. 5 Responses for sensors derived from AuNPs coated with urea thiol 1, 1-dodecanethiol, 4-methoxy-α-toluenethiol (4-MTT) or 11-mercaptoundecanoic acid (11-MUA) in detection of acetone. | ||
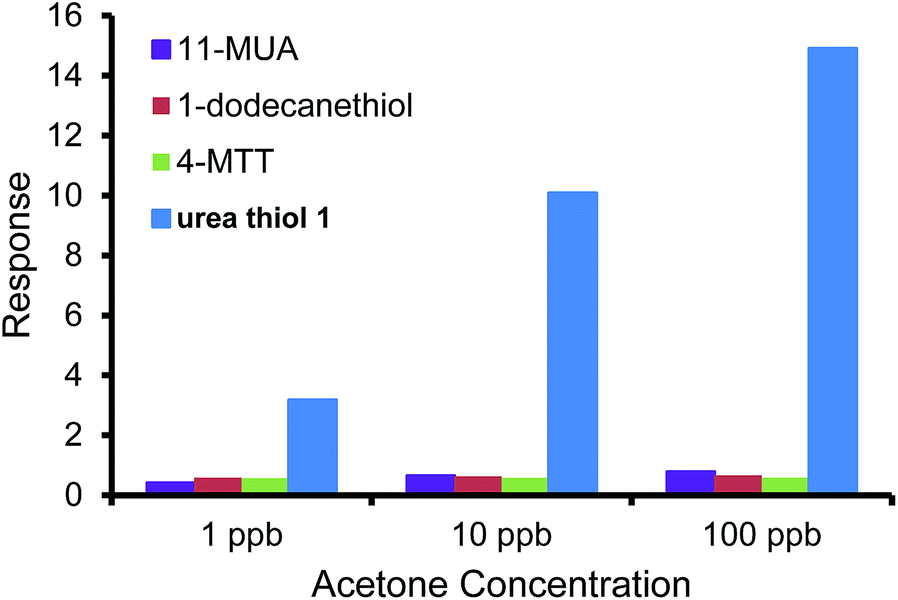 | ||
| Fig. 6 The responses of sensors derived from AuNPs coated with urea thiol 1, 1-dodecanethiol 4-methoxy-α-toluenethiol (4-MTT) and 11-mercaptoundecanoic acid (11-MUA) in detection of a mixture of 1 ppb benzene, 1 ppb ethanol and acetone varied from 1 ppb to 100 ppb. | ||
On comparing the acetone sensing performance of the urea thiol 1-derived sensor to other chemiresistors reported in the literature,2–12,23–32 we noted differences in several key parameters that underscore the advantages of the present design. First, an operating temperature under ambient conditions is preferable. Second, the present design features high sensitivity with a low limit of detection and a wide dynamic range for acetone detection, as would be needed for the challenging analytical application of breath analysis. This present study provides a paradigm using size complementary thiol-urea H-bond receptor functionalized AuNPs to enhance affinity and selectivity of a chemiresistor for sensing carbonyl VOCs.
Conclusion
The urea-functionalized thiol AuNPs sensor described herein provided a high sensitivity and good relative selectivity for detection of acetone in reversible and reproducible manner at ambient temperature. Gas sensing measurements reveal that this sensor shows the lowest limit of detection to acetone among the reported chemiresistors to date. The concept of designed thiols for selective interaction with target analytes based on functional group interactions, such as urea-carbonyl hydrogen bonding, may well serve as a powerful strategy to improve chemiresistor sensitivity and selectivity in detection of other VOC classes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support by NIH (P42 ES023716).References
- C. Turner, P. Spanel and D. Smith, Physiol. Meas., 2006, 27, 321–337 CrossRef PubMed.
- M. Righettoni, A. Tricoli and S. E. Pratsinis, Anal. Chem., 2010, 82, 3581–3587 CrossRef CAS PubMed.
- L. P. Qin, J. Q. Xu, X. W. Dong, Q. Y. Pan, Z. X. Cheng, Q. Xiang and F. Li, Nanotechnology, 2008, 19, 185705 CrossRef PubMed.
- S. H. Yan, S. Y. Ma, W. Q. Li, X. L. Xu, L. Cheng, H. S. Song and X. Y. Liang, Sens. Actuators, B, 2015, 221, 88–95 CrossRef CAS.
- B. Bhowmik, V. Manjuladevi, R. K. Gupta and P. Bhattacharyya, IEEE Sens. J., 2016, 16, 3488–3495 CAS.
- V. K. Tomer and S. Duhan, J. Mater. Chem. A, 2016, 4, 1033–1043 RSC.
- N. H. Al-Hardan, M. J. Abdullah and A. A. Aziz, Appl. Surf. Sci., 2013, 270, 480–485 CrossRef CAS.
- R. Rella, J. Spadavecchia, M. G. Manera, S. Capone, A. Taurino, M. Martino, A. P. Caricato and T. Tunno, Sens. Actuators, B, 2007, 127, 426–431 CrossRef CAS.
- L. Geng, Y. Q. Zhao, X. L. Huang, S. R. Wang, S. M. Zhang and S. H. Wu, Sens. Actuators, B, 2007, 120, 568–572 CrossRef CAS.
- J. S. Park, X. P. Shen and G. X. Wang, Sens. Actuators, B, 2009, 136, 494–498 CrossRef CAS.
- A. Teleki, S. E. Pratsinis, K. Kalyanasundaram and P. I. Gouma, Sens. Actuators, B, 2006, 119, 683–690 CrossRef CAS.
- C. Q. Ge, C. S. Xie and S. Z. Cai, Mater. Sci. Eng., B, 2007, 137, 53–58 CrossRef CAS.
- E. S. Snow, F. K. Perkins and J. A. Robinson, Chem. Soc. Rev., 2006, 35, 790–798 RSC.
- P. Qi, O. Vermesh, M. Grecu, A. Javey, O. Wang, H. J. Dai, S. Peng and K. J. Cho, Nano Lett., 2003, 3, 347–351 CrossRef CAS.
- N. J. Pinto, I. Ramos, R. Rojas, P. C. Wang and A. T. Johnson, Sens. Actuators, B, 2008, 129, 621–627 CrossRef CAS.
- C. L. Li, N. Chartuprayoon, W. Bosze, K. Low, K. H. Lee, J. Nam and N. V. Myung, Electroanalysis, 2014, 26, 711–722 CrossRef CAS.
- S. Vallejos, I. Gracia, J. Bravo, E. Figueras, J. Hubalek and C. Cane, Talanta, 2015, 139, 27–34 CrossRef CAS PubMed.
- L. Han, X. J. Shi, W. Wu, F. L. Kirk, J. Luo, L. Y. Wang, D. Mott, L. Cousineau, S. I. I. Lim, S. Lu and C. J. Zhong, Sens. Actuators, B, 2005, 106, 431–441 CrossRef CAS.
- H. Ahn, A. Chandekar, B. Kang, C. Sung and J. E. Whitten, Chem. Mater., 2004, 16, 3274–3278 CrossRef CAS.
- G. Peng, U. Tisch, O. Adams, M. Hakim, N. Shehada, Y. Y. Broza, S. Billan, R. Abdah-Bortnyak, A. Kuten and H. Haick, Nat. Nanotechnol., 2009, 4, 669–673 CrossRef CAS PubMed.
- U. Tisch and H. Haick, Rev. Chem. Eng., 2010, 26, 171–179 CAS.
- E. Chow, K. H. Muller, E. Davies, B. Raguse, L. Wieczorek, J. S. Cooper and L. J. Hubble, J. Phys. Chem. C, 2010, 114, 17529–17534 CrossRef CAS.
- H. Wohltjen and A. W. Snow, Anal. Chem., 1998, 70, 2856–2859 CrossRef CAS.
- H. L. Zhang, S. D. Evans, J. R. Henderson, R. E. Miles and T. H. Shen, Nanotechnology, 2002, 13, 439–444 CrossRef CAS.
- N. Krasteva, I. Besnard, B. Guse, R. E. Bauer, K. Mullen, A. Yasuda and T. Vossmeyer, Nano Lett., 2002, 2, 551–555 CrossRef CAS.
- F. J. Ibanez, U. Gowrishetty, M. M. Crain, K. M. Walsh and F. P. Zamborini, Anal. Chem., 2006, 78, 753–761 CrossRef CAS PubMed.
- F. J. Ibanez and F. P. Zamborini, ACS Nano, 2008, 2, 1543–1552 CrossRef CAS PubMed.
- Y. Joseph, B. Guse, T. Vossmeyer and A. Yasuda, J. Phys. Chem. C, 2008, 112, 12507–12514 CrossRef CAS.
- F. I. Bohrer, E. Covington, C. Kurdak and E. T. Zellers, Anal. Chem., 2011, 83, 3687–3695 CrossRef CAS PubMed.
- J. K. Kim, Y. S. Yang, S. C. Ha, S. M. Cho, Y. S. Kim, H. Y. Kim, H. Yang and Y. T. Kim, Sens. Actuators, B, 2005, 106, 189–198 CrossRef.
- S. M. Briglin, T. Gao and N. S. Lewis, Langmuir, 2004, 20, 299–305 CrossRef CAS PubMed.
- F. J. Ibanez and F. P. Zamborini, Small, 2012, 8, 174–202 CrossRef CAS PubMed.
- G. Konavalina and H. Haick, ACS Appl. Mater. Interfaces, 2012, 4, 317–325 CrossRef PubMed.
- G. Konvalina and H. Haick, Acc. Chem. Res., 2014, 47, 66–76 CrossRef CAS PubMed.
- X. Z. Luo, X. J. Jia, J. H. Deng, J. L. Zhong, H. J. Liu, K. J. Wang and D. C. Zhong, J. Am. Chem. Soc., 2013, 135, 11684–11687 CrossRef CAS PubMed.
- H. Wang, B. Li, H. Wu, T. L. Hu, Z. Yao, W. Zhou, S. Xiang and B. Chen, J. Am. Chem. Soc., 2015, 137, 9963–9970 CrossRef CAS PubMed.
- M. Kaftory, M. Kapon and M. Boroshansky, Chem. Mater., 1994, 6, 1245–1249 CrossRef CAS.
- Z. Xie, M. V. R Raju, B. B. Brown, A. C. Stewart, M. H. Nantz and X. A. Fu, Transducers, 2017, 1425–1428 Search PubMed.
- S. Park and M. N. Yousaf, Langmuir, 2008, 24, 6201–6207 CrossRef CAS PubMed.
- J. Willwacher, S. Rakshit and F. Glorius, Org. Biomol. Chem., 2011, 9, 4736–4740 RSC.
- N. Dendane, A. Hoang, L. Guillard, E. Defrancq, F. Vinet and P. Dumy, Bioconjugate Chem., 2007, 18, 671–676 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 7, 801–802 RSC.
- R. Paulini, B. L. Frankamp and V. M. Rotello, Langmuir, 2002, 18, 2368–2373 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |