
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and performance evaluation of nanostructured NaFexCr1−X(SO4)2 cathode materials in sodium ion batteries (SIBs)
Umair Nisar†
 a,
Mona Hersi Gulied†b,
R. A. Shakoor*a,
Rachid Essehlic,
Zubair Ahmada,
Abdullah Alashrafa,
Ramazan Kahramanb,
Siham Al-Qaradawid and
Ahmed Solimane
a,
Mona Hersi Gulied†b,
R. A. Shakoor*a,
Rachid Essehlic,
Zubair Ahmada,
Abdullah Alashrafa,
Ramazan Kahramanb,
Siham Al-Qaradawid and
Ahmed Solimane
aCenter for Advanced Materials (CAM), Qatar University, P. O. Box 2713, Doha, Qatar. E-mail: shakoor@qu.edu.qa
bDepartment of Chemical Engineering, College of Engineering, Qatar University, P. O. Box 2713, Doha, Qatar
cQatar Environment and Energy Research Institute, Hamad Bin Khalifa University, Qatar Foundation, 5825, Doha, Qatar
dDepartment of Chemistry & Earth Sciences, College of Arts and Science, Qatar University, P. O. Box 2713, Doha, Qatar
eGas Processing Center (GPC), Qatar University, P. O. Box 2713, Doha, Qatar
First published on 24th September 2018
Abstract
This research work focuses on the synthesis and performance evaluation of NaFexCr1−X(SO4)2 (X = 0, 0.8 and 1.0) cathode materials in sodium ion batteries (SIBs). The novel materials having a primary particle size of around 100–200 nm were synthesized through a sol–gel process by reacting stoichiometric amounts of the precursor materials. The structural analysis confirms the formation of crystalline, phase pure materials that adopt a monoclinic crystal structure. Thermal analysis indicates the superior thermal stability of NaFe0.8Cr0.2(SO4)2 when compared to NaFe(SO4)2 and NaCr(SO4)2. Galvanostatic charge/discharge analysis indicates that the intercalation/de-intercalation of a sodium ion (Na+) into/from NaFe(SO4)2 ensues at about 3.2 V due to the Fe2+/Fe3+ active redox couple. Moreover, ex situ XRD analysis confirms that the insertion/de-insertion of sodium into/from the host structure during charging/discharging is accompanied by a reversible single-phase reaction rather than a biphasic reaction. A similar sodium intercalation/de-intercalation mechanism has been noticed in NaFe0.8Cr0.2(SO4)2which has not been reported earlier. The galvanostatic measurements and X-ray photoelectron spectroscopy (XPS) analysis confirm that the Cr2+/Cr3+ redox couple is inactive in NaFexCr1−X(SO4)2 (X = 0, 0.8) and thus does not contribute to capacity augmentation. However, suitable carbon coating may lead to activation of the Cr2+/Cr3+ redox couple in these inactive materials.
1 Introduction
Due to the technological advancements in portable electronics, the demand for secondary batteries has increased considerably in the last decade.1,2 To date, the lithium ion battery system is considered one of the best candidates for portable energy storage applications due to its high-energy and power density.2–5 However, due to rapid technological advancements, the demand for high energy, high power, safer and cheaper batteries is increasing at a great pace.1,6–8 Therefore, new battery systems with promising chemistries are also being explored as an alternative to lithium ion batteries.9–11In the last decade, much research was devoted to the development of sodium ion batteries (SIBs) due to their similar electrochemical mechanism to lithium ion batteries. Also, the abundance of sodium resources in nature makes this technology much cheaper as compared to lithium ion batteries.12–14 Unfortunately, due to the larger size of sodium ion (Na+) as compared to lithium (Li+), it is difficult to identify new compounds capable of reversibly intercalating Na+ at fast rates. Recently considerable efforts have been devoted to develop novel potential cathode materials for Na-ion batteries.14,15 Towards this direction, several cathode materials have been identified such as Na3V2(PO4)3, NaTi2(PO4)3, Na4Fe3(PO4)2(P2O7), NaNiO2, NaMnO2, NaFePO4, Na2FeP2O7, Na3V2(PO4)2F3, Na4Co3(PO4)2P2O7 demonstrating promising energy storage performance.16–24 However, to address the future needs, development of cheaper, safe and high-performance cathode materials is still challenging.14,15,25
Recently, NaFe(SO4)2 has been reported having promising electrochemical performance. This material intercalates/de-intercalates sodium at (3.2 V) demonstrating charge/discharge capacity (80 mA h g−1) with good rate capability and cyclability.12 Being impressed with the electrochemical performance of NaFe(SO4)2 and promising merits of chromium (Cr) such as (i) reasonable high voltage and (ii) structural stability, we decided to synthesize an offshoot of NaFe(SO4)2 by substituting iron (Fe) with chromium (Cr). In this study, we report the synthesis, structural, thermal properties and electrochemical performance of NaFexCr1−X(SO4)2, where (X = 0, 0.8 and 1.0). To the best of our knowledge, this is a comprehensive report which describes the sodium insertion/de-insertion mechanism and structural alteration in NaFexCr1−X(SO4)2 during the charging/discharging process.
2 Experimental
2.1 Materials preparation
NaFexCr1−X(SO4)2 where (X = 0, 0.8,1.0) was synthesized by a sol–gel process using sodium nitrate, iron nitrate nonahydrate, chromium nitrate and ammonium sulfate (Sigma Aldrich) in stoichiometric ratios. Firstly, all the precursors were dissolved in 100 ml of distilled water with continuous stirring at around 50 °C. After 2 hours of continuous stirring, the temperature was raised to 70 °C and the solution was evaporated until a clear transparent gel was formed. The resulting gel was dried in a conventional oven at around 120 °C for 6 hours. Later, the gel was homogeneously ground into fine powder and then calcined at 420 °C for 6 hours in air to form pure NaFexCr1−X(SO4)2 (X = 0, 0.8,1.0). During the calcination process, the heating/cooling rates were set at 5 °C per minutes to ensure the formation of phase pure crystalline materials. A schematic diagram of the synthesis process is presented in Fig. 1.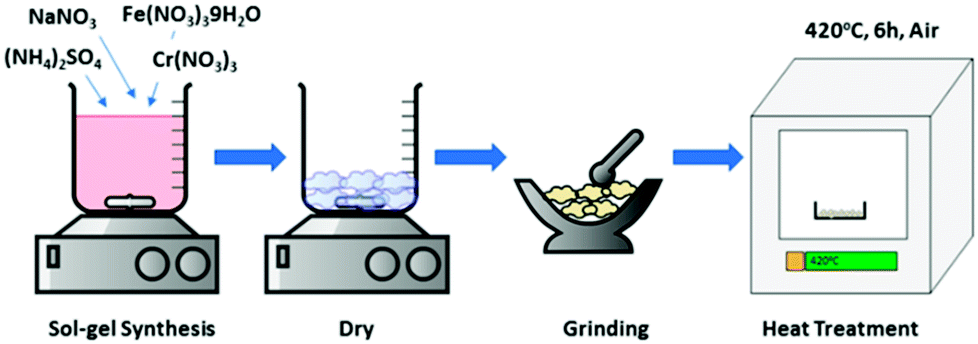 | ||
| Fig. 1 Schematic process for sol–gel synthesis of NaFexCr1−X(SO4)2 (X = 0, 0.8, 1.0). | ||
2.2 Structural characterization
Powdered XRD (Panalytical diffractometer) using Cu-Kα radiations was used to identify the phase purity and structural details of the synthesized materials. The data were collected over the 2θ angular range of 10–90° with a step size of 0.01°. The morphology (particle size, shape, distribution) and elemental mapping of the synthesize materials were under taken using scanning electron microscopy (SEM, NOVA NANOSEM 450). Thermal stability of the synthesized materials was studied using thermogravimetric analysis (Perkin Elmer, TGA 4000) from room temperature to 500 °C at the heating rate of 10 °C in a N2 atmosphere. Fourier-transform infrared spectroscopy (Perkin Elmer Frontier FT-IR) was further used to confirm the phase purity using KBr pellet method in the range of 500–2500 cm−1 wave numbers. The activity of redox couples was studied through X-ray Photoelectron spectroscopy (XPS) (Thermo-Scientific-Sigma Probe).2.3 Electrochemical characterization
The cathodes were fabricated by forming a slurry composed of 70 wt% active material NaFexCr1−X(SO4)2, 20 wt% conductive carbon (super-P) and 10.0 wt% polyvinylidene fluoride binder in 1-methyl-2-pyrrolidone (NMP). The slurry was homogeneously mixed for 10 hours and was later cast on an aluminum foil using a doctor blade. The cast electrode sheet was dried in a conventional oven for 6 hours to remove NMP. The dried cathode sheet was then rolled using a rolling machine for good compaction and then was dried in a vacuum oven for 12 hours to remove the last traces of water. The electrodes of 14.0 mm diameter were punched from vacuum dried cathode sheet and later shifted to the glove box for cell fabrication. Electrochemical measurements were performed by fabricating 2032-type coin cells in an Ar-filled glove box. Sodium metal was used as the negative electrode. The electrolyte was composed of 1 M NaClO4 dissolved in ethylene carbonate (EC) and 2.0 wt% of fluoroethylene carbonate (FEC). Galvanostatic charge/discharge tests were performed using WonAtech battery cycler (WBSC 3000L, Korea) in the voltage range of 1.5–4.5 V at room temperature.3 Results and discussion
The structural properties and phase purity of the synthesized phases was studied through XRD analysis. The XRD patterns of pristine NaFe(SO4)2, NaFe0.8Cr0.2(SO4)2 and NaCr(SO4)2 are presented in Fig. 2. It can be noticed that the synthesized materials are crystalline, and the absence of any impurity peak (s) confirm the high purity of the developed materials.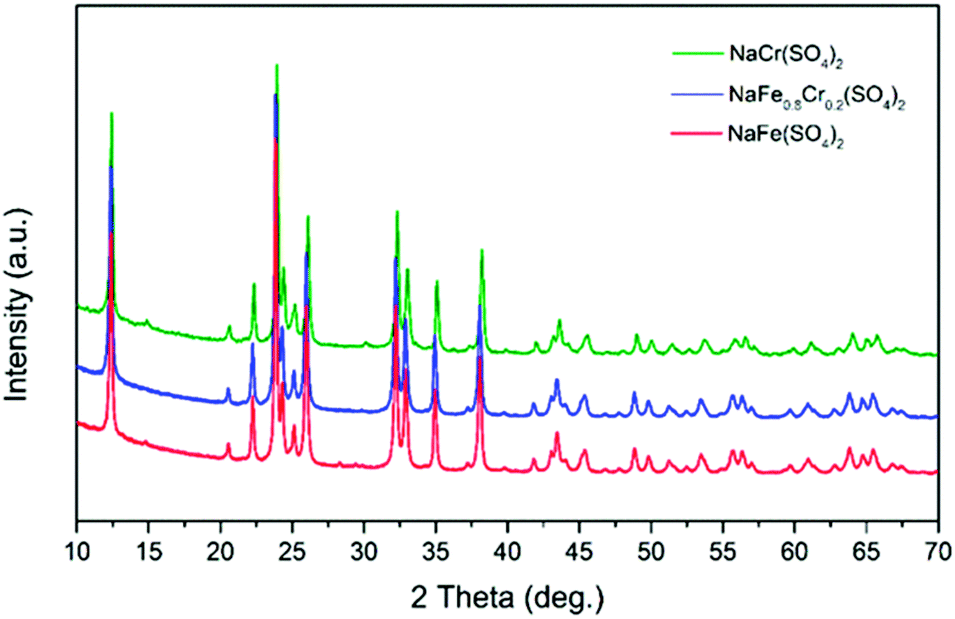 | ||
| Fig. 2 XRD patterns of pristine NaFe(SO4)2, NaFe0.8Cr(SO4)2 and NaCr(SO4). | ||
The XRD data was used to calculate the crystallite size (L) using the well-known Scherrer equation. The calculated crystallite size for the developed NaFe(SO4)2, NaFe0.8Cr0.2(SO4)2 and NaCr(SO4)2 is presented in Table 1.
| Identity | 2θ | K | λ | β | L (Å) |
|---|---|---|---|---|---|
| NaFe(SO4)2 | 23.8941 | 1.0747 | 1.547 | 0.1663 | 0.2138 |
| NaCr(SO4)2 | 23.9995 | 1.0747 | 1.547 | 0.1663 | 0.2129 |
| NaFe0.8Cr0.2(SO4)2 | 23.9046 | 1.0747 | 1.547 | 0.1919 | 0.1852 |
In order to have more insight of the structural details of the synthesized phases, Rietveld refinement was carried out. Our analysis confirms that NaFexCr1−X(SO4)2 (X = 0, 0.8 and 1.0) adopts a monoclinic crystal structure. As an example, Fig. 3 shows a Rietveld refinement of pristine NaFexCr1−X(SO4)2 where (X = 0.8) which shows good agreement between the experimental and calculated patterns.
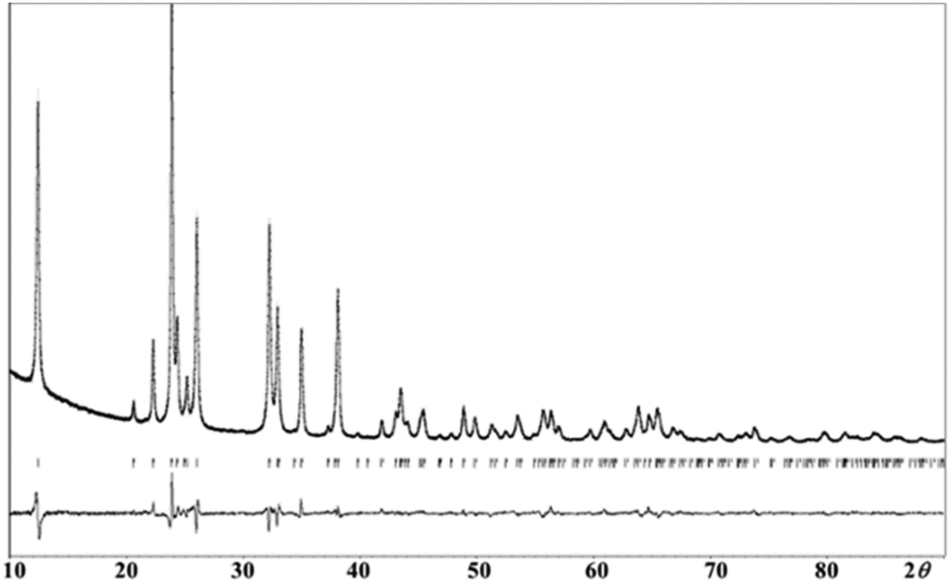 | ||
| Fig. 3 Final observed, calculated, and difference plots for PXRD (Cu-Kα radiation) Rietveld refinement of NaFe0.8Cr0.2(SO4)2. [Rp = 0.040, Rwp = 0.052, Rexp = 0.022]. | ||
The refined lattice parameters and other crystallographic information of NaFe0.8Cr0.2(SO4)2 is presented in Table 2. Moreover, atomic coordinates and isotropic displacement parameters for NaFe0.8Cr0.2(SO4)2 are also tabulated in Table 3.
| Sr. # | Description | Crystallographic |
|---|---|---|
| 1 | Chemical formula | NaFe0.8Cr0.2(SO4)2 |
| 2 | Crystal system, space group | Monocline, C2/m |
| 3 | Lattice parameter (a) | 7.98494 (Å) |
| 4 | Lattice parameter (b) | 5.12776 (Å) |
| 5 | Lattice parameter (c) | 7.13229 (Å) |
| 6 | β | 92.2166 (°) |
| 7 | Volume of unit cell | 291.81 (Å3) |
| Atom | Wyck. | Occ. | X | Y | Z | Uiso |
|---|---|---|---|---|---|---|
| Na1 | 2c | 1 | 0 | 0 | 1/2 | 0.0219 |
| Fe1 | 2a | 0.8 | 0 | 0 | 0 | 0.0119 |
| Cr1 | 2a | 0.2 | 0 | 0 | 0 | 0.0119 |
| S1 | 4i | 1 | 0.36107 | 0 | 0.22128 | 0.0096 |
| O1 | 8j | 1 | 0.46999 | 0.23434 | 0.20424 | 0.0090 |
| O2 | 4i | 1 | 0.24124 | 0 | 0.06535 | 0.0032 |
| O3 | 4i | 1 | 0.29052 | 0 | 0.40408 | 0.0080 |
The crystal structure for NaFe0.8Cr0.2(SO4)2 is shown in Fig. 4. As can be seen in this structure, the sodium (Na), iron (Fe) and chromium (Cr) ions occupy the octahedral sites in the a–b plane which are bridged together by (SO4) polyanions and creates an interplanar space for 2D Na+ diffusion.
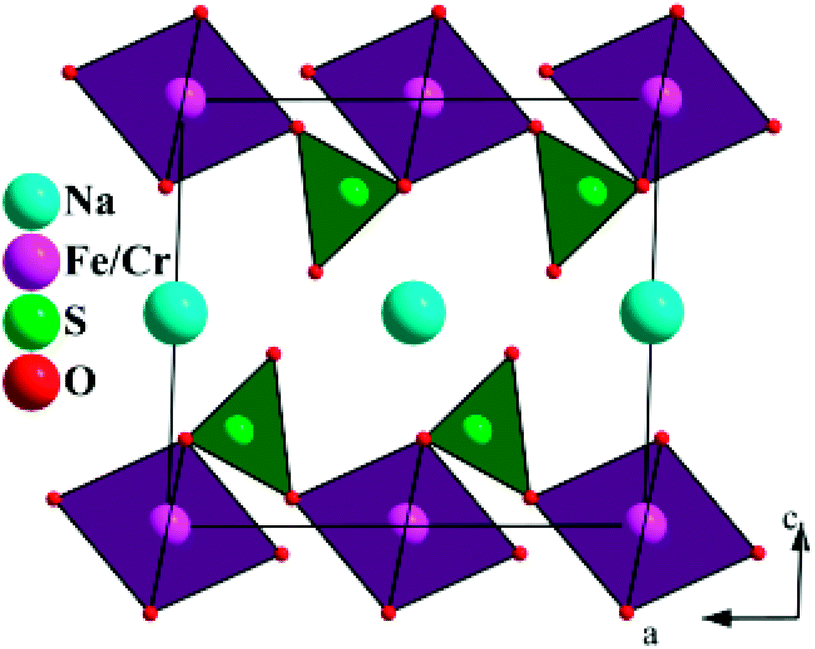 | ||
| Fig. 4 The crystal structure of NaFe0.8Cr0.2(SO4)2 projected along [010]. | ||
Fig. 5(a–c) shows the morphology of NaFe(SO4)2, NaCr(SO4)2 and NaFe0.8Cr0.2(SO4)2 respectively. It can be clearly seen that the synthesized materials have irregular particle morphology containing primary particle size between 100–200 nm. The particle agglomeration can also be clearly noticed in NaCr(SO4)2 and NaFe0.8Cr0.2(SO4)2. Fig. 5(d–f) shows the elemental mapping for NaFe0.8Cr0.2(SO4)2. It can be clearly seen from Fig. 5(e and f) that iron and chromium are homogeneously distributed throughout the synthesized materials.
 | ||
| Fig. 5 SEM images for (a) NaFe(SO4)2, (b) NaCr(SO4)2 and (c) NaFe0.8Cr0.2(SO4)2, (d–f) elemental mapping for NaFe0.8Cr0.2(SO4)2 for Fe and Cr. | ||
To investigate the thermal stability of NaFe(SO4)2, NaFe0.8Cr0.2(SO4)2 and NaCr(SO4)2, thermo-gravimetric analysis (TGA) was performed from room temperature to 500 °C in N2 atmosphere (Fig. 6). The NaFe(SO4)2 and NaCr(SO4)2 retains about 98.58 and 97.98% of their original weights up to 500 °C respectively. On the other hand, NaFe0.8Cr0.2(SO4)2 retains around 99.21% of its original weight. The results clearly indicate that the NaFe(SO4)2 and NaCr(SO4)2 materials have inferior thermal stability as compared to NaFe0.8Cr0.2(SO4)2. The inferior thermal stability of NaFe(SO4)2 and NaCr(SO4)2 as compared to NaFe0.8Cr0.2(SO4)2 indicates their inferior structural stability with increasing temperature. Furthermore, small substitution of iron with chromium to form NaFe0.8Cr0.2(SO4)2 improves the thermal stability of the material as indicated by weight loss (99.21%). The better thermal stability of chromium doped NaFe0.8Cr0.2(SO4)2 may be attributed to stronger bonding between iron (Fe) and chromium (Cr) in the crystal structure. It has been reported earlier that chromium doping enhances the thermal stability of the materials which is due to the high excess stabilization energy of Cr3+ in the structure.26–28 The stabilized structure therefore, is also expected to improve the electrochemical stability of the material specially at higher temperature.
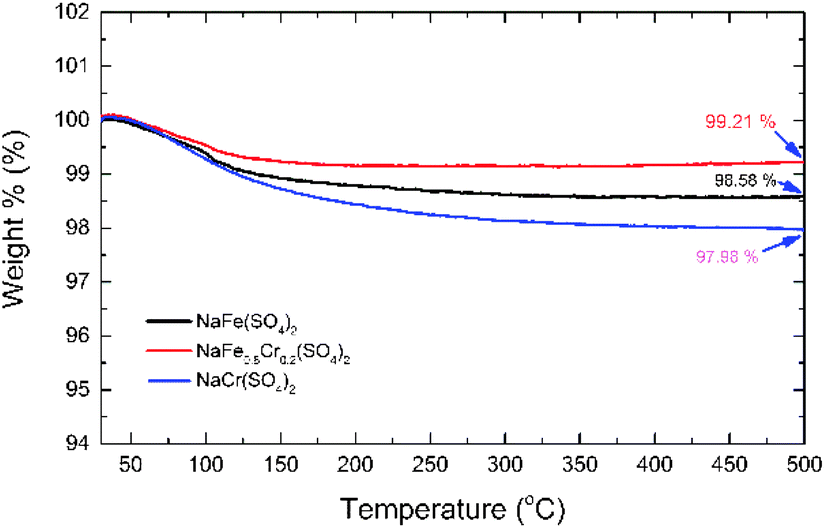 | ||
| Fig. 6 TGA data for NaFe(SO4)2, NaFe0.8Cr0.2(SO4)2 and NaCr(SO4)2. | ||
Fig. 7 shows the FTIR spectra of NaFe(SO4)2, NaFe0.8Cr0.2(SO4)2 and NaCr(SO4)2 recorded between 2500 and 500 cm−1. Four main internal modes are observed, which can be described as (i) the symmetric stretching mode ν1, (ii) the symmetric bending mode ν2, (iii) the asymmetric stretching mode ν3, and (iv) the asymmetric bending mode ν4. As displayed in Fig. 7 the split of asymmetric stretching (ν3: 997 cm−1) and bending (ν4: 688 cm−1) modes at a low wavenumber can be ascribed to the peaks for the sulfate group which confirms the formation of sulfate-based materials. These findings are consistent with previous studies.29–31
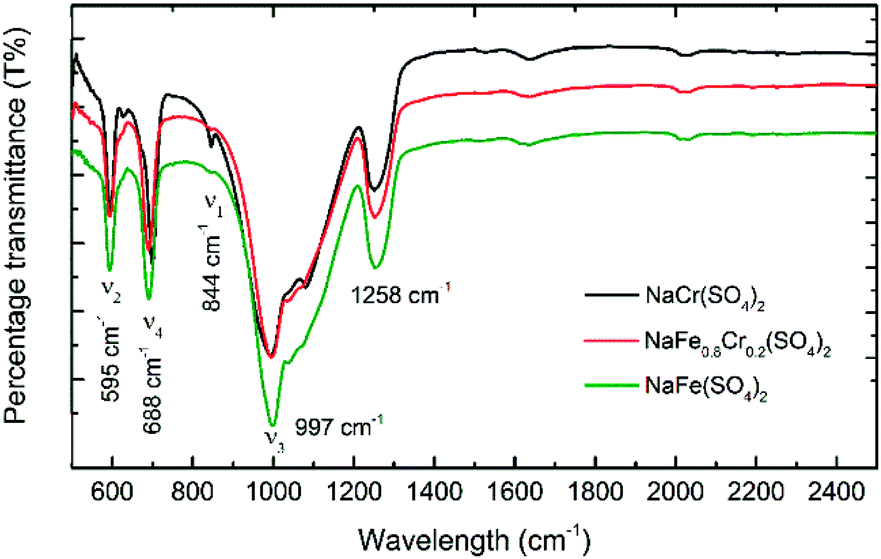 | ||
| Fig. 7 FTIR spectra for NaFe(SO4)2, NaFe0.8Cr0.2(SO4)2 and NaCr(SO4)2 for the range 500–2500 cm−1. | ||
To confirm the electrochemical activity of NaFe(SO4)2, NaFe0.8Cr0.2(SO4)2 and NaCr(SO4)2, differential capacity vs. voltage was plotted as shown in Fig. 8. It can be clearly seen from that NaFe(SO4)2 shows a peak at around 3.38 and 3.124 V during oxidation and reduction respectively which corresponds to active Fe2+/Fe3+ redox couple. On the other hand, NaFe0.8Cr0.2(SO4)2 shows peaks at around 3.169 and 3.005 V during oxidation and reduction which also corresponds to Fe2+/Fe3+ redox active couple. NaFe0.8Cr0.2(SO4)2 demonstrates lesser polarization during charging and discharging whereas NaFe(SO4)2 experiences more polarization as evident in dQ/dV curves in Fig. 8. It can be noticed that the substitution of iron with chromium reduces the redox potential of Fe2+/Fe3+ which may be attributed to multicomponent effect as reported previously.29–31 Finally, NaCr(SO4)2 shows no oxidation and reduction peaks which clearly indicated the inactive behaviour of Cr2+/Cr3+ redox couple in NaFexCr1−X(SO4)2 (X = 0, 0.8).
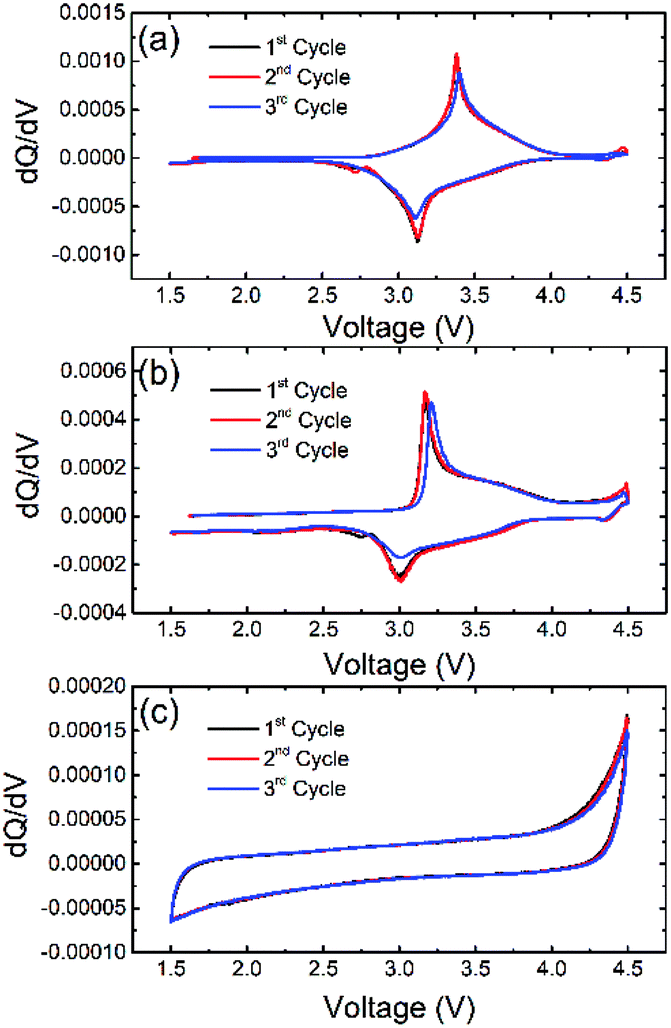 | ||
| Fig. 8 dQ/dV curves for (a) NaFe(SO4)2, (b) NaFe0.8Cr0.2(SO4)2 and (c) NaCr(SO4)2. | ||
Fig. 9(a–d) shows the electrochemical analysis of NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2 under different C-rates. It is pertinent to mention Cr2+/Cr3+ redox couple is inactive and thus NaCr(SO4)2 has not included in the further electrochemical discussion. Fig. 9(a) shows the rate capability data for NaFe(SO4)2 at different C-rates. The initial discharge capacity at 0.1C is around 78 mA h g−1 which gradually decreases with increasing C-rate i.e. the discharge capacity at 1C is 50 mA h g−1 which reduces to 24 mA h g−1 and at 5C. Fig. 9(b) shows the galvanostatic charge/discharge curves for NaFe(SO4)2 which shows a slanting discharge plateau corresponding to Fe2+/Fe3+ redox couple. It can be noticed that intercalation/de-intercalation of sodium into/from the host structure takes place at about 3.2 V due to active Fe2+/Fe3+ redox couple which is consistent with dQ/dV analysis shown in Fig. 8.
 | ||
| Fig. 9 Rate capability data, and galvanostatic charge–discharge curves at different C-rates for (a and b) NaFe(SO4)2, (c and d) NaFe0.8Cr0.2(SO4)2. | ||
Fig. 9(c) shows the rate capability data for NaFe0.8Cr0.2(SO4)2 at different C-rates. As compared with NaFe(SO4)2 the discharge capacity delivered by NaFe0.8Cr0.2(SO4)2 is inferior due to the presence of inactive Cr2+/Cr3+ redox couple as indicated in dQ/dV analysis. With the addition of chromium, the amount of active iron decrease, therefore, the discharge capacity decreases. It can be noticed that the discharge capacity delivered by NaFe0.8Cr0.2(SO4)2 at 0.1C is around 60 mA h g−1 and it also decreases with increasing C-rate similar to NaFe(SO4)2 i.e. the capacity at 1C and 5C are around 30 and 15 mA h g−1 respectively. Fig. 9(d) shows the galvanostatic charge/discharge curves for NaFe0.8Cr0.2(SO4)2 at different C-rates. The shape of the discharge curves for NaFe0.8Cr0.2(SO4)2 is slightly different as compared NaFe(SO4)2 which is mainly due to inactive chromium. The average voltage plateau due to active Fe2+/Fe3+ redox couple is comparatively lower as compared to NaFe(SO4)2 which is consistent with our dQ/dV results and may be attributed to multicomponent transition metals effect.29–31 We believe that suitable carbon coating technique to deposit a thin conductive carbon layer on NaCr(SO4)2 and NaFe0.8Cr0.2(SO4)2 may result in active Cr2+/Cr3+ redox couple leading to better energy storage performance.
To further investigate the activity of the redox couples in NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2, X-ray photoelectron spectroscopy (XPS) was carried out. Fig. 10(a and b) reveals that the Fe is present as Fe3+ (binding energy around 725.06 and 712.39 eV) in the as-prepared electrode (before discharge) in NaFe(SO4)2, whereas it is reduced to Fe2+ (binding energy around 723.5 and 710.3 eV) after the cell is discharged to 1.5 V. The results for Fe in NaFe0.8Cr0.2(SO4)2 are similar to NaFe(SO4)2 as shown in Fig. 10(c and d). Furthermore, the oxidation state for Cr in NaFe0.8Cr0.2(SO4)2 is Cr3+ (binding energy around 577.73 and 587.48 eV) in the as-prepared electrode (before discharge) which is not changed after discharging to 1.5 V which clearly shows that Cr3+/Cr2+ redox couple is inactive and is consistent with the dQ/dV curves (Fig. 8) and charge discharged curves as shown in Fig. 9(b and d).
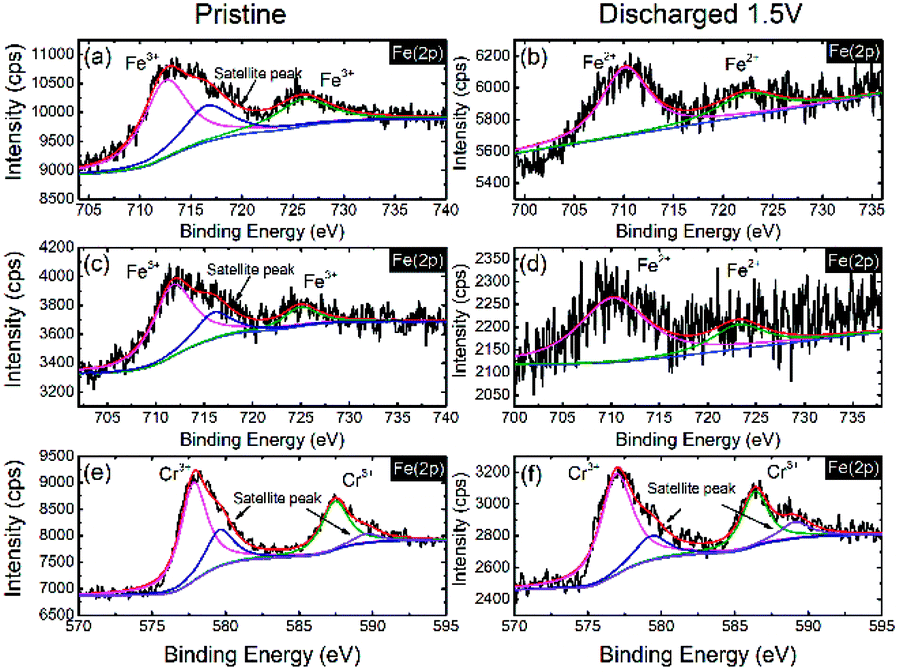 | ||
| Fig. 10 XPS spectra for Fe in NaFe(SO4)2 (a) as prepared electrode, (b) discharged to 1.5 V, Fe in NaFe0.8Cr0.2(SO4)2 (c) as prepared electrode, (d) discharged to 1.5 V and Cr in NaFe0.8Cr0.2(SO4)2 (e) as prepared electrode, (f) discharged to 1.5 V. | ||
Interestingly, the slanting voltage plateau in the charge/discharge curves of NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2 indicates the intercalation/de-intercalation of sodium in these materials through a single-phase reaction as reported in previous studies for another cathode materials.24 To confirm the interaction/de-intercalation mechanism of sodium into/from NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2 during charging and discharging, the ex situ XRD was employed. For this purpose, the cells were charged/discharged at different cut-off voltages, immediately disassembled in a glove box, washed and dried. Finally, XRD spectra were recorded to study the structural variations during intercalation/de-intercalation of sodium into/from the host structure. Fig. 11(a and b) shows the ex situ XRD data for NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2 respectively. It can be seen from that the XRD patterns that discharging the NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2 to 1.5 V results in shifting of the XRD peaks to lower 2θ values which is due to the expansion of the unit cell due to intercalation of sodium into the host lattice. Furthermore, later charging the cells to 3.0 V and 4.5 V, shifts the XRD peaks to higher 2θ values which may be ascribed to the contraction of the unit cell due to de-intercalation of sodium from the host lattice. It is important to notice that there is only peak shifting during intercalation/de-intercalation and no new peak is generated which reflects the property of single-phase reaction. These results are consistent with the galvanostatic charge/discharge curves with slanting plateau which is associated with the single-phase mode of intercalation/de-intercalation in cathode materials.
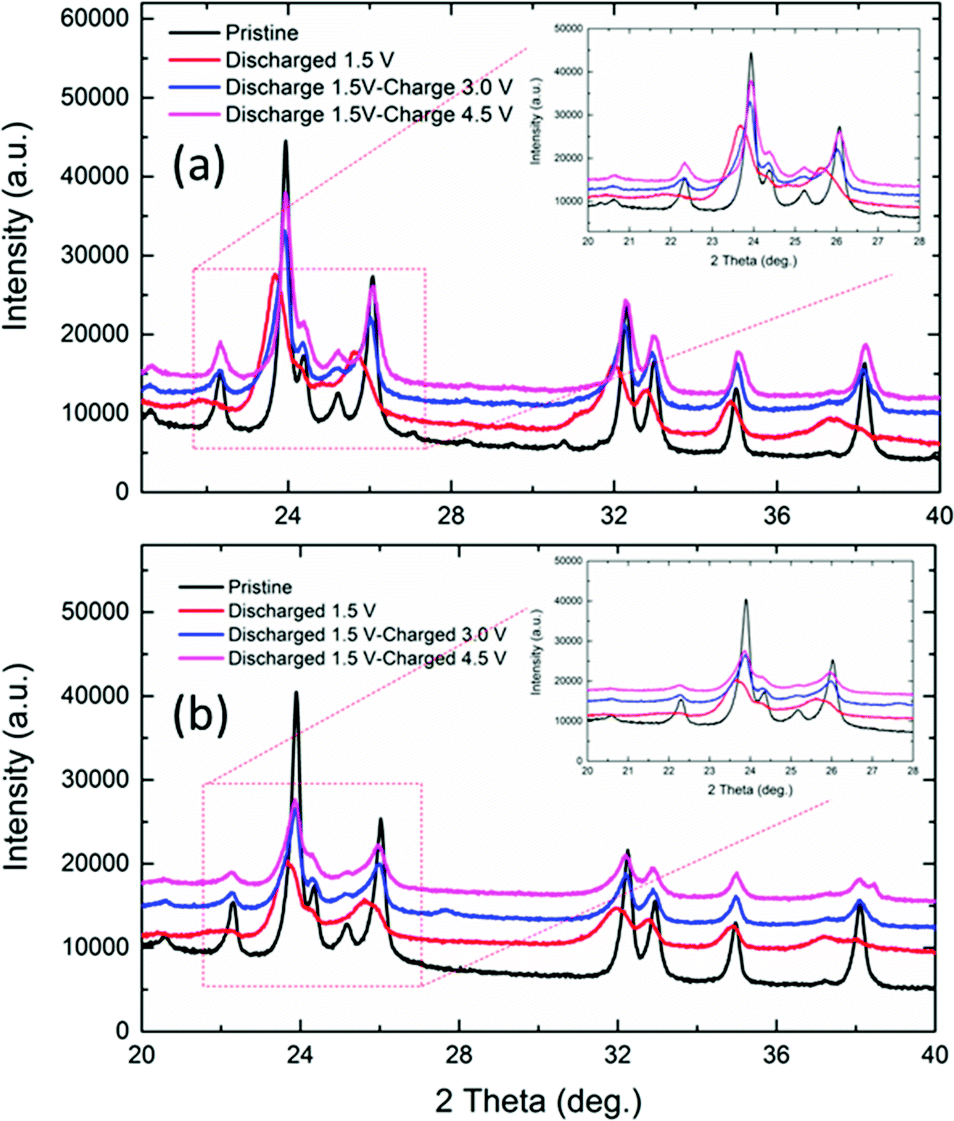 | ||
| Fig. 11 Ex situ XRD patterns for (a) NaFe(SO4)2, (b) NaFe0.8Cr0.2(SO4)2 at different voltages during charge and discharge. | ||
Fig. 12 shows the cycling performance of NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2 at 0.1C. The initial discharge capacity for NaFe(SO4)2 is around 80 mA h g−1 whereas for NaFe0.8Cr0.2(SO4)2 it is around 60 mA h g−1. After cycling for 30 cycles, the capacity for NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2 is around 73 mA h g−1 and 59 mA h g−1 respectively. It is noticed that NaFe(SO4)2 shows slow capacity decay with successive cycling. Inset in Fig. 12 shows the capacity retention of NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2 after 30 cycles. NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2 retains 92.8% and 100% of its initial capacity after 30 cycles.
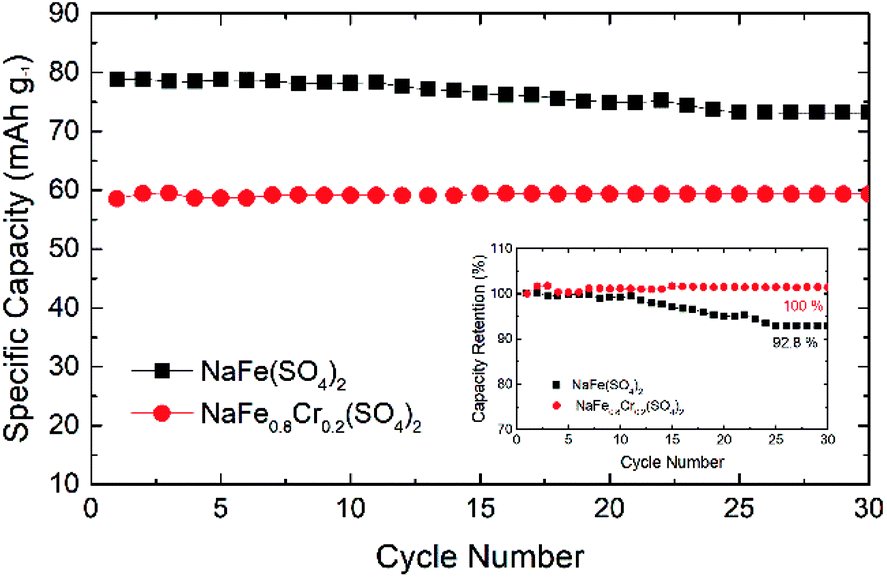 | ||
| Fig. 12 Cycling performance of NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2, inset showing percentage capacity retention. | ||
4 Conclusions
Phase pure NaFexCr1−X(SO4)2 cathode materials (where X = 0, 0.8, 1) were synthesized through a sol–gel process and their structural, thermal and electrochemical properties are evaluated. The structural analysis indicates that NaFexCr1−X(SO4)2 (X = 0, 0.8 and 1.0) adopts a monoclinic crystal structure. It is noticed that NaFe0.8Cr0.2(SO4)2 demonstrates better thermal properties as compared to NaCr(SO4)2 and NaFe(SO4)2. However, substitution of iron (Fe) with chromium (Cr) leads to inferior electrochemical performance which can be essentially ascribed to inactive Cr2+/Cr3+ redox couple. The ex situ XRD shows that the intercalation/de-intercalation mechanism in NaFe(SO4)2 and NaFe0.8Cr0.2(SO4)2 is governed by single-phase reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from internal grant awarded by Center for Advanced Materials (CAM), Qatar University, Doha 2713, Qatar.References
- G. Jeong, Y.-U. Kim, H. Kim, Y.-J. Kim and H.-J. Sohn, Energy Environ. Sci., 2011, 4, 1986 RSC
.
- B. Xu, D. Qian, Z. Wang and Y. S. Meng, Mater. Sci. Eng., R, 2012, 73, 51–65 CrossRef
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef
- J.-M. Tarascon, Philos. Trans. R. Soc., A, 2010, 368, 3227–3241 CrossRef PubMed
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef PubMed
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef PubMed
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef PubMed
- U. Nisar, R. Amin, R. Essehli, R. A. Shakoor, R. Kahraman, D. K. Kim, M. A. Khaleel and I. Belharouak, J. Power Sources, 2018, 396, 774–781 CrossRef
- S. P. Guo, J. C. Li, Q. T. Xu, Z. Ma and H. G. Xue, J. Power Sources, 2017, 361, 285–299 CrossRef
- L. P. Wang, L. Yu, X. Wang, M. Srinivasan and Z. J. Xu, J. Mater. Chem. A, 2015, 3, 9353–9378 RSC
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef PubMed
- P. Singh, K. Shiva, H. Celio and J. B. Goodenough, Energy Environ. Sci., 2015, 8, 3000–3005 RSC
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Adv. Energy Mater., 2016, 6, 1–38 Search PubMed
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef PubMed
- S. Y. Lim, H. Kim, R. A. Shakoor, Y. Jung and J. W. Choi, J. Electrochem. Soc., 2012, 159, A1393–A1397 CrossRef
- F. Lalere, V. Seznec, M. Courty, R. David, J. N. Chotard and C. Masquelier, J. Mater. Chem. A, 2015, 3, 16198–16205 RSC
- Y. Fang, L. Xiao, J. Qian, Y. Cao, X. Ai, Y. Huang and H. Yang, Adv. Energy Mater., 2016, 6, 2–9 Search PubMed
- X. Wu, G. Zhong, Z. Tang and Y. Yang, J. Power Sources, 2016, 327, 666–674 CrossRef
- M. Nose, H. Nakayama, K. Nobuhara, H. Yamaguchi, S. Nakanishi and H. Iba, J. Power Sources, 2013, 234, 175–179 CrossRef
- M. H. Han, E. Gonzalo, G. Singh and T. Rojo, Energy Environ. Sci., 2015, 8, 81–102 RSC
- G. Ali, J.-H. Lee, D. Susanto, S.-W. Choi, B. W. Cho, K.-W. Nam and K. Y. Chung, ACS Appl. Mater. Interfaces, 2016, 8, 15422–15429 CrossRef PubMed
- R. A. Shakoor, D.-H. Seo, H. Kim, Y.-U. Park, J. Kim, S.-W. Kim, H. Gwon, S. Lee and K. Kang, J. Mater. Chem., 2012, 22, 20535 RSC
- R. A. Shakoor, C. S. Park, A. A. Raja, J. Shin and R. Kahraman, Phys. Chem. Chem. Phys., 2016, 18, 3929–3935 RSC
- Y. You and A. Manthiram, Adv. Energy Mater., 2017, 1701785, 1–11 Search PubMed
- R. M. Rojas, K. Petrov, G. Avdeev, J. M. Amarilla, L. Pascual and J. M. Rojo, J. Therm. Anal. Calorim., 2007, 90, 67–72 CrossRef
- M. Aklalouch, J. M. Amarilla, R. M. Rojas, I. Saadoune and J. M. Rojo, J. Power Sources, 2008, 185, 501–511 CrossRef
- M. Aklalouch, J. M. Amarilla, I. Saadoune and J. M. Rojo, J. Power Sources, 2011, 196, 10222–10227 CrossRef
- H. Li, X. Bi, Y. Bai, Y. Yuan, R. Shahbazian-Yassar, C. Wu, F. Wu, J. Lu and K. Amine, Adv. Mater. Interfaces, 2016, 3, 1–8 Search PubMed
- Y. Meng, S. Zhang and C. Deng, J. Mater. Chem. A, 2015, 3, 4484–4492 RSC
- M. D. Lane, Am. Mineral., 2007, 92, 1–18 CrossRef
Footnote |
| † Authors with equal contributions. |
| This journal is © The Royal Society of Chemistry 2018 |