DOI:
10.1039/C8RA06866F
(Paper)
RSC Adv., 2018,
8, 35031-35041
miR-183-96-182 clusters alleviated ox-LDL-induced vascular endothelial cell apoptosis in vitro by targeting FOXO1
Received
16th August 2018
, Accepted 17th September 2018
First published on 12th October 2018
Abstract
Objective: To investigate the role of FOXO1 and miR-183-96-182 clusters in ox-LDL induced endothelial cell apoptosis. Methods: FOXO1 overexpression (OE) and knockdown (KD) as well as AKT1 OE in human umbilical vein endothelial cells (HUVECs) and human aortic endothelial cells (HAECs) were achieved by lentiviral transduction. Upregulation of miR-183-5p, miR-182-5p or miR-96-5p was mimicked by agomir treatment. FOXO1 gene transcription was monitored by FOXO1 promotor reporter assay. Cell apoptosis in culture was monitored by TiterTACS in situ detection. Regulation of FOXO1 gene expression by an miRNA targeting mechanism was monitored by AGO2-RNA immunoprecipitation assay. Results: FOXO1 mRNA and protein expression levels in ox-LDL treated HUVECs or HAECs were significantly upregulated due to transcriptional and miRNA targeting mechanisms. MiR-183-5p, miR-182-5p and miR-96-5p expression levels in HUVECs or HAECs were significantly reduced by ox-LDL treatment, the overexpression of which by agomir treatment partially reduced the FOXO1 mRNA/protein expression levels and cell apoptosis which was upregulated by ox-LDL treatment. FOXO1 overexpression antagonized the effect of the agomir treatment indicated above. MiR-183-5p, miR-182-5p and miR-96-5p agomir treatment partially rescued the FOXO1 pSer256/total FOXO1 protein ratio and the AKT1 pSer473 level that were reduced by ox-LDL treatment in the HUVECs or HAECs. AKT1 overexpression significantly reduced FOXO1 protein expression, increased miR-182-5p and miR-183-5p expression, and partially alleviated ox-LDL induced HUVEC or HAEC apoptosis in an miR-183-5p and miR-182-5p-dependent manner. Conclusion: miR-183-96-182 clusters could partially alleviate ox-LDL-induced apoptosis in HUVECs or HAECs by targeting FOXO1.
Introduction
Vascular endothelial cell dysfunction and death contribute significantly to the pathogenesis and early development of atherosclerosis, a peripheral arterial disease with higher morbidity in the elderly than others.1,2 Malfunctions in endothelial cells that line the inner wall of the artery are often characterized by uncontrolled synthesis of reactive oxygen species (ROS) molecules as well as the hyper-activation of pro-inflammatory and pro-apoptotic signaling cascades, leading to disruptive cell apoptosis or pyroptosis.1,3 Disruption of the arterial wall increases its permeability, allowing an influx of other cytotoxic and pro-inflammatory mediators, such as macrophages, T cells, oxidized low-density lipoprotein (ox-LDL), ROS and others, into the vascular smooth muscle milieu, which triggers a wider range of inflammatory responses and worsens the syndrome.1,4 Reducing or inhibiting endothelial cell apoptosis is considered a possible approach to alleviate atherogenesis at an early stage. Previous studies have demonstrated that when binding to its ligand ox-LDL, which is produced by oxidation of low-density lipoproteins from various cell types and extracellular space, the activation of the scavenger receptor lectin-type oxidized LDL receptor 1 (LOX-1) and the triggered downstream signaling cascade are the predominant causes of ROS synthesis, pro-inflammatory responses and apoptosis in endothelial cells. In particular, the activation of LOX-1 reduces the kinase activity of AKT through inhibiting the upstream kinase phosphatidyl inositol 3-kinase (PI3K) and AKT inhibition untying the transcriptional activity of FOXO1 due to a decrease in the inhibitory phosphorylation at Ser256 on the latter.5,6 Hyperactivated FOXO1 induced by LOX-1 signaling therefore increases the transcription of various pro-apoptotic genes, such as Bim, TNF-related apoptosis-inducing ligands and Fas ligands, and promotes endothelial cell apoptosis, meanwhile the idea of targeting the LOX-1/AKT/FOXO1 axis has inspired several conceivable approaches for anti-apoptotic efforts in atherosclerotic endothelial cells, one of which is the manipulation of microRNAs (miRNAs).7–9
Current knowledge on the involvement and regulatory function of miRNAs in maintaining homeostasis as well as in the pathogenesis and development of atherosclerosis has been reviewed by Schober et al..4 Briefly, by regulating the mRNA stability and protein translation of their target genes, different miRNAs may achieve either atherosclerosis-promoting or -suppressing results.7,10 Studies on the regulation of miRNAs in endothelial cell apoptosis have recently adopted high throughput techniques such as miRNA sequencing and microarray analysis to identify multiple miRNAs which are differentially expressed between atherosclerotic and healthy conditions and have not previously been noted.11–14 In the present research, we investigated the results of a previously-published study that employed such high throughput techniques, and noticed that two miRNAs, namely miR-183-5p and miR-182-5p, were sharply downregulated in atherosclerotic lesions compared to healthy counterparts derived from atherosclerotic model mice.12 The role that these two miRNAs play in atherogenesis is largely undetermined. A previous study performed by Qin et al. demonstrated that transfection with miR-182-5p mimic could partially reduce ROS production and apoptosis in an ox-LDL treated RAW264.7 macrophage cell line by targeting the Toll-like receptor 4.15 The anti-apoptotic role of an miR-183-96-182 cluster in various tumor cells has been well characterized; notably, Tang et al. reported that suppression of an miR-183-96-182 cluster enhanced ROS-induced apoptosis in glioma cells in vitro.16–19 The pro-apoptotic transcription factor FOXO1 has been identified as a co-target of miR-183-5p, miR-96-5p and miR-182-5p.20–23 A recent study by Li et al. showed that the anti-apoptotic effect of an antioxidant enzyme, glutaredoxin 1, in endothelial cells is partially mediated by its regulation on the AKT/FOXO1 axis,24 although the involvement of FOXO1 in ox-LDL induced endothelial cell apoptosis hasn’t been properly examined before. In the present study we investigated our hypothesis that miR-183-5p, miR-182-5p and miR-96-5p would decrease ox-LDL-induced endothelial cell apoptosis at least in part by targeting FOXO1. We employed agomir treatment to mimic the endogenous upregulation of the three miRNAs; endothelial cell apoptosis, as well as FOXO1 mRNA and protein expression in endothelial cells, was monitored before and after ox-LDL treatment.
Materials and methods
Cell culture and treatment
HUVEC and HAEC human vascular endothelial cell lines were purchased from the National Infrastructure of Cell Line Resource (Shanghai, China) and cultured in 200PRF medium (Invitrogen) supplemented with Low serum growth supplement (Invitrogen) following the manufacturer’s instructions. HUVECs and HAECs were cultured in a humidified incubator with 37 °C atmosphere supplemented with 5% CO2. Lentiviral vectors for constructing FOXO1 knockdown (KD), FOXO1 overexpression (OE) or AKT1 OE cell lines were purchased from Fulengen (Guangzhou, China) and were applied following the manufacturer’s instructions. Cells at about 50% confluence were subject to lentiviral transduction at a multiplicity of infection of 50 transduction units per cell. The sufficiency of gene OE or KD was verified by RT-qPCR and western blot analysis. The overexpression or inhibition of miR-183-5p, miR-182-5p or miR-96-5p in HUVECs and HAECs were mimicked by agomir or antagomir treatment, respectively. MiRNA agomirs and antagomirs involved in the present research were purchased from Genepharma (Shanghai, China) and were used for cell treatment at 50 pM with 1‰ (v/v to culture medium) lipofectamine 2000 (Thermo Fisher Scientific, Shanghai, China) for 24 hours under culture conditions. Native human ox-LDL was purchased from Solarbio (Beijing, China) and was used for cell treatment at 50 μg ml−1 for 24 hours under culture conditions. Other than the cell apoptosis assay (described later in detail), cells were cultured on a 6 well plate until 80% confluence was reached before treatment.
RT-qPCR and western blot analysis
Primers for RT-qPCR analysis semi-quantifying the transcription level of FOXO1, AKT1, miR-183-5p, miR-182-5p and miR-96-5p were purchased as a customized kit from Genecopoeia (Guangzhou, China) and applied following the manufacturer’s instructions. GAPDH was used as an internal reference for FOXO1 and AKT1 quantification, and U6 was used for the three miRNAs. Semi-quantification was performed using the 2⁁-ΔΔCt method and presented as fold change compared to an external reference (negative control). For western blot analysis, primary antibodies against FOXO1 (NB100-2312), FOXO1-pSer256 (NB100-81927), AKT1 (NBP1-51602), AKT1-pSer473 (NB100-56749), and GAPDH (NB100-56875) were purchased from Bio-techne (Shanghai, China). HRP-conjugated anti-rabbit (ab205718) and anti-mouse (ab6789) secondary antibodies were purchased from Abcam (Shanghai, China). Primary antibodies were used at a working concentration of 1 μg ml−1 and the secondary antibodies were used at a working concentration of 0.2 μg ml−1. Western blot analysis was performed under reducing conditions. Semi-quantification of protein expression by western blot analysis was performed by comparing the gray scale of the WB band of each target protein to that of beta-actin using ImageJ software (Ver. 1.51) and presented as fold change compared to an external reference (negative control). RT-qPCR and western blot experiments were performed following universal protocols.
Cell apoptosis assay
Cell apoptosis was evaluated in situ by a colorimetric method using a TiterTACS In Situ Detection Kit (Bio-techne) following the manufacturer’s instructions. About 5 × 104 cells per well cultured on a 96-well plate were treated with ox-LDL as indicated or a vehicle (PBS) of the same volume before analysis.
FOXO1 promoter reporter assay
This assay was performed to compare the FOXO1 gene transcription level in HUVECs or HAECs before and after ox-LDL treatment. Cells were transfected with pEZX-PG04 reporter plasmids carrying the secreted alkaline phosphatase gene as a reference gene and the Gaussia luciferase (GLuc) gene flanked at the 5′ position by a promoter sequence located 1239 bp upstream and 240 bp downstream of the transcription start site of the FOXO1 gene on the human genome. Cells with positive transfection were selected by puromycin (Solarbio), and GLuc activity was measured using the Secrete-Pair™ Dual Luminescence Assay Kit (Genecopoeia) following the manufacturer’s instructions.
AGO2-RNA immunoprecipitation (AGO2-RIP) assay
For AGO2-RIP, cells after ox-LDL or vehicle (PBS) treatment were lysed with a cell lysis buffer for Western and IP (Beyotime, Shanghai, China). A mouse monoclonal anti-human AGO2 antibody (H00027161-M01, Bio-techne) was incubated at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 ratio with cell lysate for 2 hours at 4 °C with gentle agitation, followed by incubation with Pierce™ Protein A/G Magnetic Beads (Thermo Fisher Scientific) for another 2 hours at 4 °C with gentle agitation. The beads were then isolated with a magnet, and the proteins or RNAs associated with the beads as indicated were detected by western blot or RT-qPCR analysis, respectively.
:100 ratio with cell lysate for 2 hours at 4 °C with gentle agitation, followed by incubation with Pierce™ Protein A/G Magnetic Beads (Thermo Fisher Scientific) for another 2 hours at 4 °C with gentle agitation. The beads were then isolated with a magnet, and the proteins or RNAs associated with the beads as indicated were detected by western blot or RT-qPCR analysis, respectively.
Statistical analysis
Statistical analysis was performed using Graphpad Prism software (Ver. 7). All data represent 6 independent replicates and were presented as mean ± SD, unless otherwise indicated. Data were normalized to a control group and were presented as fold change when applicable. Student’s t test and one-way analysis of variance with Tukey’s test as the post hoc test were used for the significance tests. A p value < 0.05 was considered significant.
Results
FOXO1 expression in HUVECs or HAECs was regulated by transcriptional and miRNA targeting mechanisms
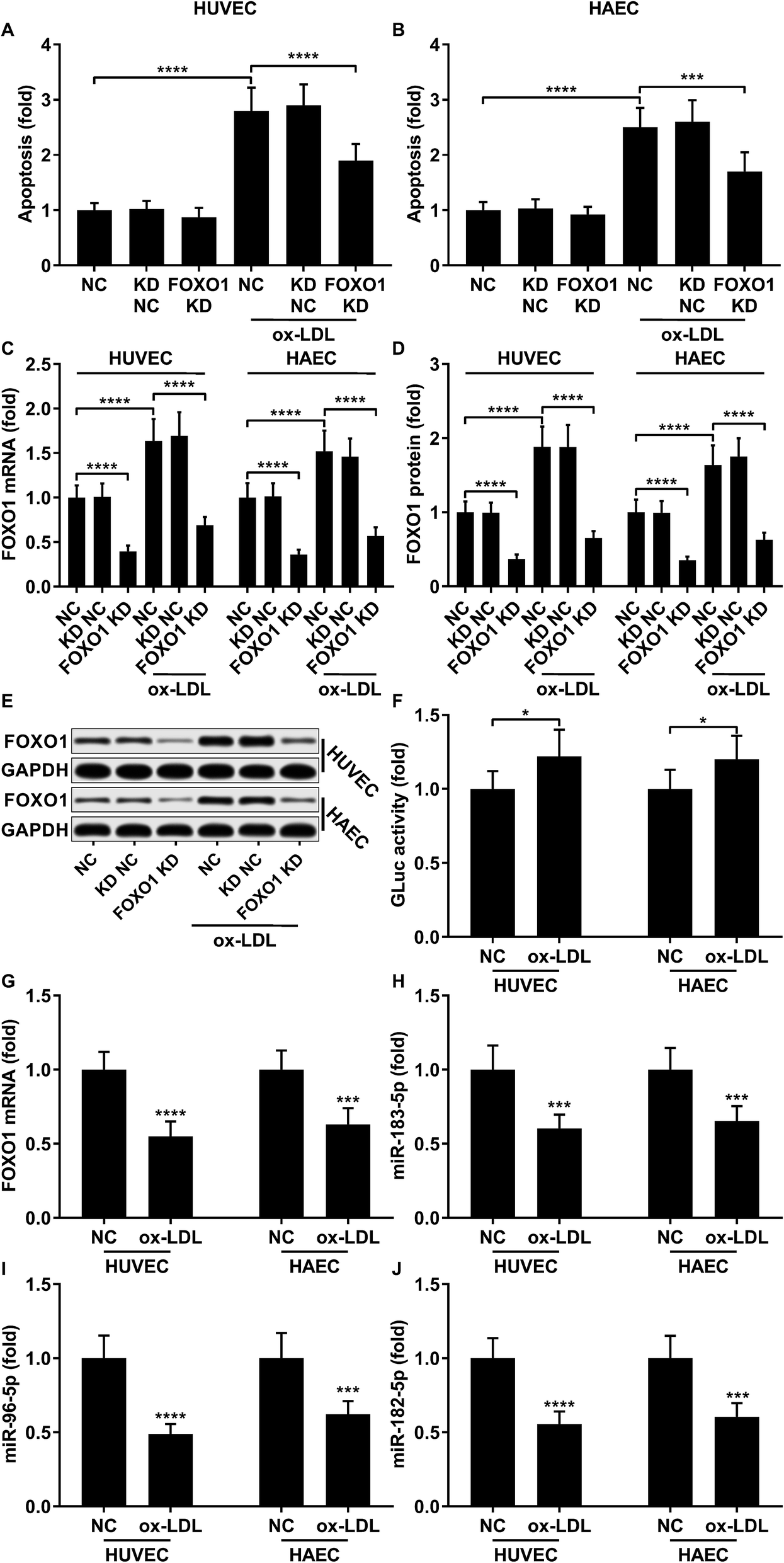
To first verify the role of FOXO1 in ox-LDL-induced endothelial cell apoptosis in vitro, we examined cell apoptosis as well as the FOXO1 mRNA and protein expression levels in ox-LDL-treated endothelial cells with or without FOXO1 knockdown. Our data show that ox-LDL treatment significantly increases HUVEC or HAEC apoptosis and FOXO1 gene expression in vitro, all of which can be significantly attenuated by FOXO1 knockdown (Fig. 1A–E), suggesting the pro-apoptotic role of FOXO1 in ox-LDL induced endothelial cell apoptosis. To our surprise, the FOXO1 promotor reporter assay results suggest that FOXO1 gene transcription in ox-LDL treated HUVECs or HAECs only moderately increases compared to that in un-treated ones, which cannot fully explain the drastic upregulation of FOXO1 mRNA and protein expression levels induced by the ox-LDL treatment (Fig. 1F). We hypothesized that FOXO1 gene expression in the HUVECs and HAECs in our experiment was regulated not only by transcriptional regulation but also by post-transcriptional regulatory mechanisms. To investigate whether FOXO1 gene expression was regulated by miRNA targeting, we performed an AGO2-RIP assay to detect FOXO1 mRNA associating with the AOG2 co-immunoprecipitated complex. The AGO2-RIP results show that the abundance of FOXO1 mRNA in the AGO2 co-immunoprecipitated complex is significantly reduced in cell lysates derived from HUVECs or HAECs after ox-LDL treatment compared to un-treated ones (Fig. 1G), suggesting that FOXO1 gene expression in these two endothelial cell lines is indeed regulated by an miRNA-targeting mechanism, which seems to be weakened by ox-LDL treatment.
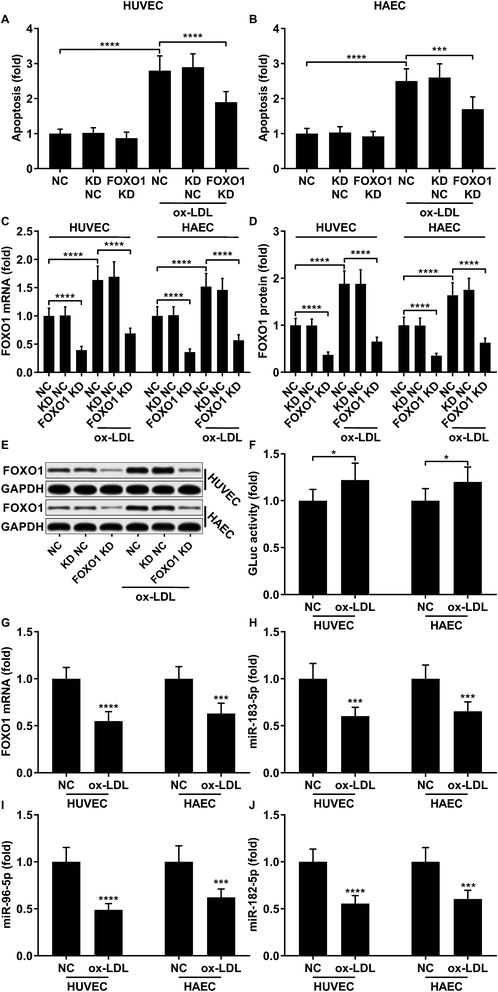 |
| | Fig. 1 Upregulation of pro-apoptotic FOXO1 in ox-LDL treated HUVECs or HAECs is due to transcriptional regulation and reduction in the miRNA targeting mechanism. (A and B) Cell apoptosis in ox-LDL treated HUVECs or HAECs could be partially relieved by FOXO1 knockdown. (C–E) FOXO1 knockdown drastically reduced FOXO1 mRNA and protein expression levels in the HUVECs or HAECs with or without ox-LDL treatment. (F) FOXO1 promoter reporter assay suggests a mild upregulation of FOXO1 gene transcription. (G–J) AGO2-RIP assay shows that FOXO1 mRNA, miR-183-5p, miR-96-5p and miR-182-5p were downregulated in AGO2 co-immunoprecipitated complexes after ox-LDL treatment. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001. | |
miR-183-5p, miR-182-5p and miR-96-5p alleviated ox-LDL induced endothelial cell apoptosis by targeting FOXO1
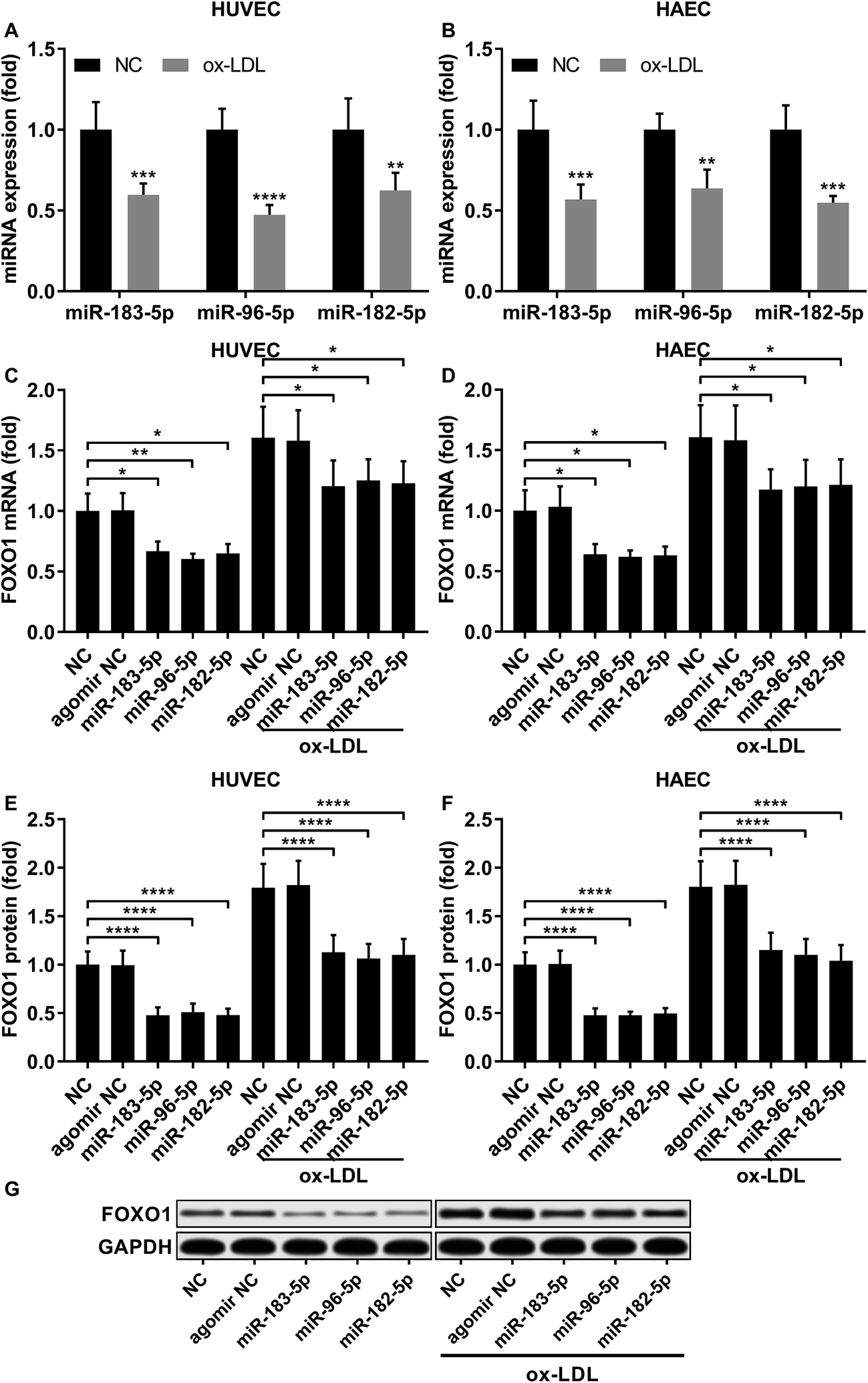
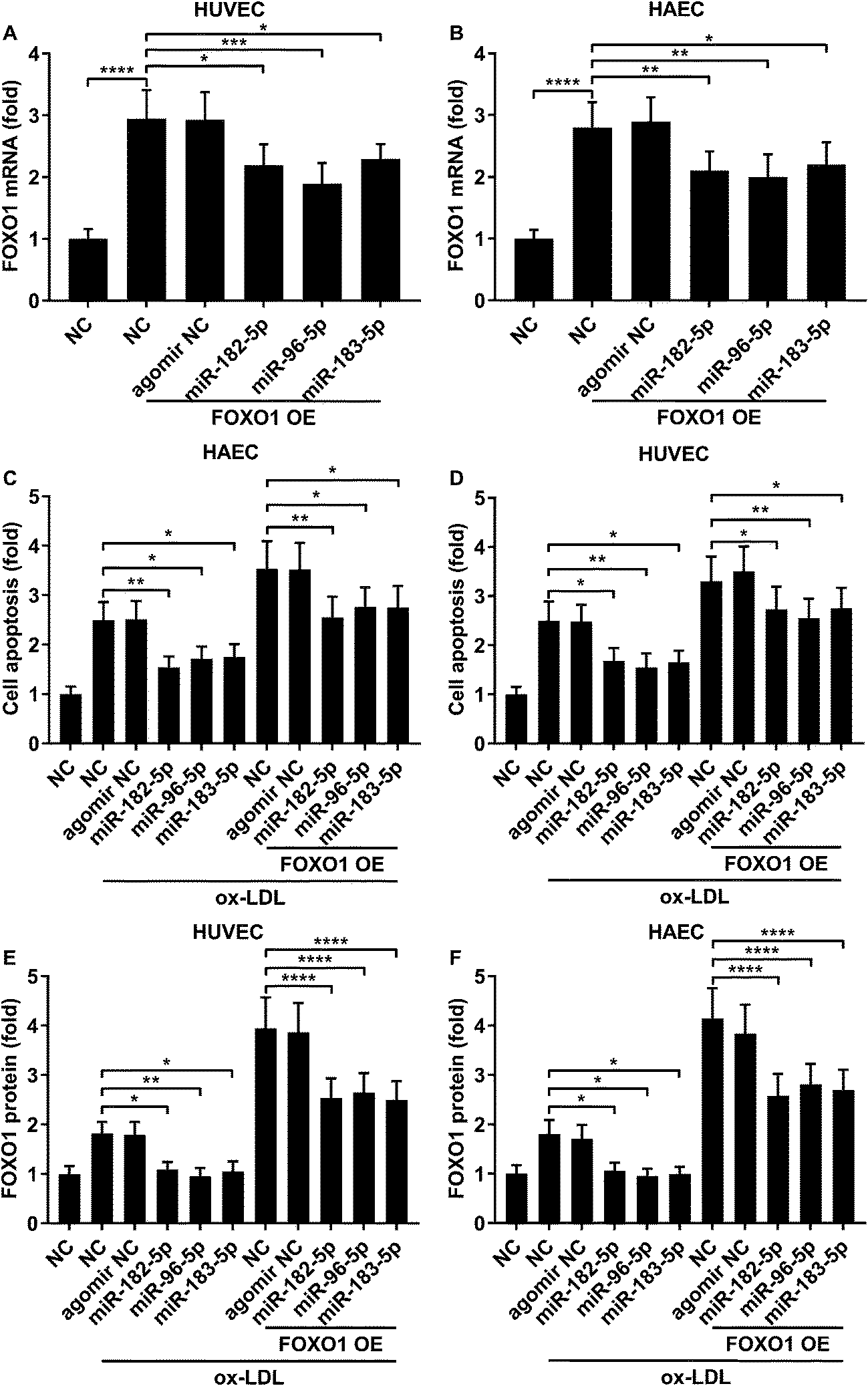
Previous studies have demonstrated that FOXO1 mRNA can be targeted by miR-183-5p, miR-182-5p and miR-96-5p, the former two of which have been found to be downregulated in atherosclerotic lesions,12 while our AGO-RIP data also demonstrated a significant decrease in miR-183-5p, miR-182-5p and miR-96-5p abundance in the AGO2 co-immunoprecipitated complexes after ox-LDL treatment (Fig. 1H–J). We therefore hypothesized that the FOXO1 expression in endothelial cells was regulated by these three miRNA, which might alleviate ox-LDL induced endothelial cell apoptosis. RT-qPCR results reveal that miR-183-5p, miR-96-5p or miR-182-5p expression levels are significantly reduced in endothelial cells after ox-LDL treatment (Fig. 2A and B). Treatment with agomir mimicking miR-183-5p, miR-96-5p or miR-182-5p overexpression significantly reduced FOXO1 mRNA and protein expression levels in the HUVECs and HAECs with or without ox-LDL treatment (Fig. 2C–G), suggesting that these three miRNAs can downregulate FOXO1 gene expression in endothelial cells. We also found that treatment with these three miRNA agomirs significantly reduced FOXO1 mRNA and protein expression in FOXO1 overexpressing HUVECs or HAECs (Fig. 3A, B and E, F). Treatment with the three miRNA agomirs significantly reduced cell apoptosis induced by ox-LDL treatment in the HUVECs and HAECs, which can be partially overturned by FOXO1 overexpression (Fig. 3C and D). The data suggested that ox-LDL treatment significantly upregulated endothelial cell apoptosis and endogenous FOXO1 gene expression while downregulating that of miR-183-5p, miR-182-5p or miR-96-5p, with the potential to partially reduce ox-LDL treatment-induced endothelial cell apoptosis by targeting FOXO1.
