
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalixarene alpha-ketoacetylenes: versatile platforms for reaction with hydrazine nucleophile†
Anton A. Muravev *a,
Svetlana E. Solovieva*ab,
Farida B. Galievab,
Olga B. Bazanovaa,
Ildar Kh. Rizvanova,
Kamil A. Ivshinb,
Olga N. Kataevaab,
Susan E. Matthewsc and
Igor S. Antipinab
*a,
Svetlana E. Solovieva*ab,
Farida B. Galievab,
Olga B. Bazanovaa,
Ildar Kh. Rizvanova,
Kamil A. Ivshinb,
Olga N. Kataevaab,
Susan E. Matthewsc and
Igor S. Antipinab
aArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center of RAS, Kazan, 420088 Russia. E-mail: antonm@iopc.ru; svsol@iopc.ru
bA.M. Butlerov Institute of Chemistry, Kazan Federal University, Kazan, 420008 Russia
cSchool of Pharmacy, University of East Anglia, Norwich, NR4 7TJ UK
First published on 21st September 2018
Abstract
Late stage diversification of calix[4]arenes and thiacalix[4]arenes with heterocycles remains a significant synthetic challenge and hampers further exploitation of the scaffolds. Here we describe the development of a short and facile synthetic route to conformationally diverse novel calix[4]arene and thiacalix[4]arene ynones using a palladium cross coupling approach (5% Pd(II) + 10% Cu(I)) with benzoyl chloride. Their successful conversion to heterocycles to afford pyrazoles was demonstrated through treatment with hydrazine. Functionalisation is calixarene conformation and linker independent enabling access to a library of structures.
Introduction
(Thia)calix[4]arenes are a widely investigated platform for molecular recognition due to the ease of their preparation and functionalisation through regio-, stereo- and iteroselective modification1–5 and the ability to immobilise in four distinct conformations.6 These macrocycles not only provide a cavity for inclusion of various guest species (such as fullerenes7 and dyes8), but also assist coordination (small ions9 to large biomolecules10) by means of receptor groups, which form a binding site for a specific substrate.11–14 The latter often leads to a synergetic effect, or multivalency,15 in such applications16 as biological recognition, sensing, luminescence, magnetism, catalysis, and membrane separation.Heterocycles, including triazoles,17 imidazoles,18 pyridines19 and pyrroles20 are some of the most extensively investigated moieties on a calixarene framework,21 however extension to more diverse rings remains challenging. Two fundamental approaches are available for inclusion of such functionality; introduction of a pre-assembled moiety (Fig. 1a) or in situ modification of an activated calixarene (Fig. 1b and c). The former route is convenient only when the heterocycle is stable under the employed reaction conditions or does not require the use of protecting groups for example terpyridines22,23 or thiophenes.24
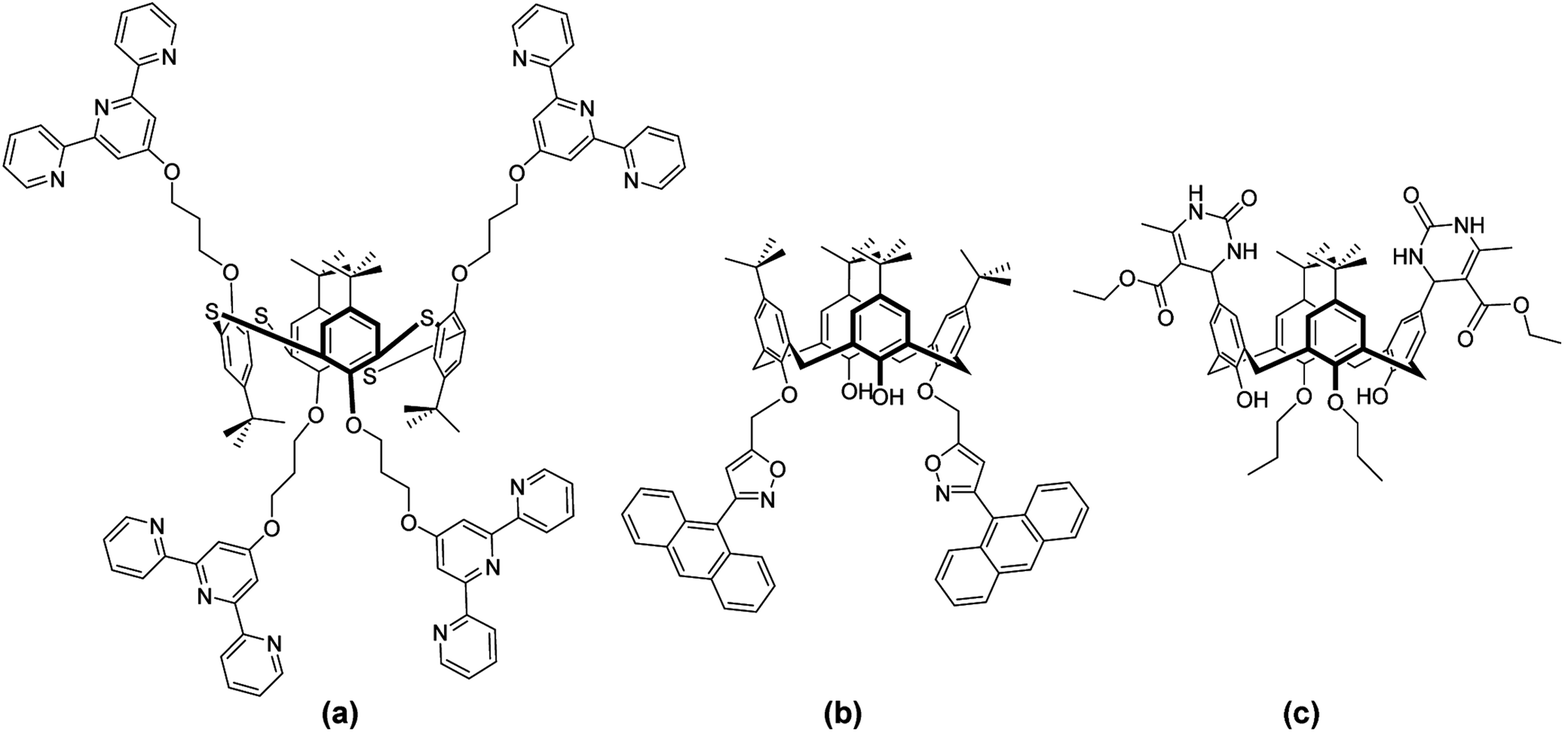 | ||
| Fig. 1 Heterocycle derivatised (thia)calixarenes. | ||
The most extensively studied example of the second approach is the functionalisation of calixarenes with alkynes. This route has offered up a number of derivatisation possibilities through participation in pericyclic reactions (Fig. 1b) with nitrile oxides25 and azides26–28 yielding isoxazoles and triazoles. Additional examples include the reaction of calixarene β-amidoamines with triethylamine to afford imidazolines29 and the three-component Biginelli reaction (Fig. 1c) of calixarene aldehydes with urea and β-ketoesters to give dihydropyrimidines.30 However, these methods offer limited diversity of heterocycle structure and a particular challenge is the avoidance of competitive inter and intramolecular reactions leading to mixed, bridged31–34 or multimeric products.
To-date conversion to ynones and subsequent nucleophilic addition–elimination reactions have not been employed on macrocycles. This approach offers a rapid entry to a variety of nitrogen- and oxygen-containing heterocycles35 through diverse cyclization pathways from a single precursor. In contrast to small molecules, a challenge to developing this approach on (thia)calixarenes is the avoidance of inter- or intramolecular crosslinking on reaction with nucleophiles; this is particularly important, when polytopic platforms are considered.
Here we describe the successful functionalisation of (thia)calixarenes with multiple ynones and demonstrate their potential for heterocycle formation through subsequent controlled reaction with hydrazine to produce pyrazoles without crosslinking.
Results and discussion
There are numerous approaches to the preparation of ketoacetylenes: the addition of lithium acetylenides to aromatic aldehydes with subsequent oxidation of benzyl alcohol by MnO2,36 alkynylation of benzaldehydes by phenylalkynyl iodides in the presence of iridium,37 and cross-coupling of carboxylic acid chloroanhydrides with terminal aryl and hetaryl acetylenes,38 to name just a few. Despite this to-date, only a single example of a calixarene ynone has been reported, where upper-rim functionalisation was achieved through coupling of an aldehyde with a Grignard salt in the presence of Jones reagent.39 Functionalisation of the more synthetically useful lower-rim of calixarenes has not been reported.In this study, the per functionalisation of the lower-rim of calix- and thiacalix[4]arenes with ynones has been investigated through derivatisation of alkyne precursors. Alkynyl derivatives of (thia)calixarenes have been extensively exploited for click chemistry and are readily accessible in a range of conformations.40
The presence of sulphide bridges, in thiacalixarenes, creates an additional challenge to functionalisation and necessitates the use of milder conditions. In particular, the sulphur bridge in thiacalixarenes excludes the possibility of general ynone synthesis using organolithium reagents and MnO2 oxidation. Additionally, the sulphur atoms may coordinate palladium and copper and require use of higher quantities in the cross-coupling reactions as catalysts along with possible steric hindrance towards the attack on the carbon–carbon triple bond.
We employed various routes to increase the reactivity of the functional groups during ynone formation (Table 1) using 1c in the 1,3-alternate stereoisomeric form as a model calix[4]arene, due to the larger distance between alkynyl groups compared to cone and partial cone stereoisomers (Scheme S1†). Interestingly, reaction of 1c40a with benzoyl chloride and cyanuric chloride (TCT) in the presence of magnesium bromide as a Lewis acid,41 to enhance the activity of carbonyl component, was unsuccessful (Table 1). Equally activation of the C–C triple bond with trimethylaluminum42 proved ineffective. Mixed results were seen with copper complexes,43,44 with the reaction either not proceeding with the DABCO complex of Cu(I)43 or yielding a hardly separable mixture of the products of cross-coupling and acetylide Glaser homocoupling in the presence of copper(I) iodide.44
| Reagent | Catalyst | t, °C | Solvent | Yield |
|---|---|---|---|---|
| a Mixture of the products of cross-coupling and acetylide Glaser homocoupling. | ||||
| PhCOONa | 2 eq. TCT/MgBr2 | 80 | MeCN | No reaction |
| PhC(O)Cl | Me3Al | 80 | TEA/PhCH3 | No reaction |
| Cu(I) | 80 | TEA/PhCH3 | —a | |
| Cu(I) + DABCO | 80 | TEA/PhCH3 | No reaction | |
| 5% Pd(0) + 10% Cu(I) | 20 | TEA/THF | Mixture | |
| Pd(0) | 20 | TEA/THF | Mixture | |
| 5% Pd(II) + 10% Cu(I) | 20 | TEA/THF | 91% of 2c | |
In contrast, employment of palladium catalysis successfully yielded the desired ynones. Low reproducibility of the yields was noted when using air- and moisture-sensitive tetrakis(triphenylphosphine)palladium(0) as a catalyst. However, the use of 5% Pd(II) and 10% Cu(I) as the catalytically system in tetrahydrofuran45 gave reproducibly high yield of the target ynone 2c. Variation of the reaction time under these conditions showed that full conversion of 1c is achieved within 1.5 h, while an increase in the time of reaction does not increase the yield.
In summary, Pd(II)/Cu(I) catalyst system is the most reactive among the conditions under study, enabling full conversion of all the alkyne groups of calixarene 1c to ynones with a reduced quantity and number of side products compared to copper(I) or Pd(0) catalysis conditions.
Thus, we carried out palladium/copper-catalysed cross-coupling of calix[4]arene in the cone, partial cone, and 1,3-alternate conformations of 1 with benzoyl chloride (an example synthesis is given in Scheme 1) giving 2a, b and c in 79%, 88% and 91% yields, respectively. The reaction was extended to the thiacalix[4]arene derivatives 3 from 1d–1g46 using the same methodology to give an extended family of derivatives in which the distance between the macrocyclic core and the acetylenic function can be varied (Scheme 2).
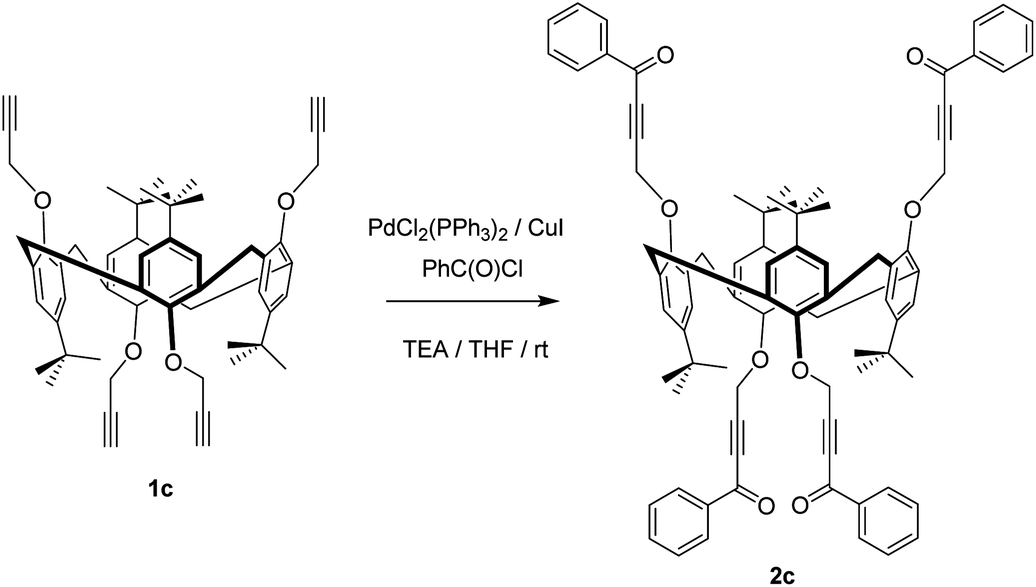 | ||
| Scheme 1 Example synthesis of ketoacetylene 2c from alkynyl precursor 1c. | ||
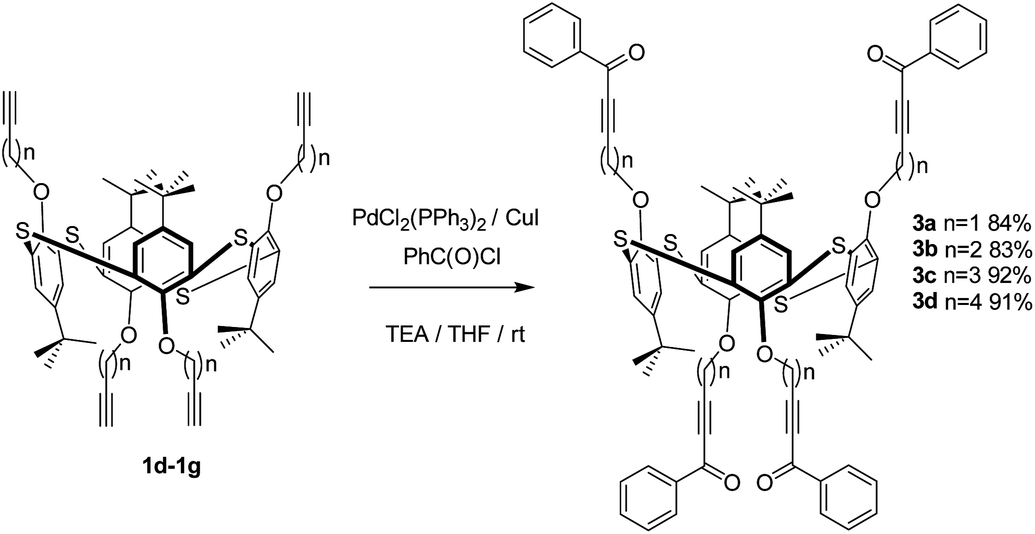 | ||
| Scheme 2 Synthesis of thiacalix[4]arene ketoacetylenes 3. | ||
Successful cross-coupling was indicated by the presence of the characteristic aromatic multiplets of the benzoyl substituents, the loss of the triplet associated with the terminal acetylenic proton and a simplification of the splitting pattern of oxymethylene groups (Fig. 4 and ESI†) in the 1H NMR spectra of products 2 and 3. Full derivatization was confirmed by HRMS data in the presence of CsCl.
The structure of compound 2c was also determined by X-ray single diffraction (Fig. 2). Compound 2c crystallizes in space group I41/a, with the asymmetric part of the unit cell containing 1/4 of the molecule. The whole molecule is obtained by a series of symmetry operations, including 4-fold rotoinversion and 2-fold axes, thus 2c adopts symmetrical 1,3-alternate conformation. The crystal packing is determined by multiple weak CH⋯O and CH⋯π interactions (Fig. 3).
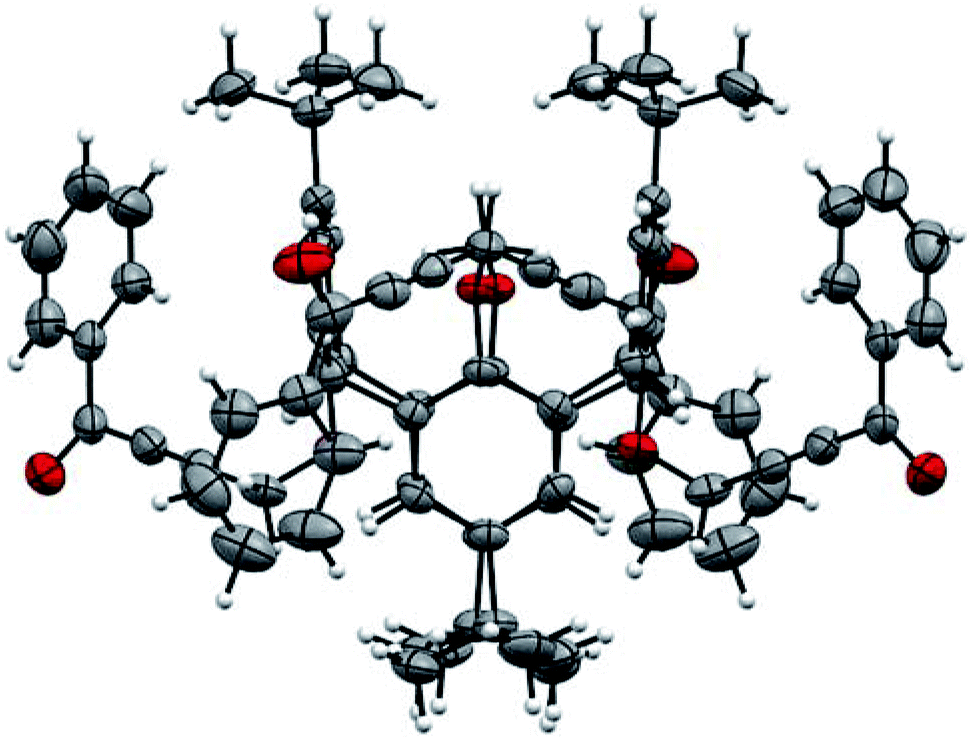 | ||
| Fig. 2 The molecular geometry of compound 2c. | ||
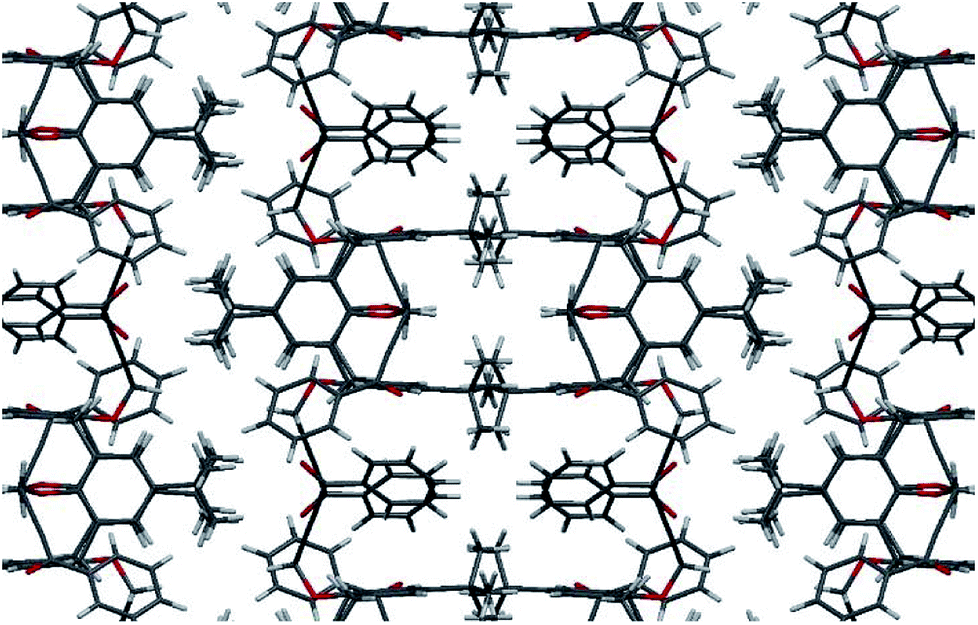 | ||
| Fig. 3 A fragment of crystal packing of compound 2c, view along b axis. | ||
Ketoacetylenes offer diverse cyclization pathways to a range of nitrogen- and oxygen-containing heterocycles35 with acidic protons including pyrazoles,47 pyrroles,48 pyrimidines.49 Treatment with the highly reactive hydrazine45 in refluxing isobutanol for 4 h (Scheme 3) to form pyrazoles was investigated on 2. Surprisingly, in spite of the close arrangement of ynone fragments on the calixarene platform and the possibility of their intramolecular crosslinking by hydrazine molecules, the reaction afforded only the pyrazole derivatives 4. These could be isolated by simple filtration, in 69–87% yields. Interestingly there is a direct dependence of the yields of pyrazoles with the distance between the ynones with the yield being lowest for the cone and highest for the 1,3-alternate.
 | ||
| Scheme 3 Example synthesis of pyrazoles 4. | ||
Pyrazole ring formation instead of crosslinking could be related to at least one of three factors.
(1) Kinetic effect, where the attack of the NH2 fragment on the terminal carbon of the carbon–carbon triple bond is faster than rearrangement of the neighboring carbonyl group from the same or another calixarene molecule.
(2) Thermodynamic effect, in which pyrazole ring formation is entropically more favorable and has a lower energy than any of crosslinked products.
(3) That the amino group in hydrazine is more nucleophilic than the amino group near the imine nitrogen formed after nucleophilic attack of hydrazine on carbonyl group of ynone fragment and dehydration. Assuming a large excess of hydrazine with respect to ynone fragments, this would result in a two-stage mechanism, namely, attack of four hydrazine molecules on the four carbonyl groups of the ynone derivative of calixarene followed by ring closures into four pyrazole fragments.
We postulate that the third factor is the most important as it is less dependent on the temperature of the reaction and concentrations of the reagents than the former two factors.
The structure assignment of pyrazoles 4a and 4c, fixed in the cone and 1,3-alternate conformations is straightforward due to high symmetry resulting in equivalency of the phenol rings. (ESI, Fig. S30 and S41†). The key signals for formation of the pyrazole being found at 6.33 and 6.26 ppm, respectively. IR spectra of all the pyrazoles 4 display absorption bands at ≈3350 and ≈1480 cm−1 corresponding to stretching vibrations of N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds of the pyrazole ring (Fig. S29, S34 and S40†). High-resolution MALDI mass spectra of the pyrazoles 4 mixed with CsCl salt (Fig. S33, S39 and S43†) show the only peak of the product at m/z 1405.5927 (4a), 1405.5915 (4b), and 1405.6020 (4c), while no peaks of the compounds with terminal alkyne groups were observed, which indicates full conversion of the alkynone precursors 2.
N bonds of the pyrazole ring (Fig. S29, S34 and S40†). High-resolution MALDI mass spectra of the pyrazoles 4 mixed with CsCl salt (Fig. S33, S39 and S43†) show the only peak of the product at m/z 1405.5927 (4a), 1405.5915 (4b), and 1405.6020 (4c), while no peaks of the compounds with terminal alkyne groups were observed, which indicates full conversion of the alkynone precursors 2.
In the case of partial cone stereoisomer 4b, there is a significantly more complex splitting pattern in the 1H NMR spectrum due to the presence of three different phenolic rings (I, II and III) (Fig. 4). The conversion to the pyrazole is accompanied by large shifts in the oxymethylene (H8) protons and the appearance of three peaks for the pyrazole proton (H10) at 6.34, 5.98 and 5.75 ppm in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio. Retention of the partial cone conformation is clearly indicated by the presence of two partially overlapped AB quadruplets in the region 4.02–3.90 and at 3.03 ppm. Full characterization was achieved through 13C/1H HMBC, HSQC and described in Suppl. Information (Fig. S37 and S38†).
:1:1 ratio. Retention of the partial cone conformation is clearly indicated by the presence of two partially overlapped AB quadruplets in the region 4.02–3.90 and at 3.03 ppm. Full characterization was achieved through 13C/1H HMBC, HSQC and described in Suppl. Information (Fig. S37 and S38†).
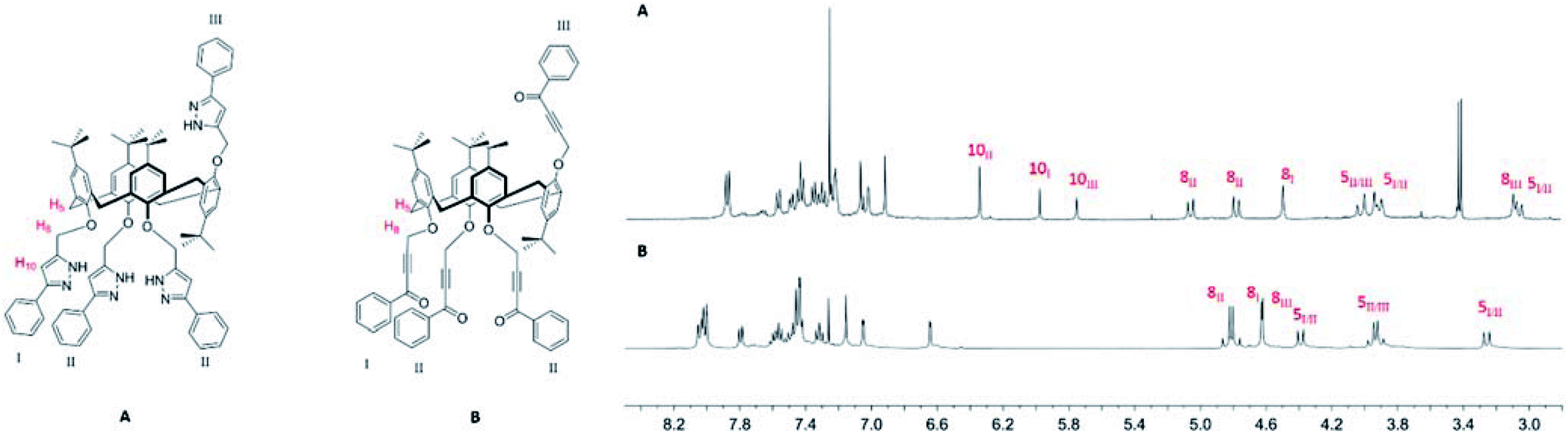 | ||
| Fig. 4 Partial 1H NMR spectra of compounds 4b (A) and 2b (B) (CDCl3, 400 MHz, 298 K). | ||
Conclusions
An optimal palladium catalysed cross-coupling method has been developed for the preparation of ynones on (thia)calixarenes from the common alkyne precursors. Treatment with hydrazine, as a model nucleophile, yielded the desired pyrazoles in high yield, without inter or intra molecular crosslinking, demonstrating the suitability of the ynone approach for heterocycle formation on these platforms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Russian Foundation for Basic Research (project no. 17-03-00389-a). X-ray diffraction studies were funded by the subsidy allocated to Kazan Federal University for the state assignment in the sphere of scientific activities (4.1493.2017/4.6 and 4.5151.2017/6.7). The authors thank the Royal Society (IEC\R2\170142) for funding work undertaken in S. E. M.’s laboratory.Notes and references
- Calixarenes and Beyond, ed. P.Neri, J. L.Sessler and M.-X.Wang, Springer, New York, 2016 Search PubMed.
