
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe streptazolin- and obscurolide-type metabolites from soil-derived Streptomyces alboniger YIM20533 and the mechanism of influence of γ-butyrolactone on the growth of Streptomyces by their non-enzymatic reaction biosynthesis†
Na Luo‡
a,
Ya-Bin Yang‡ b,
Xue-Qiong Yangb,
Cui-Ping Miaoa,
Yi-Qing Lia,
Li-Hua Xua,
Zhong-Tao Ding*b and
Li-Xing Zhao*a
b,
Xue-Qiong Yangb,
Cui-Ping Miaoa,
Yi-Qing Lia,
Li-Hua Xua,
Zhong-Tao Ding*b and
Li-Xing Zhao*a
aYunnan Institute of Microbiology, College of Life Science, Yunnan University, 2 Cuihu North Road, Kunming, People's Republic of China 650091. E-mail: zlx70@163.com
bFunctional Molecules Analysis and Biotransformation Key Laboratory of Universities in Yunnan Province, School of Chemical Science and Technology, Yunnan University, 2 Cuihu North Road, Kunming, People's Republic of China 650091. E-mail: ztding@ynu.edu.cn
First published on 12th October 2018
Abstract
Eleven new compounds with streptazolin- and obscurolide-type skeletons were isolated from soil-derived Streptomyces alboniger obtained from Tibet, China. Two types of unprecedented skeletons of obscurolide dimer and an obscurolide-type compound with an aromatic polyketide of pentanone substituted at the benzene ring were determined by spectral data analysis. Compound 11 was the first evidence of two nitrogens in streptazolin-type structures. Compound 1 indicated an inhibitory effect on nitric oxide production in LPS-activated macrophages with an inhibition ratio of 51.7% at 50 μM, and on anticoagulant activity on platelet activating factor (PAF)-induced platelet aggregation with an inhibition ratio of 26.0 ± 9.1% at 200 μg mL−1. 11 had anti-acetylcholinesterase activity with an inhibition ratio of 27.2% at a concentration of 50 μM. Mechanistic aspects of the non-enzymatic reaction as well as a more detailed picture of the biosynthetic relationships of the streptazolin- and obscurolide-type metabolites are described. Acidic and basic conditions can inhibit the growth of Streptomyces, and γ-butyrolactones were found to be hormones controlling antibiotic production in Streptomyces. In the pH fermentation tests, acylation of γ-butyrolactones was successfully used to explain the mechanism of influence on the growth of Streptomyces.
Introduction
Streptomyces spp. have been thoroughly investigated for more than 70 years as they produce a number of diverse bioactive compounds, especially clinical chemotherapeutics, such as antibacterial, anticancer, and immunosuppressive agents. It has been proposed that there are more genes encoding the potential production of small molecules than have been found, indicating that in-depth research should be developed to activate the silent genes.1,2 New biotechnological techniques, such as gene mining and gene-regulating methods, have been applied to stimulate Streptomyces strains to produce new molecules.3,4 At the same time, bioactive compound production also is related to nutritional regulators, morphological development, physiological processes and other hormone or hormone-like production in the Streptomyces culture.5 Mining novel bioactive compounds from microorganisms harbored in under-explored niches has proved an efficient method in pharmaceutical research and development.6,7 Streptazolin, mainly produced by members of the genus Streptomyces, presents antibiotic and antifungal activities.8,9 Some synthesis research on the compound and its analogues has also been developed.10–12 Recent research has proved that the compound can also increase bacterial killing and elaboration of immunostimulatory cytokines by macrophages.13 The diene and oxazolidinone moieties are considered as the pharmacophore in streptazolin and its analogues.13,14 Obscurolides, a series of butyrolactone derivatives produced by Streptomyces viridochromogenes (strain Tü 2580), present weak inhibitory activity against phosphodiesterases.15,16 γ-Butyrolactones play an autoregulation role in antibiotic production and differentiation for many members of the genus Streptomyces by modulating the DNA binding activity of cognate receptor proteins.17 Novel butyrolactones as signaling molecules should be involved in many functional aspects in microbial life, which would be useful for synthetic biology.18 In our effort to search for new biological compounds from Streptomyces harbored in different niches, eight new butyrolactones (obscurolide-type), two new streptazolin-type compounds, a new metabolite and two new natural products were isolated from the culture broth of Streptomyces alboniger YIM20533, an isolate obtained from a soil sample from Tibet, China. Their structures were determined and interpreted by 1D NMR, 2D NMR, and HR-ESIMS data (Fig. 1). In this research, we report the isolation, structural elucidation, bioactivities and the mechanism of influence on the growth of Streptomyces via the biosynthesis of butyrolactone derivatives.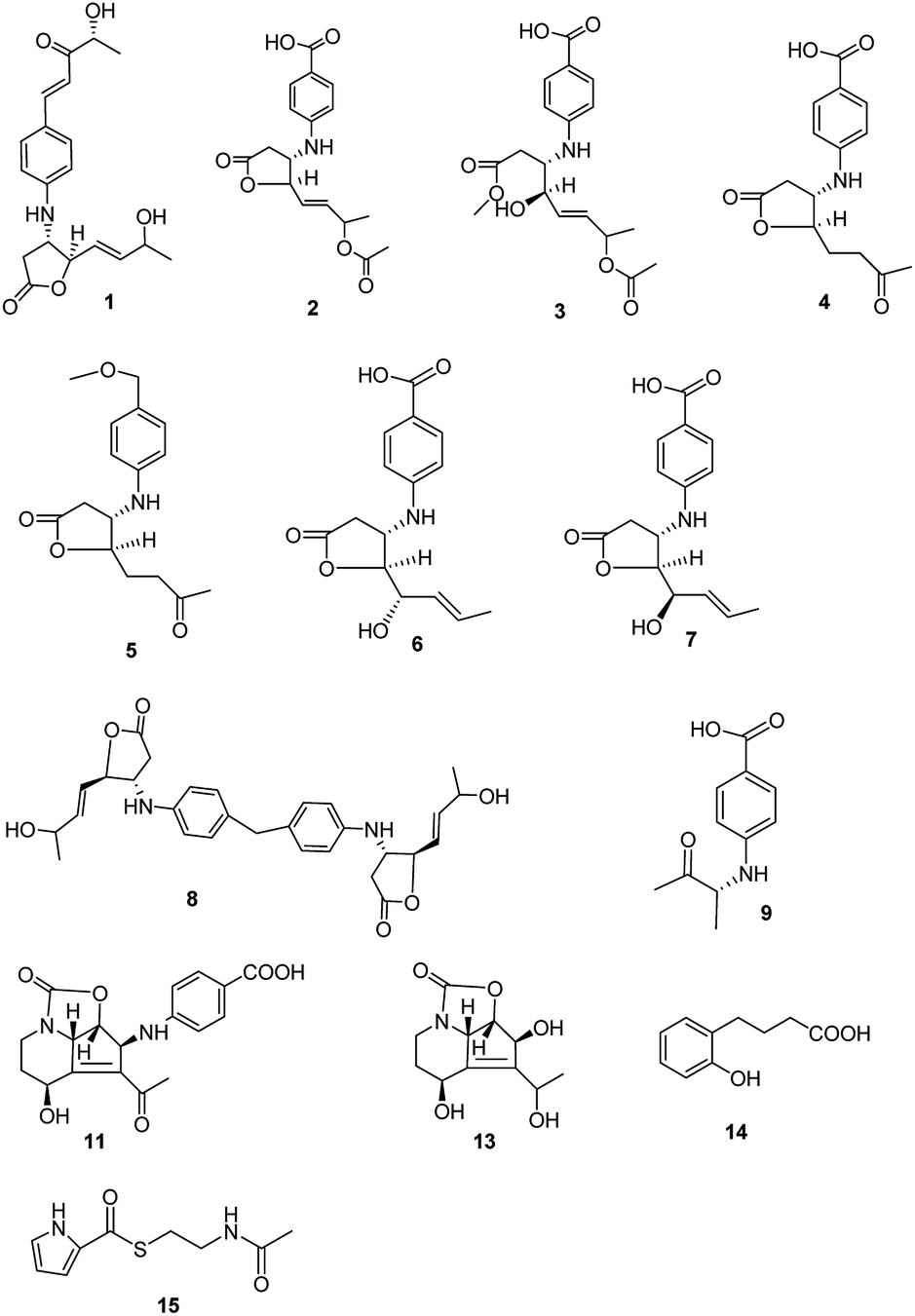 | ||
| Fig. 1 Structures of compounds 1–9, 11, and 13–15 isolated from Streptomyces alboniger YIM20533. | ||
Results and discussion
The molecular formula of streptalbonin A (1) was determined as C19H23NO5 from HR-ESIMS analysis. The 1H and 13C NMR spectroscopic analysis, including DEPT, clearly showed two methyls, one methylene, twelve methines, two olefinic quaternary carbons, and two carbonyl carbons, which indicated the skeleton of obscurolide.15 An additional pentanone should be substituted at the benzene ring, which was confirmed by the COSY correlations between H-7′ and H-8′, H-10′ and H-11′, and the HMBC correlations from H-11′ to C-9′ and C-10′; H-7′ and H-8′ to C-9′; and H-7′ to C-3′ and C-5′. Compound 1 has an unprecedented skeleton with a pentanone substituted at the benzene ring in obscurolide-type compounds. The fraction structure of this compound, an aromatic polyketide, is rarely found in natural products. The other COSY correlations of H-2′/H-3′; H-5′/H-6′; and H-2/H-3/H-4/H-5/H-6/H-7/H-8, and the HMBC correlations from H-2 to C-1, C-3 and C-4; H-4 to C-1, C-5, and C-6; and H-8 to C-6 and C-7 also confirmed the skeleton of obscurolide (Fig. 2). The trans orientation at C-7′ and C-8′ was determined by the coupling constant at 15.6 Hz. The relative configuration between H-3 and H-4 of 1 was determined as trans by the ROESY correlation of H-3/H-5. The configurations (3S, 4R) of streptalbonin A at C-3 and C-4 and trans orientation at C-5 and C-6 were determined by comparing the NMR spectrum with those for other obscurolides and pseudonocardides also found in actinomyces and their biogenesis.15,19 Streptalbonin A exists in two diastereomers at C-7 just as the obscurolide A2 reported in previous work.15 The configuration of C-10′ of compound 1 was determined as R by comparing the NMR and optical rotation with those for (R)-2-hydroxy-5-phenylpent-4-en-3-one formed by microorganism enzymes.20 The fraction structure (R)-2-hydroxy-5-phenylpent-4-en-3-one existed in compound 1.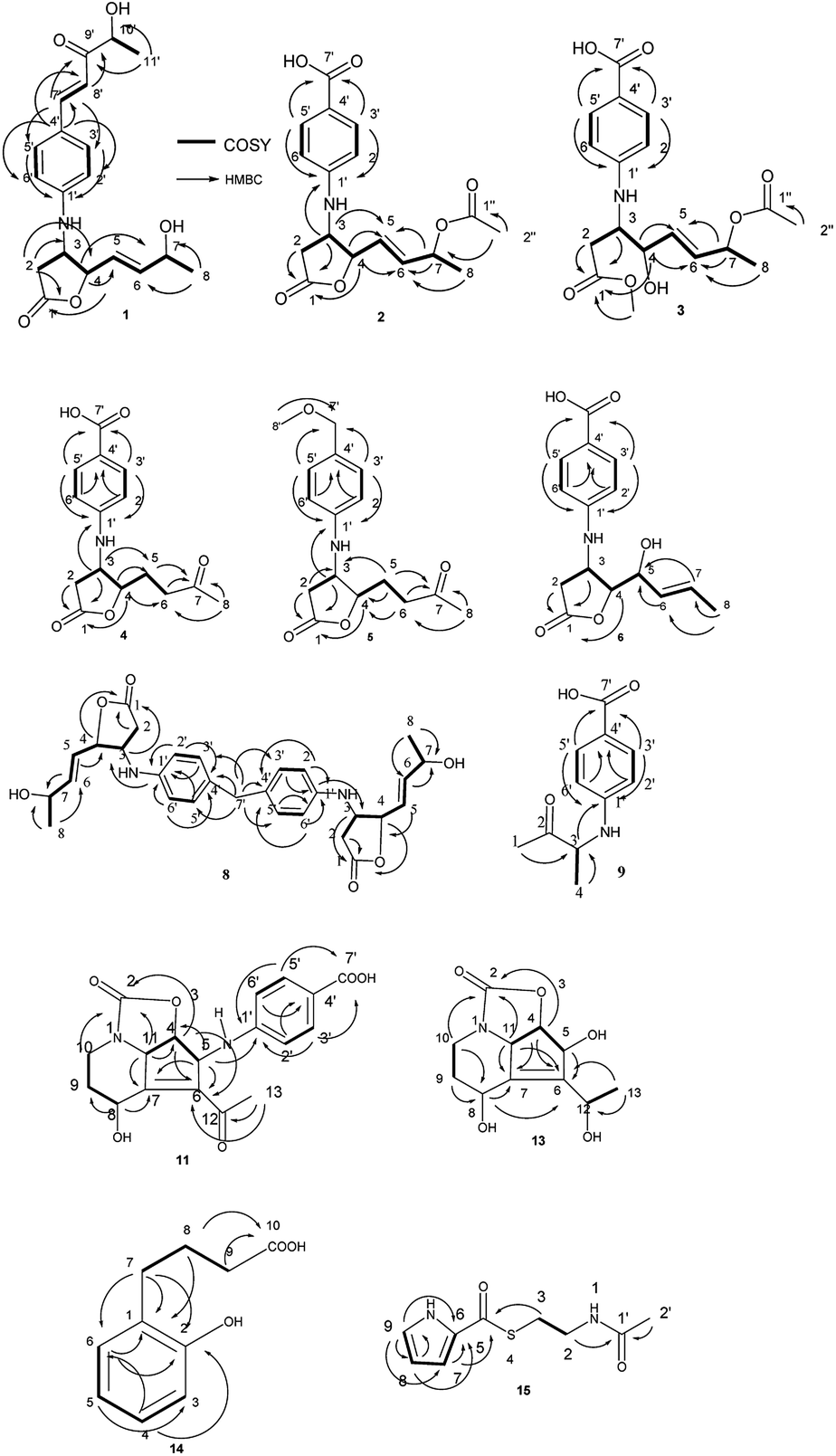 | ||
| Fig. 2 The COSY and HMBC correlations in compounds 1–6, and 8, 9, 11, and 13–15. | ||
The molecular formula of streptalbonin B (2) was determined as C17H19NO6 from HR-ESIMS analysis. The spectroscopic analysis also showed the skeleton of obscurolide. The COSY correlations of H-2′/H-3′; H-5′/H-6′; and H-2/H-3/H-4/H-5/H-6/H-7/H-8, and the HMBC correlations from H-2, H-3 and H-4 to C-1; H-4, H-7 and H-8 to C-6; H-3, H-4 and H-7 to C-5; H-3 to C-1′; and H-3′ and H-5′ to C-1′ and C-7′ confirmed this speculation. The structure difference compared with other known obscurolides was the acetyl at C-7, which was determined by the weak HMBC correlation from H-2′′ to C-7 (Fig. 2). The relative configurations between H-3 and H-4 were determined as trans by the ROESY correlations of H-2′/H-4; H-3/H-5. The configurations of streptalbonin B and the trans orientation at C-5 and C-6 were determined by comparing the NMR results with those for compound 1.