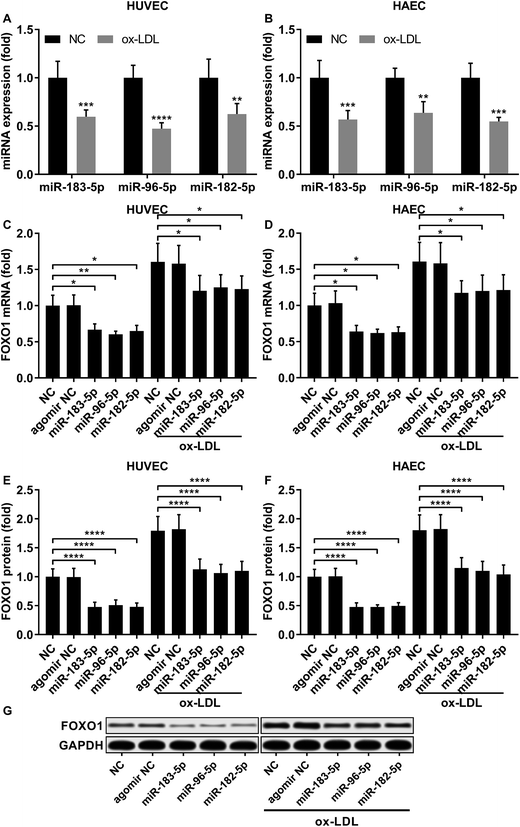 |
| | Fig. 2 FOXO1 gene expression was reduced by miR-183-5p, miR-96-5p or miR-182-5p agomir treatment in HUVECs or HAECs with or without ox-LDL treatment. (A and B) Expression levels of the three miRNAs significantly decrease after ox-LDL treatment in HUVECs or HAECs. (C–G) FOXO1 mRNA and protein expression levels significantly reduce after miR-183-5p, miR-96-5p or miR-182-5p agomir treatment in HUVECs or HAECs with or without ox-LDL treatment. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. | |
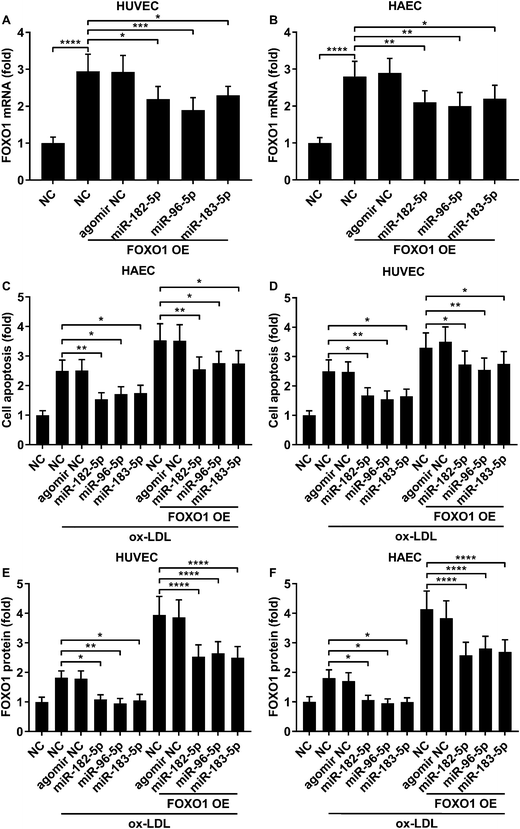 |
| | Fig. 3 FOXO1 overexpression enhanced ox-LDL-induced apoptosis in HUVECs or HAECs, which could be reduced by miR-183-5p, miR-96-5p or miR-182-5p agomir treatment. (A and B) Upregulation of FOXO1 mRNA expression by FOXO1 overexpression could be partially reduced by the indicated agomir treatment. (C and D) HUVEC or HAEC apoptosis induced by ox-LDL treatment and further enhanced by FOXO1 overexpression could be reduced by the indicated agomir treatment. (E and F) FOXO1 protein expression level change correlated to the cell apoptosis level in the HUVECs or HAECs treated in (C and D). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. | |
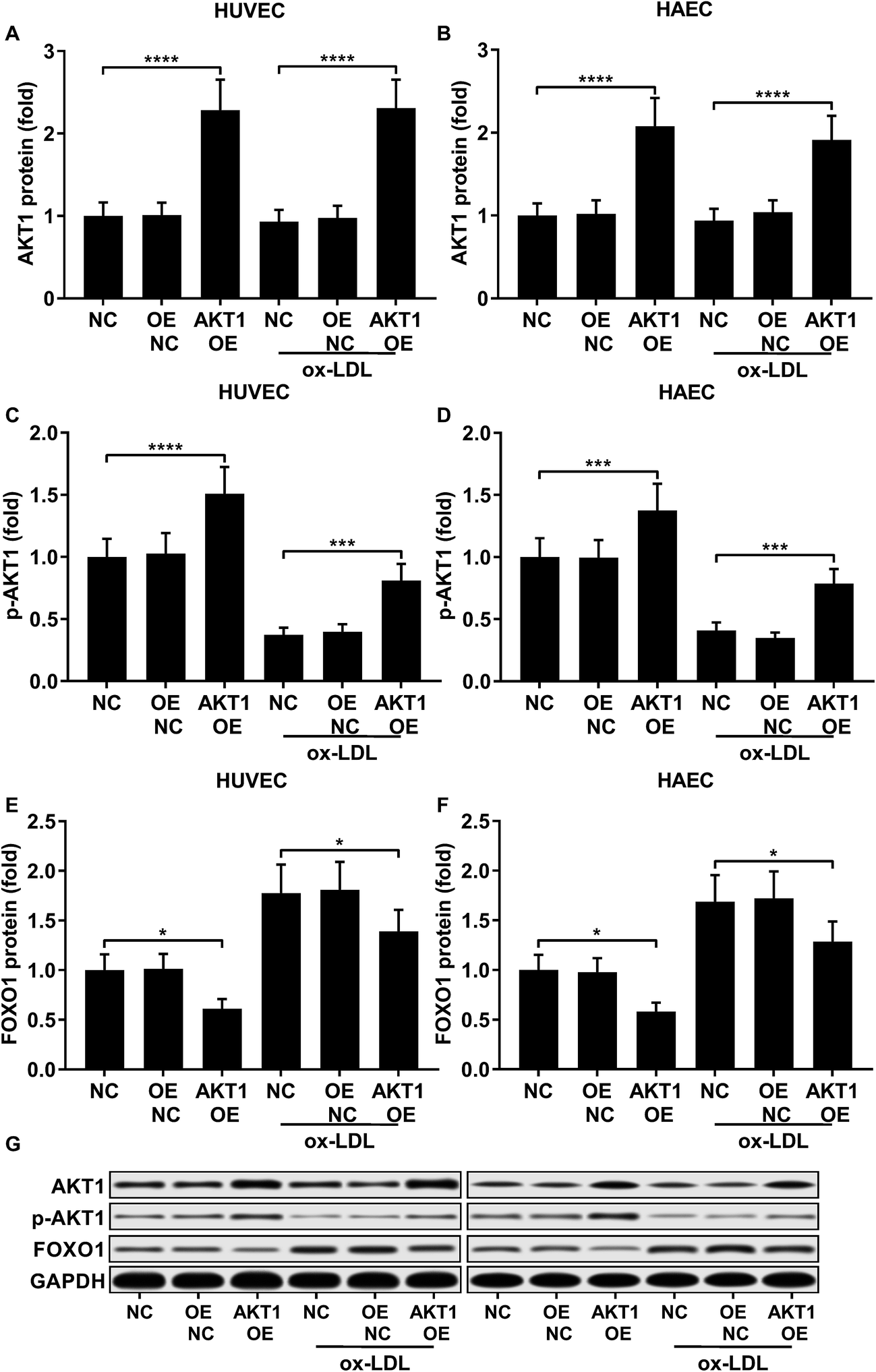
AKT overexpression reduced FOXO1 gene expression by upregulating miR-183-5p and miR-182-5p
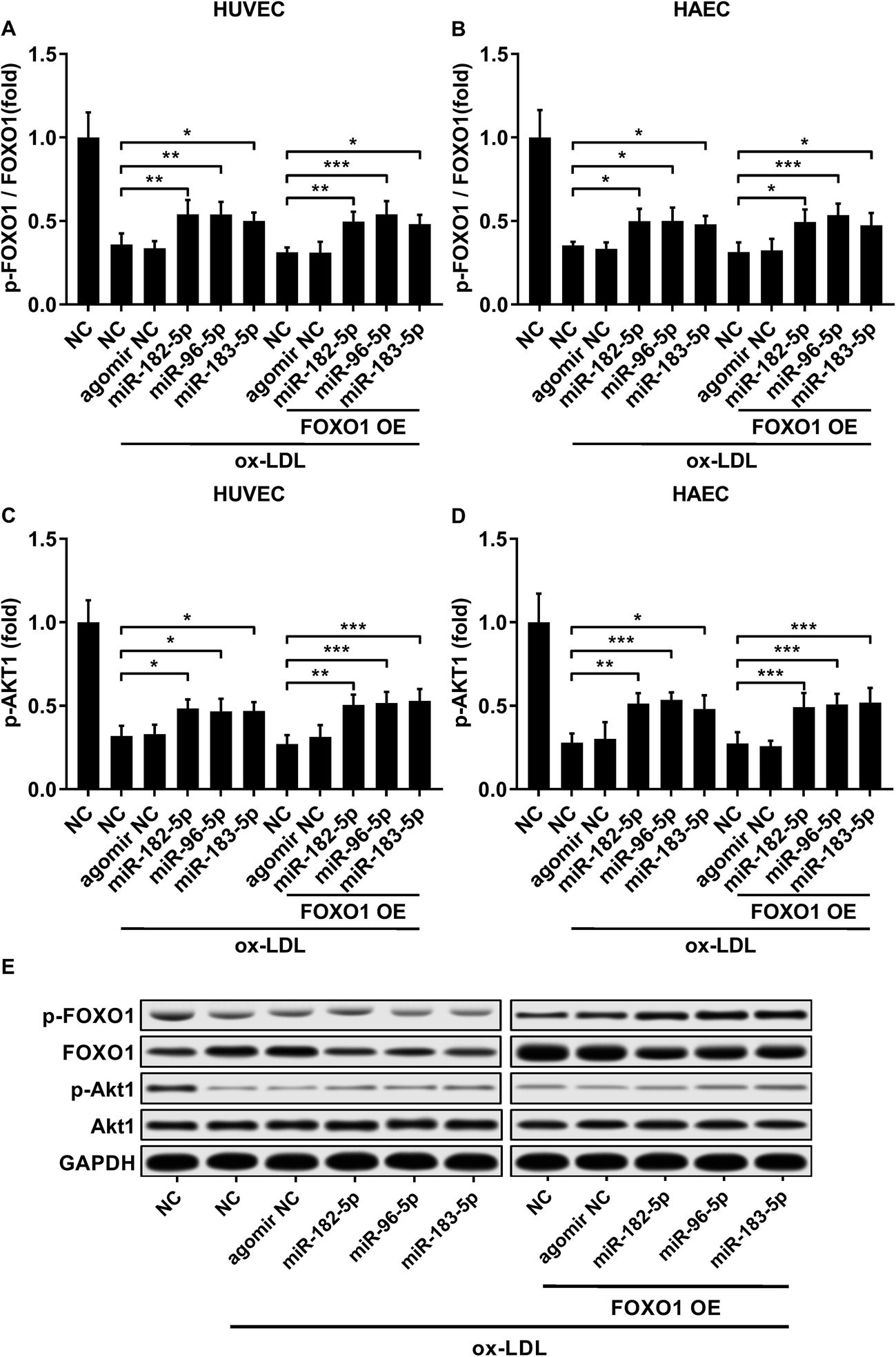
FOXO1 protein de-phosphorylation favors its nuclear localization and pro-apoptotic transcriptional activity. We employed western blot analysis to evaluate the FOXO1 phosphorylation level and calculated the phosphorylated FOXO1 versus total FOXO1 ratio in the treated HUVECs and HAECs shown in Fig. 3C and D. Our data suggested that the FOXO1 phosphorylation level was significantly reduced by ox-LDL treatment in HUVECs or HAECs, while FOXO1 overexpression drastically increased the FOXO1 phosphorylation level in the two endothelial cells; we found no significant influence of the three miRNA agomir treatment on the FOXO1 phosphorylation level in the HUVECs or HAECs (Fig. 4E). After normalizing the FOXO1 phosphorylation level to the total FOXO1 protein expression level, we found that ox-LDL treatment significantly reduced the p-FOXO1/total FOXO1 ratio in HUVECs or in HAECs, which could be partially reversed by the three miRNA agomir treatment. Notably, FOXO1 overexpression showed no significant influence on the p-FOXO1/total FOXO1 ratio (Fig. 4A, B and E). Serine/threonine kinase AKT has been found to phosphorylate and deactivate FOXO1 in endothelial cells. AKT is activated by phosphorylation after binding with PI (3, 4, 5) P3. Our western blot results showed that AKT phosphorylation was significantly reduced in HUVECs or HAECs by ox-LDL treatment and partially rescued by the three miRNA agomir treatment, which is consistent with the p-FOXO1/total FOXO1 ratio change, while ox-LDL or miRNA agomir treatment showed no detectable influence on AKT1 protein expression level (Fig. 4C–E). To further investigate whether AKT1 is involved in regulating the FOXO1 protein expression level besides catalyzing FOXO1 phosphorylation, we constructed AKT1 overexpression in HUVECs and HAECs, and the partially rescued AKT1 phosphorylation level decreased by ox-LDL treatment in the two endothelial cell lines (Fig. 5A–D and G). Our western blot results also indicated a significant downregulation of the FOXO1 protein expression level in AKT1 overexpressing HUVECs or HAECs despite the ox-LDL treatment (Fig. 5E–G). Previous research suggested that miR-183-5p expression could be induced by AKT1, we therefore examined whether AKT1 overexpression would influence the expression levels of miR-183-5p, miR-96-5p or miR-182-5p. Our RT-qPCR results suggested that miR-183-5p and miR-182-5p expression levels could be significantly increased by AKT1 overexpression in HUVECs or HAECs with or without ox-LDL treatment, while the miR-96-5p expression levels in the two endothelial cell lines were not significantly affected by AKT1 overexpression (Fig. 6A–F). Cell apoptosis assay results further demonstrated that treatment with antagomir against miR-183-5p or miR-182-5p significantly attenuated the protective effect of AKT overexpression against ox-LDL treatment-induced endothelial cell apoptosis (Fig. 6G and H). Collectively, the data suggest that AKT1 overexpression attenuated ox-LDL treatment-induced endothelial cell apoptosis not only by phosphorylating FOXO1 but also by reducing FOXO1 protein expression through upregulating miR-183-5p and miR-182-5p. A decrease in the AKT activity might therefore be a pivotal step in ox-LDL-induced endothelial cell apoptosis.
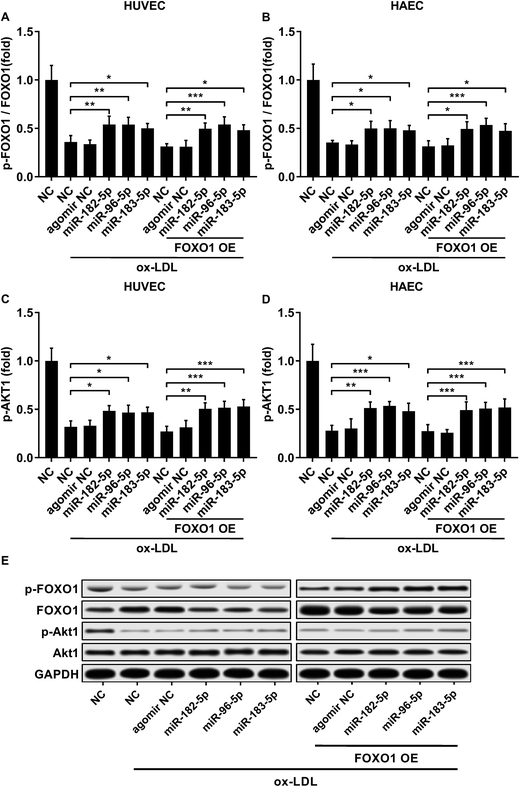 |
| | Fig. 4 Downregulation in the FOXO1 pSer256/total FOXO1 ratio and AKT1 pSer473 level caused by ox-LDL treatment in HUVECs or HAECs could be partially rescued by miR-183-5p, miR-96-5p or miR-182-5p agomir treatment, which doesn’t affect the AKT1 total protein expression level (A–E). *, p < 0.05; **, p < 0.01; ***, p < 0.001. | |
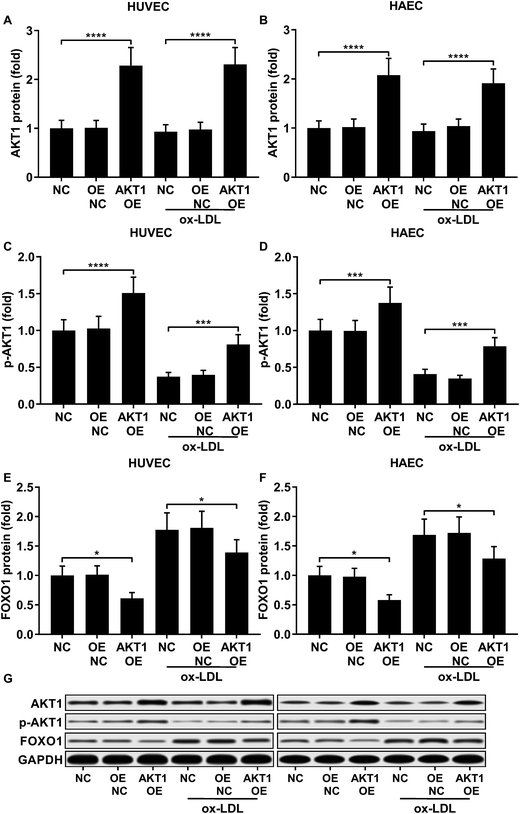 |
| | Fig. 5 AKT1 overexpression significantly increased the AKT1 protein expression level and Ser473 phosphorylation level while reducing FOXO1 protein expression in HUVECs or HAECs with or without ox-LDL treatment (A–G). *, p < 0.05; ***, p < 0.001; ****, p < 0.0001. | |
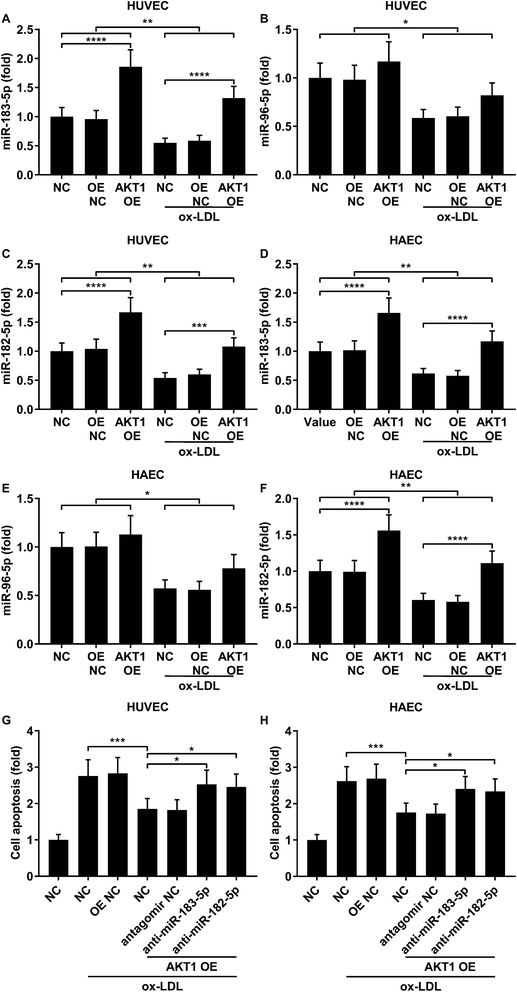 |
| | Fig. 6 AKT1 overexpression partially reduced ox-LDL-induced apoptosis in HUVECs or HAECs by upregulating miR-183-5p and miR-182-5p. (A–F) miR-183-5p and miR-182-5p were upregulated by AKT1 overexpression in HUVECs or HAECs with or without ox-LDL treatment, while miR-96-5p was not significantly influenced. (G and H) Antagomir treatment reducing the endogenous miR-183-5p or miR-182-5p expression levels partially reduced the anti-apoptotic effect of AKT1 overexpression in the HUVECs or HAECs treated by ox-LDL. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. | |
Discussion
Endothelial cell apoptosis induced by pro-inflammatory or pro-apoptotic signals is a key step in atherosclerosis development, resulting in the destruction and permeabilization of the vascular endothelium, leading to an influx of pro-inflammatory macrophages and T cells together with other biomolecules and severe secondary damage around the lesion. Investigations into the pro- and anti-apoptotic signaling events in the vascular endothelial cells under atherosclerotic conditions may therefore improve treatment strategies and benefit patients.
The purpose of this research was to find miRNAs that could potentially attenuate atherosclerosis development by reducing endothelial cell apoptosis. During a review of the literature we found that in publications by Desjarlais et al. miR-183-5p and miR-182-5p were both significantly reduced during atherogenesis, implying that by restoring or increasing the expression of these two miRNAs there might be a possibility of alleviating atherosclerosis progression.12 Although these two microRNAs haven’t been investigated in atherosclerosis, and their function in endothelial cells under other conditions remains largely unknown, the anti-apoptotic function of miR-183-5p and miR-182-5p in different cell types has been characterized multiple times; due to their pro-survival role, these two miRNAs were suggested as potential therapeutic targets against disease caused by abnormal cell proliferation, such as cancer.25–28
In the present research we also included miR-96-5p in our scope primarily because the miR-96 gene clusters with miR-183 and miR-182 in the human genome and the three mature miRNAs play similar roles under different pathologic conditions.29 We noticed that multiple studies have linked, with circumstantial evidence, the anti-apoptotic effect of miR-183-5p, miR-182-5p and miR-96-5p to the down-regulation of FOXO1 in cancer cells.23,30–32 Functioning as a transcription factor, FOXO1 induces apoptosis principally by mediating the transcription of apoptosis-executing or -promoting genes, such as Bim, TRAIL and Fas ligands.33–35 Given that the pro-apoptotic role of the FOXO1/Bim axis in endothelial cells and the atherosclerosis promoting effect of FOXO1 have been well-demonstrated,9,24,36 based on these previous reports we hypothesized that miR-183-5p, miR-96-5p and miR-182-5p might reduce atherosclerosis development by targeting FOXO1, while a decrease in their expression level in endothelial cells might be an important step in atherogenesis.
This hypothesis was verified in the present research by cell apoptosis assays and AGO-RIP. Our results confirm that FOXO1 mRNA can be targeted by the three miRNAs mentioned above in endothelial cells, and this mechanism can be reduced by ox-LDL treatment. Treatment with agomir mimicking the endogenous upregulation of the three miRNAs could decrease FOXO1 expression, significantly attenuating the increase in cell apoptosis induced by ox-LDL treatment, a key substance triggering apoptotic signaling cascades in endothelial cells when bound by its cell-surface receptor LOX-1 and frequently used to establish an atherosclerotic endothelial cell model in vitro.37 In particular, Li and Mehta have summarized in detail that inhibition of AKT kinase activity due to an inhibition of PI3K mediated by the LOX-1 downstream signaling cascade is a central step towards ox-LDL triggered endothelial cell apoptosis, and the inhibitory mechanism of AKT on the pro-apoptotic role of FOXO1 in atherosclerotic endothelial cells has been well demonstrated,24,37,38 although we have failed to find any other report directly confirming the FOXO1 activating effect of LOX-1 activation or ox-LDL treatment. In the present research, our results also demonstrate that ox-LDL treatment significantly increases FOXO1 mRNA and protein expression levels in endothelial cells both by downregulating the three miRNAs and by increasing FOXO1 gene transcription. We speculate that this transcriptional upregulation of FOXO1 in ox-LDL treated endothelial cells is perhaps a positive feed-back mechanism consolidating the pro-apoptotic signaling cascade triggered by LOX-1. Currently the molecular details of regulation of FOXO1 in atherosclerotic cells remain largely unknown, and require further investigation. Interestingly, in the present research we also found that AKT overexpression could upregulate miR-183-5p and miR-182-5p expression in endothelial cells with or without ox-LDL treatment, and AKT not only reduced FOXO1 activation but also indirectly downregulated FOXO1 protein expression by upregulating miR-183-5p and miR-182-5p. Treatment with miR-183-5p, miR-182-5p or miR-96-5p agomir didn’t affect the AKT protein expression level but partially rescued its phosphorylation level which was downregulated by ox-LDL treatment in the endothelial cells. The data seem to imply the existence of some positive feedback mechanisms in regulating AKT activation. Molecular mechanisms underlying these effects should be investigated in future research.
Conclusion
Overall, the present research represents the first insight into miRNA regulating FOXO1 expression in atherosclerotic endothelial cells. Our results suggest the anti-apoptotic and anti-atherogenic roles of miR-183-96-182 clusters in endothelial cells with potential therapeutic value. Further studies on these miRNAs, such as their other potential target genes and biosafety, are needed to promote the research of these miRNAs for applicable treatment options.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81501139).
References
- M. A. Gimbrone Jr and G. Garcia-Cardena, Circ. Res., 2016, 118, 620–636 CrossRef PubMed.
- P. N. Hopkins, Physiol. Rev., 2013, 93, 1317–1542 CrossRef CAS PubMed.
- U. Forstermann, N. Xia and H. Li, Circ. Res., 2017, 120, 713–735 CrossRef PubMed.
- A. Schober and C. Weber, Annu. Rev. Phytopathol., 2016, 11, 583–616 CrossRef PubMed.
- N. V. K. Pothineni, S. K. Karathanasis, Z. Ding, A. Arulandu, K. I. Varughese and J. L. Mehta, J. Am. Coll. Cardiol., 2017, 69, 2759–2768 CrossRef CAS PubMed.
- L. Li and G. Renier, Atherosclerosis, 2009, 204, 40–46 CrossRef CAS PubMed.
- A. Wojciechowska, A. Braniewska and K. Kozar-Kaminska, Adv. Clin. Exp. Med., 2017, 26, 865–874 Search PubMed.
- C. S. Nabzdyk, L. Pradhan-Nabzdyk and F. W. LoGerfo, J. Transl. Med., 2017, 15, 164 CrossRef PubMed.
- K. Tsuchiya and Y. Ogawa, J. Diabetes Invest., 2017, 8, 726–734 CrossRef CAS PubMed.
- E. Araldi and Y. Suarez, Biochim. Biophys. Acta, 2016, 1861, 2094–2103 CrossRef CAS PubMed.
- V. C. Ganta, M. H. Choi, A. Kutateladze, T. E. Fox, C. R. Farber and B. H. Annex, Circulation, 2017, 135, 2403–2425 CrossRef CAS PubMed.
- M. Desjarlais, S. Dussault, W. Dhahri, R. Mathieu and A. Rivard, Arterioscler., Thromb., Vasc. Biol., 2017, 37, 900–908 CrossRef CAS PubMed.
- Y. Fang, C. Shi, E. Manduchi, M. Civelek and P. F. Davies, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13450–13455 CrossRef CAS PubMed.
- R. Menghini, V. Casagrande, M. Cardellini, E. Martelli, A. Terrinoni, F. Amati, M. Vasa-Nicotera, A. Ippoliti, G. Novelli, G. Melino, R. Lauro and M. Federici, Circulation, 2009, 120, 1524–1532 CrossRef CAS PubMed.
- S. B. Qin, D. Y. Peng, J. M. Lu and Z. P. Ke, J. Cell. Physiol., 2017, 233, 6630–6637 CrossRef PubMed.
- H. Tang, Y. Bian, C. Tu, Z. Wang, Z. Yu, Q. Liu, G. Xu, M. Wu and G. Li, Curr. Cancer Drug Targets, 2013, 13, 221–231 CrossRef CAS PubMed.
- E. Pavlakis, A. B. Tonchev, A. Kaprelyan, Y. Enchev and A. Stoykova, Oncol. Rep., 2017, 37, 1579–1592 CrossRef CAS PubMed.
- X. Tang, D. Zheng, P. Hu, Z. Zeng, M. Li, L. Tucker, R. Monahan, M. B. Resnick, M. Liu and B. Ramratnam, Nucleic Acids Res., 2014, 42, 2988–2998 CrossRef CAS PubMed.
- Y. Liu, Y. Han, H. Zhang, L. Nie, Z. Jiang, P. Fa, Y. Gui and Z. Cai, PLoS One, 2012, 7, e52280 CrossRef CAS PubMed.
- H. M. Song, Y. Luo, D. F. Li, C. K. Wei, K. Y. Hua, J. L. Song, H. Xu, N. Maskey and L. Fang, Int. J. Clin. Exp. Pathol., 2015, 8, 9889–9900 CAS.
- Y. Guo, H. Liu, H. Zhang, C. Shang and Y. Song, Oncol. Lett., 2012, 4, 561–565 CrossRef CAS PubMed.
- Y. Huang, X. Fan, R. Tao, Q. Song, L. Wang, H. Zhang, H. Kong and J. Huang, J. Cell. Physiol., 2018, 233, 6693–6704 CrossRef CAS PubMed.
- L. Zhang, H. Quan, S. Wang, X. Li and X. Che, Tumor Biol., 2015, 36, 8121–8126 CrossRef CAS PubMed.
- Y. Li, M. Ren, X. Wang, X. Cui, H. Zhao, C. Zhao, J. Zhou, Y. Guo, Y. Hu, C. Yan, B. Berk and J. Wang, Sci. Rep., 2017, 7, 15539 CrossRef PubMed.
- D. P. Bi, C. H. Yin, X. Y. Zhang, N. N. Yang and J. Y. Xu, Oncol. Rep., 2016, 35, 2873–2879 CrossRef CAS PubMed.
- J. Chen, L. Gu, J. Ni, P. Hu, K. Hu and Y. L. Shi, BioMed Res. Int., 2015, 2015, 340218 Search PubMed.
- Q. Zhang, W. Di, Y. Dong, G. Lu, J. Yu, J. Li and P. Li, Tumor Biol., 2015, 36, 9245–9249 CrossRef CAS PubMed.
- C. G. Kim, H. Lee, N. Gupta, S. Ramachandran, I. Kaushik, S. Srivastava, S. H. Kim and S. K. Srivastava, Semin. Cancer Biol., 2018, 50, 142–151 CrossRef CAS PubMed.
- S. Dambal, M. Shah, B. Mihelich and L. Nonn, Nucleic Acids Res., 2015, 43, 7173–7188 CrossRef CAS PubMed.
- H. S. McLoughlin, J. Wan, R. M. Spengler, Y. Xing and B. L. Davidson, Hum. Mol. Genet., 2014, 23, 2593–2603 CrossRef CAS PubMed.
- L. Xie, A. Ushmorov, F. Leithauser, H. Guan, C. Steidl, J. Farbinger, C. Pelzer, M. J. Vogel, H. J. Maier, R. D. Gascoyne, P. Moller and T. Wirth, Blood, 2012, 119, 3503–3511 CrossRef CAS PubMed.
- S. S. Myatt, J. Wang, L. J. Monteiro, M. Christian, K. K. Ho, L. Fusi, R. E. Dina, J. J. Brosens, S. Ghaem-Maghami and E. W. Lam, Cancer Res., 2010, 70, 367–377 CrossRef CAS PubMed.
- C. Urbich, A. Knau, S. Fichtlscherer, D. H. Walter, T. Bruhl, M. Potente, W. K. Hofmann, S. de Vos, A. M. Zeiher and S. Dimmeler, FASEB J., 2005, 19, 974–976 CrossRef CAS PubMed.
- C. Daly, V. Wong, E. Burova, Y. Wei, S. Zabski, J. Griffiths, K. M. Lai, H. C. Lin, E. Ioffe, G. D. Yancopoulos and J. S. Rudge, Genes Dev., 2004, 18, 1060–1071 CrossRef CAS PubMed.
- G. Marfe, M. Tafani, F. Fiorito, U. Pagnini, G. Iovane and L. De Martino, PLoS One, 2011, 6, e27313 CrossRef CAS PubMed.
- L. Qiang, K. Tsuchiya, J. Y. Kim-Muller, H. V. Lin, C. Welch and D. Accili, J. Biol. Chem., 2012, 287, 13944–13951 CrossRef CAS PubMed.
- D. Li and J. L. Mehta, Circ. Res., 2009, 104, 566–568 CrossRef CAS PubMed.
- W. Liu, T. Wang, X. He, X. Liu, B. Wang, Y. Liu, Z. Li, R. Tan, C. Ding, H. Wang and H. Zeng, J. Cardiovasc. Pharmacol., 2016, 67, 491–502 CrossRef CAS PubMed.
Footnote |
| † These authors contributed to this work equally. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d
*d