- C. D. Gutsche, Calixarenes. An Introduction, Royal Society of Chemistry, Cambridge, 2008 Search PubMed.
- R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824–4870 RSC.
- R. Lavendomme, S. Zahim, G. de Leener, A. Inthasot, A. Mattiuzzi, M. Luhmer, O. Reinaud and I. Jabin, Asian J. Org. Chem., 2015, 4, 710–722 CrossRef.
- S. E. Solovieva, V. A. Burilov and I. S. Antipin, Macroheterocycles, 2017, 10, 134–146 CrossRef.
- K. Iwamoto, K. Araki and S. Shinkai, J. Org. Chem., 1991, 56, 4955–4962 CrossRef.
- X. Chen, R. A. Boulos, A. S. Slattery, J. L. Atwood and C. L. Raston, Chem. Commun., 2015, 51, 11413–11416 RSC.
- A. Credi, S. Dumas, S. Silvi, M. Venturi, A. Arduini, A. Pochini and A. Secchi, J. Org. Chem., 2004, 69, 5881–5887 CrossRef PubMed.
- J. Vicens, J. Inclusion Phenom. Macrocyclic Chem., 2006, 55, 193–196 CrossRef.
- T. Grandjean, E. Gillon, S. Cecioni, C. Abderrahmen, K. Faure, D. Redelberger, E. Kipnis, E. Dessein, S. Havet, B. Darblade, S. E. Matthews, S. de Bentzmann, B. Guery, B. Cournoyer, A. Imberty and S. Vidal, J. Med. Chem., 2014, 57, 10275–10289 CrossRef PubMed.
- B. S. Creaven, D. F. Donlon and J. McGinley, Coord. Chem. Rev., 2009, 253, 893–962 CrossRef.
- Y. Bi, S. Du and W. Liao, Coord. Chem. Rev., 2014, 276, 61–72 CrossRef.
- A. Ovsyannikov, S. Solovieva, I. Antipin and S. Ferlay, Coord. Chem. Rev., 2017, 352, 151–186 CrossRef.
- A. Acharya, K. Samanta and C. P. Rao, Coord. Chem. Rev., 2012, 256, 2096–2125 CrossRef.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. -W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef PubMed.
- Multivalency: Concepts, Research & Applications, ed. J.Huskens, L. J.Prins, R.Haag and B. J.Ravoo, Wiley, Chichester, 2018 Search PubMed.
- R. Takahashi, S. Matsumoto, S. Fujii, T. Narayanan and K. Sakurai, Angew. Chem., Int. Ed., 2017, 56, 6734–6738 CrossRef PubMed.
- O. Bistri, B. Colasson and O. Reinaud, Chem. Sci., 2012, 3, 811–818 RSC.
- G. Thiabaud, G. Guillemot, I. Schmitz-Afonso, B. Colasson and O. Reinaud, Angew. Chem., Int. Ed., 2009, 48, 7383–7386 CrossRef PubMed.
- S. K. Kim, V. M. Lynch, N. J. Young, B. P. Hay, C.-H. Lee, J. S. Kim, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2012, 134, 20837–20843 CrossRef PubMed.
- S. Kumar, V. Luxami and H. Singh, Adv. Heterocycl. Chem., 2009, 97, 219–290 CrossRef.
- A. A. Muravev, F. B. Galieva, O. B. Bazanova, D. R. Sharafutdinova, S. E. Solovieva, I. S. Antipin and A. I. Konovalov, Supramol. Chem., 2016, 28, 589–600 CrossRef.
- A. A. Muravev, V. A. Burilov, S. E. Solov’eva, A. G. Strel’nik, S. K. Latypov, O. B. Bazanova, D. R. Sharafutdinova, I. S. Antipin and A. I. Konovalov, Russ. Chem. Bull., 2014, 63, 214 CrossRef.
- C. Song and T. M. Swager, Org. Lett., 2008, 10, 3575–3578 CrossRef PubMed.
- N. J. Maher, H. Diao, J. O'Sullivan, E. Fadda, F. Heaney and J. McGinley, Tetrahedron, 2015, 71, 9223–9233 CrossRef.
- A. A. Muravev, A. I. Laishevtsev, F. B. Galieva, O. B. Bazanova, I. K. Rizvanov, A. Korany, S. E. Solovieva, I. S. Antipin and A. I. Konovalov, Macroheterocycles, 2017, 10, 203–214 CrossRef.
- V. A. Burilov, D. A. Mironova, R. R. Ibragimova, S. E. Solovieva, I. S. Antipin and B. Konig, RSC Adv., 2015, 5, 101177–101185 RSC.
- V. Burilov, A. Valiyakhmetova, D. Mironova, R. Safiullin, M. Kadirov, K. Ivshin, O. Kataeva, S. Solovieva and I. S. Antipin, RSC Adv., 2016, 6, 44873–44877 RSC.
- M. Lamouchi, E. Jeanneau, J. Couim, A. Brioude and C. Desroches, C R Chim., 2013, 16, 1073–1078 CrossRef.
- A. Savithri, C. N. Chinnan and L. Varma, Tetrahedron, 2015, 71, 9667–9672 CrossRef.
- S. E. Solovieva, A. A. Muravev, R. T. Zakirzyanov, S. K. Latypov, I. S. Antipin and A. I. Konovalov, Macroheterocycles, 2012, 5, 17–22 CrossRef.
- A. A. Muravev, S. E. Solovieva, S. K. Latypov, I. S. Antipin and A. I. Konovalov, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 499–502 CrossRef.
- A. A. Muravev, S. E. Solovieva, E. N. Kochetkov, N. B. Mel’nikova, R. A. Safiullin, M. K. Kadirov, S. K. Latypov, I. S. Antipin and A. I. Konovalov, Macroheterocycles, 2013, 6, 302–307 CrossRef.
- F. Yang, F. Yin, H. Guo, Z. Huang and X. Zhang, J. Inclusion Phenom. Macrocyclic Chem., 2010, 67, 49–54 CrossRef.
- S. F. Vasilevsky, M. P. Davydova and G. A. Tolstikov, Chem. Heterocycl. Compd., 2008, 44, 1257–1261 CrossRef.
- S. M. Bromidge, D. A. Entwistle, J. Goldstein and B. S. Orlek, Synth. Commun., 1993, 23, 487–494 CrossRef.
- H. Wang, F. Xie, Z. Qi and X. Li, Org. Lett., 2015, 17, 920–923 CrossRef PubMed.
- A. C. Gotzinger and T. J. J. Muller, Org. Biomol. Chem., 2016, 14, 3498–3500 RSC.
- J. Wang and C. D. Gutsche, J. Org. Chem., 2002, 67, 4423–4429 CrossRef PubMed.
- (a) S. Cecioni, R. Lalor, B. Blanchard, J. P. Praly, A. Imberty, S. E. Matthews and S. Vidal, Chem.–Eur. J., 2009, 15, 13232–13240 CrossRef PubMed; (b) N. A. Epifanova, E. V. Popova, S. E. Solovieva, Sh. K. Latypov, I. S. Antipin and A. I. Konovalov, Macroheterocycles, 2013, 6, 47–52 CrossRef.
- M. N. S. Rad and S. Behrouz, Synlett, 2011, 17, 2562–2566 Search PubMed.
- B. Wang, M. Bonin and L. Micouin, J. Org. Chem., 2005, 70, 6126–6128 CrossRef PubMed.
- W. Yin, H. He, Y. Zhang, D. Luo and H. He, Synthesis, 2014, 46, 2617–2621 CrossRef.
- J.-X. Wang, B. Wei, Z. Liu and Y. Fu, Synth. Commun., 2001, 31, 3527–3532 CrossRef.
- S. Roy, M. P. Davydova, R. Pal, K. Gilmore, G. A. Tolstikov, S. F. Vasilevsky and I. V. Alabugin, J. Org. Chem., 2011, 76, 7482–7490 CrossRef PubMed.
- A. A. Muravev, F. B. Galieva, A. G. Strel’nik, R. I. Nugmanov, M. Gruner, S. E. Solov’eva, S. K. Latypov, I. S. Antipin and A. I. Konovalov, Russ. J. Org. Chem., 2015, 51, 1334–1342 CrossRef.
- C. Boersch, E. Merkul and T. J. J. Muller, Angew. Chem., Int. Ed., 2011, 50, 10448–10452 CrossRef PubMed.
- J. Shen, G. Cheng and X. Cui, Chem. Commun., 2013, 49, 10641 RSC.
- A. S. Karpov and T. J. J. Muller, Org. Lett., 2003, 5, 3451–3454 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, physical data including 1D/2D NMR and HRMS, and crystallographic data. CCDC 1579205. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06349d |
| This journal is © The Royal Society of Chemistry 2018 |