The molecular formula of streptalbonin C (3) was determined as C18H23NO7 by HR-ESIMS analysis. The spectroscopic analysis clearly showed the skeleton of obscurolide. The COSY correlations of H-2′/H-3′; H-5′/H-6′; and H-2/H-3/H-4/H-5/H-6/H-7/H-8, and the HMBC correlations from H-2, H-3, and OCH3 to C-1; H-4, H-7, H-8 to C-6; H-4, H-7 to C-5; H-3′, H-5′ to C-1′ and C-7′ confirmed this skeleton (Fig. 2). The acetyl at C-7 was confirmed by the chemical shift of H-7 compared with those of compound 2. The configurations of streptalbonin C at C-3, C-4 were determined by comparing the NMR spectrum with that of compound 2.
The molecular formula of streptalbonin D (4) was determined as C15H17NO5 from HR-ESIMS analysis. The 1H and 13C NMR spectroscopic analysis clearly showed the skeleton of obscurolide as in compounds 1–3. The COSY correlations of H-2′/H-3′; H-5′/H-6′; and H-2/H-3/H-4/H-5/H-6, and the HMBC correlations from H-2, H-3 and H-4 to C-1; H-5, H-6 and H-8 to C-7; H-3 and H-4 to C-5; H-3′ and H-5′ to C-1′ and C-7′; and H-2′ and H-6′ to C-4′ also confirmed this structure (Fig. 2). The relative configurations between H-3 and H-4 of 4 were determined as trans by the NOESY correlations of H-3/H-5. The configurations of streptalbonin D at C-3 and C-4 were determined as 3S, 4R by comparing the NMR spectrum with that for compound 3 and biogenesis.
The molecular formula of streptalbonin E (5) was determined as C16H21NO4 from HR-ESIMS analysis. The 1H and 13C NMR spectroscopic analysis clearly showed the skeleton of obscurolide. The COSY correlations of H-2′/H-3′; H-5′/H-6′; H-2/H-3; and H-4/H-5/H-6, and the HMBC correlations from H-2, H-3 and H-4 to C-1; H-5, H-6 and H-8 to C-7; H-5 to C-3 and C-4; H-3′ and H-5′ to C-1′ and C-7′; H-8′ to C-7′; and H-2′ and H-6′ to C-4′ also confirmed this structure (Fig. 2). The relative configurations between H-3 and H-4 were determined as trans by the NOESY correlations of H-3/H-5. The configurations of streptalbonin E were determined to be the same as those found for the other compounds.
The molecular formula of streptalbonin F (6) was determined as C15H17NO5 from HR-ESIMS analysis. The 1H and 13C NMR spectroscopic analysis clearly showed the skeleton of obscurolide. The COSY correlations of H-2′/H-3′; H-5′/H-6′; and H-2/H-3/H-4/H-5/H-6/H-7/H-8, and the HMBC correlations from H-2, H-3 and H-4 to C-1; H-8 to C-6 and C-7; H-3′ and H-5′ to C-1′ and C-7′; and H-2′ and H-6′ to C-4′ also confirmed this structure (Fig. 2). The trans orientation at C-6 and C-7 was determined by the NOESY correlations between H-5 and H-7, and H-6 and H-8. The relative configurations between H-3 and H-4 of 6 were determined as trans by the NOESY correlations of H-3/H-5. The configurations of streptalbonin F at C-3 and C-4 were determined as 3S and 4R by comparing the NMR spectrum with that for compound 5. The configuration of C-5 was elucidated as S by comparing the NMR data with those of musacins also isolated from Streptomyces.21 Streptalbonin G (7) was determined to be the diastereoisomer of streptalbonin F (6) by NMR, 2D-NMR and CD analysis. The difference between compounds 6 and 7 was the configuration of C-5, which was found to be S for 6 and R for 7 by NMR analysis.
The molecular formula of streptalbonin H (8) was determined as C29H34N2O6 from HR-ESIMS analysis. The 1H and 13C NMR spectroscopic analysis clearly showed the skeleton of obscurolide. The COSY correlations of H-2′/H-3′; H-5′/H-6′; and H-2/H-3/H-4/H-5/H-6/H-7/H-8, and the HMBC correlations from H-2, H-3 and H-4 to C-1; H-8 to C-6 and C-7; H-3′ and H-5′ to C-1′; and H-2′ and H-6′ to C-4′ also confirmed this structure. The key correlations from H-7′ to C-3′, 4′, and 5′ indicated that the C-7′ is connected to C-4′ (Fig. 2), which showed an unprecedented streptalbonin dimer. The trans orientation at C-5 and C-6 was determined by comparing the NMR results with those for other compounds isolated in this strain. The relative configurations between H-3 and H-4 of 8 were determined as trans by the NOESY correlations of H-3/H-5. The configurations of streptalbonin H at C-3 and C-4 were determined as 3S and 4R by comparing the NMR results with those for compound 3.
The molecular formula of streptalbonin I (9) was determined as C11H13NO3 from HR-ESIMS analysis. The 1H and 13C NMR spectroscopic analysis clearly showed two methyls, five methines, two olefinic quaternary carbons, and two carbonyl carbons. The COSY correlations of H-2′/H-3′; H-5′/H-6′; and H-3/H-4, and the HMBC correlations from H-1 and H-4 to C-3; H-3 to C-1′; H-3′ and H-5′ to C-1′ and C-7′; and H-2′ and H-6′ to C-4′ also confirmed this structure (Fig. 2). The configuration of this compound was determined as R by comparing the optical rotation for this compound with that for (R)-3-hydroxy-butan-2-one,22 which is also found in Streptomyces.23
Compound 10 was determined as 8α-hydroxystreptazolone by spectroscopic analysis.11 The molecular formula of strepalbin A (11) was determined as C18H18N2O6 from HR-ESIMS analysis. Comparing the NMR data of compounds 10, and 11, these compounds have similar structures, except for strepalbin A (11) with a p-aminobenzoic acid substituted at C-5, which was confirmed by the HMBC correlation from H-5 to C-1′. The COSY correlations of H-11/H-4/H-5 and H-8/H-9/H-10, and the HMBC correlations from H-4, H-10, and H-11 to C-2; H-5, H-8, and H-11 to C-7; H-4, H-5, and H-13 to C-6; H-8 to C-9; H-3′ and H-5′ to C-1′ and C-7′; and H-2′ and H-6′ to C-4′ also confirmed this structure (Fig. 2). The configurations of this compound were determined to be the same as those for 8α-hydroxystreptazolone by comparing the NMR spectra, and the NOESY correlations of H-4/H-11; H-4/H-2′, and H-5/H-8 also confirmed this elucidation.
4a,5-Dihydrostreptazolin (12) was determined by spectroscopic analysis24 and it was the first time that it was isolated from a natural source. Strepalbin B (13) was determined as C11H15NO5 from HR-ESIMS analysis. Comparing with the NMR data of 8α-hydroxy-streptazolone,11 strepalbin B (13) had a hydroxyl at C-12 instead of the carbonyl in 8α-hydroxy-streptazolone, and this structure was determined by the COSY correlations of H-9/H-10 and H-11/H-4/H-5, and the HMBC correlations of H-4, H-10, H-11/C-2; H-4, H-5, H-8, H-11/C-6, C-7; H-9, H-10/C-8; and H-13/C-6, C-12 (Fig. 2). The configurations of this compound were determined to be the same as for 8α-hydroxy-streptazolone by comparing the NMR spectra,11 and the NOESY correlations also confirmed it. The configuration of C-12 in compound 13 was not determined owing to the low amount.
Compound 14 was determined as C10H12O3 from HR-ESIMS analysis. The 1H and 13C NMR spectroscopic analysis clearly showed three methylenes, four methines, two olefinic quaternary carbons, and one carbonyl carbon. There were also a disubstituted benzene and a butyric acid in the structure of 14, named as 4-(2-hydroxyphenyl)butyric acid, which was determined by the COSY correlations of H-3/H-4/H-5/H-6 and H-7/H-8/H-9, and HMBC correlations of H-8, H-9/C-10; H-7, H-8/C-1; H-7/C-2, C-6; H-6/C-1, C-2; and H-4/C-2, C-6 (Fig. 2). This structure was first isolated in natural products.
Compound 15 was determined as C9H12N2O2S from HR-ESIMS analysis. The 1H and 13C NMR spectroscopic analysis clearly showed two methylenes, three methines, one olefinic quaternary carbon, and two carbonyl carbons. The structure of 15 was determined by the COSY correlations of H-2/H-3 and H-7/H-8/H-9, and HMBC correlations of H-2, H-2′/C-1′; H-3, H-7/C-5; and H-7, H-8, H-9/C-6 (Fig. 2). This structure was first isolated in natural products.
Some new compounds were evaluated for their nitric oxide inhibition (1, 2, 3, 4, 5, 6, 7, 8, 9, 11, obscurolide A1,15 obscurolide A2 (ref. 15)) cytotoxicity (11), anticoagulant activity (1, 8, 9, and 11), and anti-acetylcholinesterase activity (1, 8, 9, and 11). Compound 1 indicated an inhibitory effect on nitric oxide production in LPS-activated macrophages with an inhibition ratio of 51.7% at 50 μM, and anticoagulant activity on platelet activating factor (PAF)-induced platelet aggregation with an inhibition ratio of 26.0 ± 9.1% at 200 μg mL−1. 11 had anti-acetylcholinesterase activity with an inhibition ratio of 27.2% at a concentration of 50 μM. The other tested compounds had no obvious activities in the corresponding assays.
The obscurolides were isolated from Streptomyces viridochromogenes and turned out to be inhibitors of cyclic AMP phosphodiesterase. Previous work has focused on their structure and bioactivity. Only one paper described the manipulation of fermentation pattern to initiate obscurolide biosynthesis,25 and no detailed process was discussed. Unexpectedly, a non-enzymatic reaction was found to be a key step in the production of these obscurolide- and streptazolin-type compounds. The common precursor 4-aminobenzoic acid was also isolated from this strain, and 4,4′-methylenedianiline is a common industrial chemical, so they might be the bioprecursors in the biosynthesis of corresponding metabolites. Structure 1a should be an aromatic polyketide produced by this strain. The non-enzymatic reaction of these obscurolide- and streptazolin-type compounds is presented in Fig. 3. Structure 1b is γ-butyrolactone, and γ-butyrolactones are signalling molecules regulating antibiotic production and differentiation in Streptomyces.17 The production of important antibiotics in Streptomyces is often regulated by low-molecular-weight bacterial hormones called autoregulators. Although 60% of Streptomyces strains may use γ-butyrolactone-type molecules as autoregulators and some use furan-type molecules, little is known about the signaling molecules used to regulate antibiotic production in many other members of this genus.5 We found that the acidity and alkalinity influenced the growth of this Streptomyces. In this research, some acylate products of γ-butyrolactone were isolated, and the acidity can accelerate the acylation, so we deduced that the conversion reaction changed the structure of γ-butyrolactone. The activity of γ-butyrolactone from Streptomyces as a bacterial hormone was declined so the change of the γ-butyrolactone structure catalyzed by acid inhibited the growth of Streptomyces.
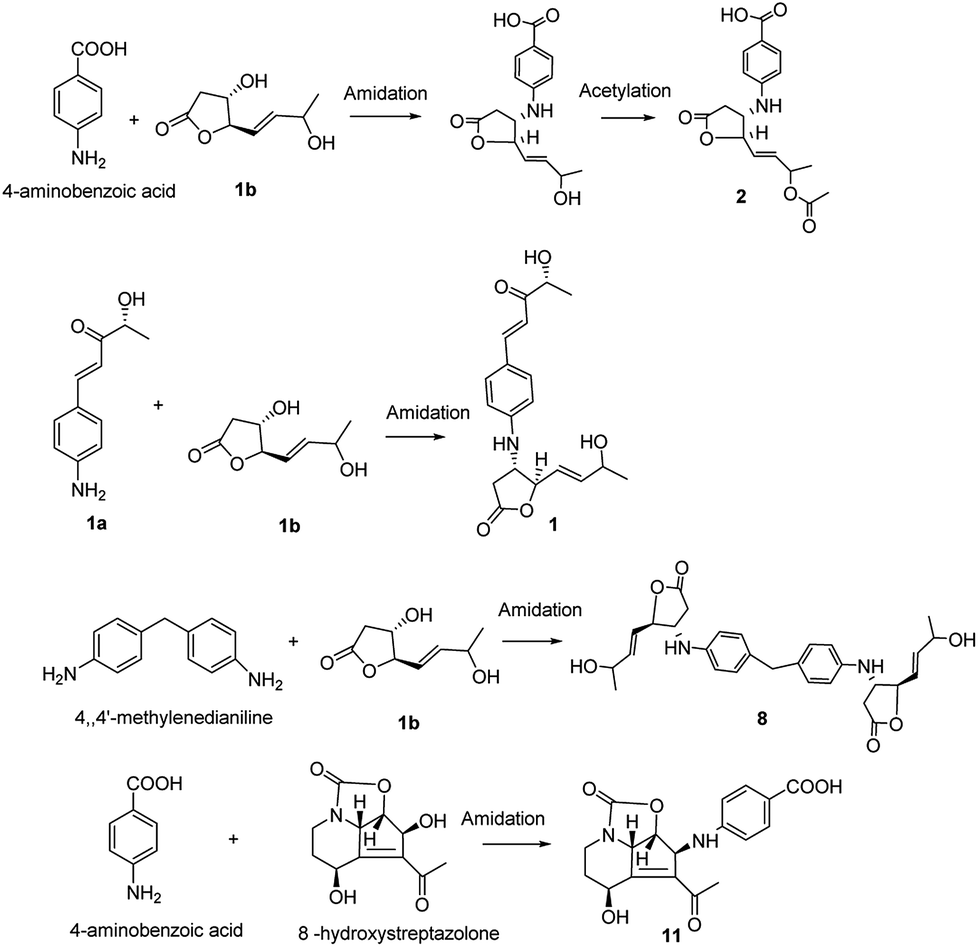 | ||
| Fig. 3 Proposed mechanism of the non-enzymatic synthesis of compounds 1, 2, 8, and 11. | ||
Conclusions
Eleven new compounds were isolated from Streptomyces alboniger, which was derived from soil in Tibet, China. Two types of unprecedented skeletons of obscurolide dimer and an obscurolide-type compound with a pentanone substituted at the benzene ring were found in this work. Compound 1 indicated an inhibitory effect on nitric oxide production in LPS-activated macrophages and anticoagulant activity on platelet activating factors (PAF)-induced platelet aggregation. 11 exhbitied anti-acetylcholinesterase activity. Mechanistic aspects of the non-enzymatic reactions of the biosynthetic relationships of the streptazolin and obscurolide-type metabolites were exhibited. The acylation of γ-butyrolactones was successfully used to explain the mechanism of influence of acid–base properties on the growth of Streptomyces.Experimental
General experimental procedure
Silica gel (200–300 mesh; Qingdao Marine Chemical Group Co.), Lichroprep RP-18 (Beijing Greenherbs and Technology and Development Co.) and Sephadex LH-20 (GE Healthcare Co.) were used for column chromatography. 1D and 2D NMR spectra were obtained on a Bruker AVANCE 500, 600 MHz NMR instrument (Bruker). MS spectra were recorded with an Agilent G3250AA (Agilent) and an AutoSpec Premier P776 spectrometer (Waters). ORs were obtained on a Jasco P-1020 polarimeter. Circular dichroism spectra were obtained using an Applied Photophysics Chirascan spectrometer (Applied Photophysics Ltd.).Biological material and cultivation of actinomycetic strain
The actinomycetic strain YIM20533 was isolated from the soil in Tibet, China. The species was identified as Streptomyces alboniger based on morphological and genetic (16S rRNA gene sequence) analyses. A voucher specimen was deposited at the Yunnan Institute of Microbiology, Kunming, P.R. China. This bacterium was cultivated on 80 L scale using 8 L seed medium (yeast extract 0.4%, glucose 0.4%, malt extract 0.3%, decavitamin 0.01%, pH 7.2) and the fermentation medium (soluble starch 2.4%, glucose 0.1%, peptone 0.3%, yeast extract 0.3%, CaCO3 0.3%, pH 7.0) at 28 °C for 7 days on a rotary shaker (130 rpm).Extraction and isolation of compounds
After 7 days of growth, the mycelia were removed from the cultures (80 L) by filtration. The filtrate was extracted with isometric ethyl acetate (EtOAc) 3 times, and the solvent was removed under vacuum to obtain the EtOAc extract (20.0 g). The EtOAc extract was separated into five fractions (Fr. 1–Fr. 5) by column chromatography on silica gel (200–300 mesh), eluting with a stepwise CHCl3/MeOH gradient (CHCl3; CHCl3/MeOH 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v; CHCl3/MeOH 25:1 v/v; CHCl3/MeOH 12.5:1 v/v; MeOH). Fr. 3 was divided into three parts (Fr. 3–1 to Fr. 3–3) using a Sephadex LH-20 column with MeOH. Fr. 3–1 was isolated using a Lichroprep RP-18 column with H2O/MeOH (60:30 v/v) and further purified using a silica gel column with petroleum ether/EtOAc gradient (4:1 to 2:1) to afford compounds 9 (10.5 mg) and 14 (4.6 mg). Fr. 3–2 was isolated by a Lichroprep RP-18 column with H2O/MeOH gradient (v/v 90:10 to 50:50) to give four subfractions, Compounds 10 (2.7 mg), 11 (2.4 mg), 12 (2.1 mg), and 15 (3.6 mg) were obtained from Fr. 3.2.2 using a silica gel column with petroleum ether/EtOAc gradient (4:1 to 1:1). Fr. 4 was separated into four fractions (Fr. 4–1 to Fr. 4–4) using a Sephadex LH-20 column with MeOH, then Fr 4–1 was isolated using a Lichroprep RP-18 column with H2O/MeOH gradient (v/v 90:10 to 50:50) and further purified by a silica gel column with a petroleum ether/EtOAc gradient (8:1 to 3:1) to afford compounds 4 (3.2 mg), 5 (3.6 mg), 6 (3.0 mg) and 7 (2.5 mg). Fr. 4–3 was isolated using a Lichroprep RP-18 column with H2O/MeOH gradient (v/v 90:10 to 50:50) and further purified using a silica gel column with a petroleum ether/EtOAc gradient (5:1 to 2:1) to afford compounds 1 (10.2 mg), 2 (4.3 mg) and 3 (2.2 mg). Fr. 4–4 was isolated using a Lichroprep RP-18 column with H2O/MeOH (v/v 80:20) and further purified using a silica gel column with a petroleum ether/EtOAc gradient (5:1 to 2:1) to afford compound 13 (3.2 mg). Fr. 5 was divided into three parts (Fr. 5–1 to Fr. 5–3) using a Sephadex LH-20 column with MeOH and compound 8 (11.8 mg) was obtained from Fr 5.3 using a silica gel column with a petroleum ether/EtOAc gradient elution (3:1 to 0:1).
:1 v/v; CHCl3/MeOH 25:1 v/v; CHCl3/MeOH 12.5:1 v/v; MeOH). Fr. 3 was divided into three parts (Fr. 3–1 to Fr. 3–3) using a Sephadex LH-20 column with MeOH. Fr. 3–1 was isolated using a Lichroprep RP-18 column with H2O/MeOH (60:30 v/v) and further purified using a silica gel column with petroleum ether/EtOAc gradient (4:1 to 2:1) to afford compounds 9 (10.5 mg) and 14 (4.6 mg). Fr. 3–2 was isolated by a Lichroprep RP-18 column with H2O/MeOH gradient (v/v 90:10 to 50:50) to give four subfractions, Compounds 10 (2.7 mg), 11 (2.4 mg), 12 (2.1 mg), and 15 (3.6 mg) were obtained from Fr. 3.2.2 using a silica gel column with petroleum ether/EtOAc gradient (4:1 to 1:1). Fr. 4 was separated into four fractions (Fr. 4–1 to Fr. 4–4) using a Sephadex LH-20 column with MeOH, then Fr 4–1 was isolated using a Lichroprep RP-18 column with H2O/MeOH gradient (v/v 90:10 to 50:50) and further purified by a silica gel column with a petroleum ether/EtOAc gradient (8:1 to 3:1) to afford compounds 4 (3.2 mg), 5 (3.6 mg), 6 (3.0 mg) and 7 (2.5 mg). Fr. 4–3 was isolated using a Lichroprep RP-18 column with H2O/MeOH gradient (v/v 90:10 to 50:50) and further purified using a silica gel column with a petroleum ether/EtOAc gradient (5:1 to 2:1) to afford compounds 1 (10.2 mg), 2 (4.3 mg) and 3 (2.2 mg). Fr. 4–4 was isolated using a Lichroprep RP-18 column with H2O/MeOH (v/v 80:20) and further purified using a silica gel column with a petroleum ether/EtOAc gradient (5:1 to 2:1) to afford compound 13 (3.2 mg). Fr. 5 was divided into three parts (Fr. 5–1 to Fr. 5–3) using a Sephadex LH-20 column with MeOH and compound 8 (11.8 mg) was obtained from Fr 5.3 using a silica gel column with a petroleum ether/EtOAc gradient elution (3:1 to 0:1).
| Pos. | 1 | Pos. | 2 | 3 | |||
|---|---|---|---|---|---|---|---|
| δH | δc | δH | δc | δH | δc | ||
| 1 | 176.0 | 1 | 175.7 | 172.9 | |||
| 2 | 2.37, 2.99 (m) | 34.3 | 2 | 2.39, 2.97 (m) | 34.2 | 2.39, 2.64 (m) | 35.4 |
| 3 | 4.09 (m) | 54.5 | 3 | 4.12 (m) | 54.3 | 3.88 (m) | 54.2 |
| 4 | 4.73 (m) | 85.2 | 4 | 4.70 (m) | 84.8 | 4.05 (m) | 72.9 |
| 5 | 5.74, 5.83 (m) | 124.5 | 5 | 5.80 (m) | 127.2 | 5.65 (m) | 131.0 |
| 6 | 5.74, 5.83 (m) | 139.0 | 6 | 5.80 (m) | 133.9 | 5.65 (m) | 131.7 |
| 7 | 4.34 (m) | 71.7 | 7 | 5.26 (m) | 69.8 | 5.16 (m) | 70.5 |
| 8 | 1.12 (d, J = 6.6 Hz) | 21.9 | 8 | 1.18 (d, J = 6.6 Hz) | 18.8 | 1.07 (d, J = 6.6 Hz) | 18.9 |
| 1′ | 149.9 | 1′ | 151.3 | 152.1 | |||
| 2′ | 6.56 (d, J = 8.4 Hz) | 112.8 | 2′ | 6.53 (d, J = 8.4 Hz) | 111.7 | 6.53 (d, J = 8.4 Hz) | 111.6 |
| 3′ | 7.40 (d, J = 8.4 Hz) | 130.5 | 3′ | 7.71 (d, J = 8.4 Hz) | 131.4 | 7.66 (d, J = 8.4 Hz) | 131.3 |
| 4′ | 123.7 | 4′ | 118.7 | 117.5 | |||
| 5′ | 7.40 (d, J = 8.4 Hz) | 130.5 | 5′ | 7.71 (d, J = 8.4 Hz) | 131.4 | 7.66 (d, J = 8.4 Hz) | 131.3 |
| 6′ | 6.56 (d, J = 8.4 Hz) | 112.8 | 6′ | 6.53 (d, J = 8.4 Hz) | 111.7 | 6.53 (d, J = 8.4 Hz) | 111.6 |
| 7′ | 7.55 (d, J = 15.6 Hz) | 145.0 | 7′ | 169.2 | 169.2 | ||
| 8′ | 6.80 (d, J = 15.6 Hz) | 115.7 | 1′′ | 170.7 | 170.7 | ||
| 9′ | 202.5 | 2′′ | 1.93 (s) | 19.7 | 1.94 (s) | 19.7 | |
| 10′ | 4.21 (m) | 66.8 | 3.51 (s, OCH3) | 50.7 (OCH3) | |||
| 11′ | 1.26 (d, J = 7.2 Hz) | 19.2 | |||||
| Pos. | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δH | δc | δH | δc | δH | δc | |
| 1 | 175.9 | 176.3 | 176.7 | |||
| 2 | 2.45(dd, J = 5.4 Hz, 18.0 Hz) | 34.8 | 2.39 (dd, J = 5.4 Hz, 18.0 Hz) | 35.0 | 2.43 ( dd, J = 1.8 Hz, 18.0 Hz) | 35.6 |
| 3.10 (dd, J = 7.2 Hz, 18.0 Hz) | 3.07 (dd, J = 7.2 Hz, 18.0 Hz) | 3.11 ( dd, J = 7.2 Hz, 18.0 Hz) | ||||
| 3 | 4.13 (m) | 53.7 | 4.04 (m) | 54.4 | 4.34 (m) | 50.6 |
| 4 | 4.40 (m) | 84.6 | 4.37 (m) | 84.9 | 4.34 (m) | 88.3 |
| 5 | 1.93, 2.09 (m) | 27.2 | 1.91, 2.07 (m) | 27.2 | 4.24 (m) | 72.4 |
| 6 | 2.70 (m) | 38.3 | 2.68 (m) | 38.4 | 5.57 (m) | 129.2 |
| 7 | 208.7 | 208.8 | 5.82 (m) | 128.9 | ||
| 8 | 2.14 (s) | 28.4 | 2.13 (s) | 28.4 | 1.69 (d, J = 6.0 Hz) | 16.5 |
| 1′ | 151.3 | 146.9 | 151.2 | |||
| 2′ | 6.64 (d, J = 8.0 Hz) | 111.6 | 6.63 (d, J = 8.4 Hz) | 112.9 | 6.66 (d, J = 9.0 Hz) | 111.6 |
| 3′ | 7.82 (d, J = 8.0 Hz) | 131.4 | 7.13 (d, J = 8.4 Hz) | 129.4 | 7.81 (d, J = 9.0 Hz) | 131.4 |
| 4′ | 118.6 | 126.9 | 118.5 | |||
| 5′ | 7.82 (d, J = 8.0 Hz) | 131.4 | 7.13 (d, J = 8.4 Hz) | 129.4 | 7.81 (d, J = 9.0 Hz) | 131.4 |
| 6′ | 6.64 (d, J = 8.0 Hz) | 111.6 | 6.63 (d, J = 8.4 Hz) | 112.9 | 6.66 (d, J = 9.0 Hz) | 111.6 |
| 7′ | 169.1 | 4.31 (s) | 74.3 | 169.2 | ||
| 8′ | 3.31 (s) | 56.3 | ||||
| Pos. | 7 | 8 | 9 | |||
|---|---|---|---|---|---|---|
| δH | δc | δH | δc | δH | δc | |
| 1 | 177.2 | 178.0 | 2.15 (s) | 24.0 | ||
| 2 | 2.39 (d, J = 18.0 Hz) | 35.8 | 2.41 (dd, J = 1.6 Hz, 20.0 Hz) | 35.8 | 211.4 | |
| 3.12 (dd, 7.8 Hz, 18.0 Hz) | 3.04 (dd, J = 8.0 Hz, 18.0 Hz) | |||||
| 3 | 4.32 (brs) | 49.0 | 4.05 (m) | 56.6 | 4.11 (m) | 57.9 |
| 4 | 4.37 (brs) | 88.3 | 4.81 (m) | 86.7 | 1.40 (d, J = 7.2 Hz) | 16.0 |
| 5 | 4.32 (brs) | 71.7 | 5.79 (m) | 126.3 | ||
| 6 | 5.56 (m) | 128.8 | 5.90 (m) | 139.9 | ||
| 7 | 5.91 (m) | 128.7 | 4.28 (m) | 68.3 | ||
| 8 | 1.68 (d, J = 6.6 Hz) | 16.6 | 1.22 (d, J = 6.4 Hz) | 23.3 | ||
| 1′ | 151.0 | 146.3 | 151.7 | |||
| 2′ | 6.64 (d, J = 8.0 Hz) | 111.7 | 6.56 (d, J = 8.0 Hz) | 114.8 | 6.56 (d, J = 8.4 Hz) | 111.2 |
| 3′ | 7.80 (d, J = 8.0 Hz) | 131.3 | 6.95 (d, J = 8.0 Hz) | 130.6 | 7.79 (d, J = 8.4 Hz) | 131.4 |
| 4′ | 118.9 | 133.0 | 118.0 | |||
| 5′ | 7.80 (d, J = 8.0 Hz) | 131.3 | 6.95 (d, J = 8.0 Hz) | 130.6 | 7.79 (d, J = 8.4 Hz) | 131.4 |
| 6′ | 6.64 (d, J = 8.0 Hz) | 111.7 | 6.56 (d, J = 8.0 Hz) | 114.8 | 6.56 (d, J = 8.4 Hz) | 111.2 |
| 7′ | 169.5 | 3.71 (s) | 41.1 | 169.1 | ||
| Pos. | 11 | 13 | ||
|---|---|---|---|---|
| δH | δc | δH | δc | |
| 1 | ||||
| 2 | 157.8 | 158.0 | ||
| 3 | ||||
| 4 | 4.63 (m) | 78.0 | 4.47 (m) | 80.1 |
| 5 | 4.95 (brs) | 66.0 | 4.54 (m) | 80.7 |
| 6 | 132.7 | 138.3 | ||
| 7 | 151.7 | 139.8 | ||
| 8 | 4.66 (m) | 68.7 | 4.43 (m) | 68.4 |
| 9 | 1.49, 2.12 (m) | 36.4 | 1.33, 1.93 (m) | 36.3 |
| 10 | 3.22, 3.85 (m) | 40.4 | 3.11, 3.73 (m) | 40.5 |
| 11 | 4.75 (d, J = 6.0 Hz) | 65.5 | 4.47 (m) | 65.2 |
| 12 | 203.1 | 4.71 (m) | 63.3 | |
| 13 | 2.40 (s) | 29.7 | 1.27 (d, J = 7.0 Hz) | 21.9 |
| 1′ | 150.7 | |||
| 2′ | 6.74 (d, J = 8.0 Hz) | 111.8 | ||
| 3′ | 7.87 (d, J = 8.0 Hz) | 131.5 | ||
| 4′ | 118.7 | |||
| 5′ | 7.87 (d, J = 8.0 Hz) | 131.5 | ||
| 6′ | 6.74 (d, J = 8.0 Hz) | 118.5 | ||
| 7′ | 169.6 | |||
Bioactivity assay
The AChE inhibitory activities of the compounds were assayed by the spectrophotometric method developed by Ellman et al.26 with slight modification. Tacrine (Sigma, purity > 99%) was used as a positive control with a final concentration of 0.333 μM with an inhibition ratio of 57.4%. The NO inhibitory activity of these compounds was determined using the Griess reagent assay for NO production. L-NMMA was used as the positive control with an inhibition ratio of 55.04 ± 2.80% at a concentration of 50 μM. The in vitro anticoagulant activities were investigated using the prothrombin time (PT) method for compounds 1, 9, and 11, the activated partial thromboplastin time (APTT) for compounds 1, 8, 9, and 11, thrombin time (TT) for compounds 1, 9, and 11, adenosine diphosphate (ADP) induced platelet aggregation for compound 1 and PAF-induced platelet aggregation for compound 1. Heparin was used as the positive control with PT at 37.3 ± 1.08 s at the tested concentration of 160 μg mL−1. Low molecular weight heparin (LMWH) was used as the positive control for APTT at 79.7 ± 0.36 s, and TT at 18.5 ± 2.06 s at the tested concentration of 35.6 μM. Ticagrelor and GB were used as positive controls in ADP and PAF-induced platelet aggregation for the inhibition ratios at 69.8 ± 7.8% (final concentration: 5 μg mL−1) and 93.4 ± 5.1 (final concentration: 37 μg mL−1), respectively. The cytotoxicity of the compounds against HL-60, SMMC-7721, A-549, MCF-7, and SW480 cell lines was determined in vitro by the 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethoxyphenyl)-2-(4-sulfopheny)-2H-tetrazolium (MTS) method. Taxol was used as a positive control with IC50 < 0.008 μM.pH-static fermentation
The fermentation process was analyzed for a total of 7 days. 48 hours after inoculation, the pH-value was adjusted to pH 2.5, 3.5, 4.5, 6.0, 7.0, 8.0 with citric acid and NaOH. The growth of Streptomyces was observed at different times.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research work was financially supported by the National Natural Science Foundation of China (81560571, 21562045, 81660582, 31660532), and a project of Yunling Scholars of Yunnan Province.References
- J. Begani, J. Lakhani and D. Harwani, Ann. Microbiol., 2018, 68, 419–432 CrossRef.
- Y. P. Chen, Q. Liu, H. Gao, H. P. Lin, H. Y. Tian, K. Hong, J. Li, R. W. Jiang, X. S. Yao and J. S. Tang, RSC Adv., 2014, 4, 63324–63327 RSC.
- D. Nichols, N. Cahoon, E. M. Trakhtenberg, L. Pham, A. Mehta, A. Belanger, T. Kanigan, K. Lewis and S. S. Epstein, Appl. Environ. Microbiol., 2010, 76, 2445–2450 CrossRef CAS PubMed.
- B. B. Hou, L. Y. Tao, X. Y. Zhu, W. Wu, M. J. Guo, J. Ye, H. Z. Wu and H. Z. Zhang, Appl. Microbiol. Biotechnol., 2018, 102, 4101–4115 CrossRef CAS PubMed.
- S. Kitani, K. T. Miyamoto, S. Takamatsu, E. Herawati, H. Iguchi, K. Nishitomi, M. Uchida, T. Nagamitsu, S. Omura, H. Ikeda and T. P. Nihira, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16410–16415 CrossRef CAS PubMed.
- K. Tiwari and R. K. Gupta, Crit. Rev. Biotechnol., 2012, 32, 108–132 CrossRef CAS PubMed.
- Y. Takahashi and T. Nakashima, Antibiotics, 2018, 7, 45 CrossRef PubMed.
- I. Djinni, W. Djoudi, S. Souagui, F. Rabia, S. Rahmouni, I. Mancini and M. Kecha, J. Microbiol. Methods, 2018, 148, 161–168 CrossRef CAS PubMed.
- C. L. Yang, Y. S. Wang, C. L. Liu, Y. J. Zeng, P. Cheng, R. H. Jiao, S. X. Bao, H. Q. Huang, R. X. Tan and H. M. Ge, Mar. Drugs, 2017, 15, 244 CrossRef PubMed.
- F. Z. Li, N. C. Warshakoon and M. J. Miller, J. Org. Chem., 2004, 69, 8836–8841 CrossRef CAS PubMed.
- I. Nomura and C. Mukai, Org. Lett., 2002, 4, 4301–4304 CrossRef CAS PubMed.
- B. M. Trost, C. K. Chung and A. B. Pinkerton, Angew. Chem., Int. Ed., 2004, 43, 4327–4329 CrossRef CAS PubMed.
- J. A. Perry, K. Koteva, C. P. Verschoor, W. Wang, A. M. E. Bowdish and G. D. Wright, J. Antibiot., 2015, 68, 40–46 CrossRef CAS PubMed.
- G. Zappia, P. Menendez, G. D. Monache, D. Misiti, L. Nevola and B. Botta, Mini-Rev. Med. Chem., 2007, 7, 389–409 CrossRef CAS PubMed.
- H. Hoff, H. Drautz, H. P. Fiedler, H. Zahner, J. E. Schultz, W. Keller-Schierlein, S. Philipps, M. Ritzau and A. Zeeck, J. Antibiot., 1992, 45, 1096–1107 CrossRef CAS PubMed.
- M. Ritzau, S. Philipps, A. Zeeck, H. Hoff and H. Zahner, J. Antibiot., 1993, 46, 1625–1628 CrossRef CAS PubMed.
- E. Takano, Curr. Opin. Microbiol., 2006, 9, 287–294 CrossRef CAS PubMed.
- M. Biarnes-Carrera, R. Breitling and E. Takano, Curr. Opin. Chem. Biol., 2015, 28, 91–98 CrossRef CAS PubMed.
- X. M. Zhang, D. F. Zhang, W. J. Li and C. H. Lu, Helv. Chim. Acta, 2016, 99, 191–196 CrossRef CAS.
- A. Cosp, C. Dresen, M. Pohl, L. Walter, C. Röhr and M. Müller, Adv. Synth. Catal., 2008, 350, 759–771 CrossRef CAS.
- A. Schneider, J. Spath, S. BreidingMack, A. Zeeck, S. Grabley and R. Thiericke, J. Antibiot., 1996, 49, 438–446 CrossRef CAS PubMed.
- B. M. Nestl, A. Bodlenner, R. Stuermer, B. Hauer, W. Kroutila and K. Faber, Tetrahedron: Asymmetry, 2007, 18, 1465–1474 CrossRef CAS.
- F. C. Pollak and R. G. Berger, Appl. Environ. Microbiol., 1996, 62, 1295–1299 CAS.
- H. Yamada, S. Aoyagi and C. Kibayashi, Tetrahedron Lett., 1996, 37, 8787–8790 CrossRef CAS.
- R. Thiericke and M. Zerlin, Nat. Prod. Lett., 1996, 8, 163–16726 CrossRef CAS.
- G. L. Ellman, K. D. Courtney, V. J. Andres and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88–95 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR, HSQC, HMBC, 1H–1H COSY, ROESY and HR-ESIMS spectra of compounds 1–9, 11, and 13. CD spectra of 6 and 7. See DOI: 10.1039/c8ra06690f |
| ‡ N. Luo and Y. B. Yang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |