
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePurification and structural elucidation of chondroitin sulfate/dermatan sulfate from Atlantic bluefin tuna (Thunnus thynnus) skins and their anticoagulant and ACE inhibitory activities
Fatma Krichena,
Hajer Bougatefa,
Federica Capitanib,
Ikram Ben Amorc,
Imed Koubaad,
Jalel Gargouric,
Francesca Maccarib,
Veronica Mantovanib,
Fabio Galeottib,
Nicola Volpib and
Ali Bougatef *ae
*ae
aLaboratory of Plant Improvement and Valorization of Agroressources, National School of Engineering of Sfax (ENIS), University of Sfax, Sfax 3038, Tunisia. E-mail: ali.bougatef@isbs.usf.tn; Fax: +216 74 674 364; Tel: +216 74 674 354
bDepartment of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy
cRegional Centre for Blood Transfusion of Sfax, El-Ain Road Km 0.5, CP 3003 Sfax, Tunisia
dFaculty of Science of Sfax, Laboratory of Chemistry of Natural Products, University of Sfax, Tunisia
eHigh Institute of Biotechnology of Sfax, BP 1175, 3038, Sfax, Tunisia
First published on 12th November 2018
Abstract
Chondroitin sulfate/dermatan sulfate (CS/DS) was extracted from Atlantic bluefin tuna (Thunnus thynnus) skin (SGAT) and was purified and characterized. SGAT was characterized by acetate cellulose electrophoresis, FTIR spectroscopy, 13C NMR spectroscopy and SAX-HPLC. According to the results obtained for specific chondroitinases (ABC and AC) and the SAX-HPLC separation of generated unsaturated repeating disaccharides, the polymer was found to contain a disaccharide monosulfated in positions 6 and 4 of GalNAc and disulfated disaccharides in different percentages. These results were confirmed by 13C NMR experiments. The average molecular mass was 24.07 kDa, as determined by PAGE analysis. SGAT was evaluated for its in vitro anticoagulant activity via activated partial thromboplastin time, thrombin time and prothrombin time tests. The polymer showed strong inhibitory activity against angiotensin I-converting enzyme (IC50 = 0.25 mg mL−1). Overall, the results suggest that this newly extracted CS/DS can be useful for pharmacological applications.
1. Introduction
Glycosaminoglycans (GAGs) constitute a group of complex polydisperse carbohydrates that play important roles in numerous biological processes.1,2 GAGs are linear polysaccharide chains composed of repeating disaccharide units, each consisting of a hexosamine (N-acetylgalactosamine or N-acetylglucosamine) and a uronic acid (D-glucuronic acid or L-iduronic acid) or galactose, which can be differently sulfated depending on the GAG family.3 There are many types of GAGs, and they are generally divided into four groups: (1) hyaluronic acid or hyaluronan; (2) keratan sulfate; (3) chondroitin sulfate (CS)/dermatan sulfate (DS); and (4) heparan sulfate (HS)/heparin (H).These anionic polymers are widespread in nature, occurring in a great variety of organisms, such as vertebrates and terrestrial or marine invertebrates. The extraction of GAGs from marine invertebrates has been well established; however, their relative amounts, molecular masses and sulfation vary. These marine animal sources include Norway lobster shell,4 grey triggerfish and smooth hound skins,5 bony fishes,6 sturgeon cartilage,7,8 ray skin9 and shark cartilage.10
Sulfated GAGs are extremely diverse in their molecular weights and degrees and positions of sulfation, among other properties. In particular, CS/DS isolated from the skin of marine vertebrates such as corb, grey triggerfish, smooth hound and ray have variable structures and disaccharide compositions.11–13 Indeed, CS/DS isolated from the skin of the corb and smooth hound is predominately composed of monosulfated disaccharides bearing sulfate groups at position C-4 of N-acetygalactosamine and disulfated disaccharides bearing esterified sulfate groups at position C-2 of uronic acid and position C-4 of N-acetygalactosamine.11,12 However, DS from Raja clavata ray skin has low sulfate content and is composed of non-sulfated and monosulfated disaccharides bearing sulfate groups at positions C-4 or C-6 of N-acetyl galactosamine (GalNAc).14 Moreover, GAGs possess various molecular masses and polydispersities depending on the source. In this context, CS isolated from ‘terrestrial’ origins, such as bovine, porcine and avian cartilage, generally has molecular weight values ranging from 13 to 26 kDa.15,16 In contrast, CS/DS from fish skin has molecular mass values between 15.46 and 41.72 kDa.11,12 In the same context, DS isolated from the skin of the rays Raja clavata and Raja radula has molecular masses of 31.2 kDa and 33 kDa, respectively.13,17 According to their specific structures, GAGs are involved in various biological processes, such as antihypertensive,18 anticoagulant and antithrombotic,19,20 anti-inflammatory,21 cell proliferation, cell differentiation, cancer metastasis, nerve regeneration, and differentiation of stem cells through intermolecular interactions with different functional proteins.
Anticoagulant activity is among the most widely studied properties of GAGs. Anticoagulants are extensively used in the pharmaceutical field for treating major deadly diseases such as stroke and myocardial infarction. In the treatment of these conditions, medicines with antithrombotic, anticoagulant and anti-platelet activities are mainly used. This is because they inhibit enzymes of the coagulation pathway in the blood circulation system or inhibit the activation and aggregation of platelets. Actually, anticoagulants are derived from mammalian animal sources, in particular H. Hence, there is always a threat of various mammalian animal diseases which can affect human health, such as mad cow disease.22 In order to develop and extract anticoagulants from safer sources, seafood processing by-products are considered to be a good choice.
Furthermore, hypertension is a major chronic disease which affects 30% of the adult population worldwide.23 It is well known that angiotensin I-converting enzyme (ACE) plays an important role in the fluid and salt balance in mammals as well as the renin–angiotensin and the kallikrein–kinin systems for the regulation of blood pressure.24 However, ACE catalyzes the conversion of angiotensin I (an inactive decapeptide) into angiotensin II by cleaving a dipeptide from its C-terminus. Synthetic ACE inhibitors have side effects that include skin rashes, allergic reactions, cough, headache and renal impairment.25 Therefore, the replacement of synthetic ACE inhibitors by natural ones can have benefits in terms of health implications and functionality in food systems.
The present work was carried out to characterize and to assess the in vitro anticoagulant and antihypertensive activities of sulfated glycosaminoglycan extracted from the skin of Atlantic bluefin tuna (Thunnus thynnus) (SGAT).
2. Materials and methods
2.1. Reagents
The chemicals and solvents used in the present study were purchased at analytical grade or the highest level of purity available. Alcalase® 2.4 L serine-protease from Bacillus licheniformis was obtained from Novozymes® (Bagsvaerd, Denmark). GAGs standard, CS from bovine trachea, DS from porcine intestinal mucosa, and HS (heparan sulfate) from bovine kidney were obtained from Sigma-Aldrich (St. Louis, MO, USA). Chondroitinase ABC from Proteus vulgaris (EC 4.2.2.4), specific activity of 0.5 to 2 units per mg, was obtained from Sigma-Aldrich. Chondroitinase AC from Flavobacterium heparinum (EC 4.2.2.5), specific activity of 0.5 to 2 units per mg, was obtained from Sigma-Aldrich. Unsaturated chondro/dermato disaccharides [ΔDi0S (ΔUA-[1→3]-GalNAc), ΔDi4S (ΔUA-[1→3]-GalNAc-4s), ΔDi6S (ΔUA-[1→3]-GalNAc-6S), ΔDi2S(ΔUA-2S-[1→3]-GalNAc), ΔDi2,4diS (ΔDi-dis B, ΔUA-2S-[1→3]-GalNAc-4S), ΔDi2,6diS (ΔDi-diS D, ΔUA-2S-[1→3] GalNAc-6S), Δdi4,6diS (ΔDi-diS E, ΔUA-[1→3]-GalNAc-4,6diS), and ΔDi2,4,6triS (ΔDitriS, ΔUA-2S-[1→3]-GalNAc-4S,6S)] were obtained from Seikagaku Corporation (Tokyo City, Japan) and Sigma-Aldrich. Diethylaminoethyl (DEAE)-cellulose was purchased from Pharmacia (Uppsala, Sweden). Angiotensin I-converting enzyme from rabbit lung and the ACE synthetic substrate hippuryl-L-histidyl-L-leucine (HHL) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Heparin sodium Choay was obtained from Sanofi-Aventis (France).2.2. Preparation of fish skin
Fish by-products were obtained from a local fish market in Sfax, Tunisia. The samples were packed in polyethylene bags, placed in ice with a sample/ice ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (w/w), and transported to the research laboratory within 30 min. Upon arrival, the samples were washed twice with water and separated. Only the outer skin of the fish was collected; the skin was then stored in sealed plastic bags at −20 °C until further use for the extraction and analysis of GAGs.
:3 (w/w), and transported to the research laboratory within 30 min. Upon arrival, the samples were washed twice with water and separated. Only the outer skin of the fish was collected; the skin was then stored in sealed plastic bags at −20 °C until further use for the extraction and analysis of GAGs.
2.3. Enzymatic extraction of sulfated glycosaminoglycans
GAGs were extracted according to a slightly modified version of the method described by Ben Mansour et al.9 In brief, the fish skins were cut into small pieces and homogenized using a Moulinex R62 homogenizer (Organotechnie, Courneuve, France). A 5 g amount of sample was dissolved in 250 mL sodium acetate (0.1 M), EDTA (5 mM), and cysteine (5 mM) at pH 6. Alacalase® was added, and the mixture was maintained for 24 h at 50 °C. The mixture was then allowed to cool to room temperature and then filtered. The residue was washed with distilled water and filtered again. The filtrates were mixed, and the polysaccharides were precipitated with cetylpyridinium chloride 10% (w/v). The mixture was maintained for 24 h at room temperature and centrifuged for 30 min at 5000 RPM and 4 °C using a refrigerated centrifuge (Hettich Zentrifugen, ROTINA 380R, Germany). The pellet was washed with cetylpyridinium chloride 0.05% (w/v) and then dissolved in 200 mL NaCl solution in ethanol (100:15, v/v). 700 mL of ethanol was added. The polysaccharide-containing solution was maintained for 24 h at 4 °C and then centrifuged for 30 min at 5000 RPM and 4 °C. The pellet was washed twice with 80% ethanol and then once with absolute ethanol. After that, the pellet was redissolved in deionised water and lyophilized in a freeze dryer (Martin Christ, Alpha 1-2 LDplus, Germany).
2.4. Purification of fish skin GAGs
The lyophilized sulfated GAGs were suspended in distilled water and then fractionated by anion-exchange chromatography on a DEAE-cellulose column (2 cm × 6 cm) equilibrated with 50 mM NaCl. The GAGs were eluted with a linear gradient of NaCl from 50 mM to 2 M from 0 to 150 min at a flow of 1 mL min−1. Two volumes of ethanol were added to the collected fractions corresponding to single species of GAGs evaluated by uronic acid assay,26 and the GAGs were precipitated at 4 °C. After centrifugation at 10000 g for 10 min, the pellet was dried at 50 °C and solubilized in distilled water.
2.5. Chemical composition analysis
Total carbohydrates were determined by the phenol-sulphuric acid method.27 Total uronic acid content was quantified colorimetrically according to the method described by Cesaretti et al.26 The sulfate content in the GAGs was assessed by liquid-ion chromatography (HPLC) on a Metrohm chromatograph equipped with CI SUPER-SEP columns using phthalic acid and acetonitrile as eluents. The test precision of the instrument was about ±2%. All measurements were performed in triplicate.2.6. Determination of colour
The GAGs were placed between two steel dishes with a hole 5.7 cm in diameter. The colour of the film was determined with a tristimulus colorimeter (CHROMA METER CR-400/410. KONICA MINOLTA, Japan) using the CIE Lab scale (C/2°), where L*, a* and b* are the parameters that measure lightness, redness and yellowness, respectively. A standard white plate was used as a reference. The results were the average of three measurements taken at ambient temperature at different points on the samples.2.7. Cellulose acetate electrophoresis
The cellulose acetate electrophoresis was performed as follows: 3.5 μg of the GAG standard solution containing DS, CS and HS and 3.5 μg of the purified sulfated GAGs from fish skins were placed at the origin (10 mm from the cathode side) of a cellulose acetate strip (Sartorius). Electrophoresis was carried out in Zn-acetate 0.1 M pH 6 buffer run at 60 V at room temperature for 2 h. After electrophoresis, the cellulose acetate strip was stained with alcian blue.282.8. UV spectroscopy analysis
The CS/DS solution was prepared by suspending the sample in distilled water to a concentration of 1.0 mg mL−1 for UV measurement in the wavelength range of 190 to 700 nm. Distilled water was used as a control in all processes.2.9. CS/DS molecular mass determination
The molecular mass of CS/DS obtained from Atlantic bluefin tuna skins (SGAT) was determined by PAGE according to Edens et al.29 15 μg of the purified CS/DS determined by uronic acid assay were layered on the gel. The related calibration curve was constructed using oligosaccharide standards of known molecular mass prepared from CS Buzzega et al.30 After a run of 40 min at 100 V, the gel was stained with toluidine blue (0.1% in 1% acetic acid) for 30 min, followed by destaining in 1% acetic acid. Molecular mass evaluation was performed by densitometric acquisition of the bands and comparison of their migration times on the calibration curve constructed by plotting the retention times of the standards against the logarithms of their molecular mass values.2.10. Infra-red spectroscopic analysis
The absorption spectra of the samples were obtained using FTIR spectroscopy (Analect Instruments fx-6160). The FTIR spectra of the prepared materials were recorded between 450 and 4000 cm−1 in a NICOET spectrometer. The transmission spectra of the samples were recorded using KBr pellets containing 0.1% of sample.2.11. NMR analysis
The 13C NMR spectrum of purified CS/DS was recorded at 298.1 K on a Bruker ASX300 instrument equipped with a 5 mm diameter tunable probe using Bruker software. The sample was prepared by dissolving 30 mg in 1 mL of D2O at a high level of deuteration (99.997%) to avoid the presence of relatively high water. The final solution was transferred to an NMR tube. The spectra were recorded at temperatures of 25 °C to 28 °C at 100.62 MHz unless otherwise specified. 13C chemical shifts (d, ppm) are quoted with respect to external sodium 4,4-dimethyl-4-silapentane-1-sulfonate (0.0 ppm).2.12. Enzymatic treatments and disaccharide composition evaluation
After treatment of the purified CS/DS samples with chondroitinase ABC or AC, the generated unsaturated disaccharides were separated and quantified by anion-exchange (SAX) by means of HPLC equipment from Jasco equipped with a 150 mm × 4.6 mm Spherisorb 5-SAX stainless-steel column (5 μm, trimethylammoniopropyl groups Si–CH2–CH2–CH2–N+(CH3)3 in Cl− form, Phase Separations Limited, Deeside Industrial Park, Deeside Clwyd, U.K.) and detection at 232 nm. Isocratic separation was performed using 50 mM NaCl at pH 4.00 for 5 min followed by a linear gradient from 5 to 60 min of 50 mM NaCl to 1.2 M NaCl at pH 4.00 at a flow rate of 1.2 mL min−1. Authentic unsaturated standard disaccharides were used for qualitative and quantitative purposes.2.13. Anticoagulant activity of sulfated glycosaminoglycans
The activated partial thromboplastin time (aPTT), thrombin time (TT) and prothrombin time (PT) assays were performed using a semi-automatic line STA (Diagnostica Stago). The samples were dissolved in distilled water. All analyses were performed in triplicate, and mean values were considered.2.14. Hemolytic activity
The hemolytic activities of the purified CS/DS were determined by a slightly modified version of the method described by Dathe et al.312.15. Angiotensin-I-converting enzyme inhibitory activity
The ACE inhibitory activity was measured in triplicate as previously described by Nakamura et al.32 A sample solution (80 μL) containing different concentrations (0.2 to 0.8 mg mL−1) of purified CS/DS from the skin of Atlantic bluefin tuna (SGAT) was mixed with 200 μL of 5 mM HHL and then preincubated for 3 min at 37 °C. SGAT and HHL were prepared in 100 mM borate buffer (pH 8.3) containing 300 mM NaCl. The reactions were then initiated by adding 20 μL of 0.1 U mL−1 ACE from rabbit lung prepared in the same buffer. After incubation for 30 min at 37 °C, the enzyme reactions were stopped by the addition of 250 μL of 0.05 M HCl. The liberated hippuric acid (HA) was extracted with ethyl acetate (1.7 mL) and then evaporated at 90 °C for 10 min by rotary evaporation under reduced pressure (rotary evaporator, Heidolph, Germany). The residue was dissolved in 1 mL of distilled water, and the absorbance of the extract at 228 nm was determined using a UV-visible spectrophotometer (UV mini 1240 UV/VIS spectrophotometer, Shimadzu, China). The average value from three determinations at each concentration was used to calculate the ACE inhibition rate as follows:where A refers to the absorbance of HA generated in the presence of ACE inhibitor, B is the absorbance of HA generated without ACE inhibitor (100 mM borate buffer at pH 8.3 was used instead of purified CS/DS), and C is the absorbance of HA generated without ACE (corresponding to HHL autolysis in the course of the enzymatic assay).
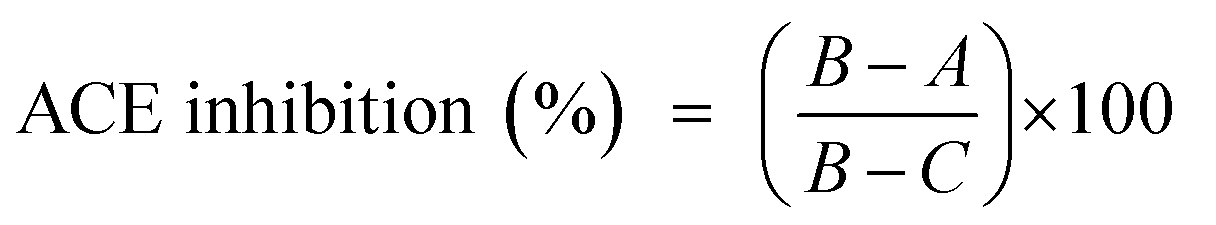
The IC50 value, defined as the concentration of purified CS/DS(mg mL−1) required to inhibit 50% of ACE activity, was calculated for each sample using non-linear regression from a plot of the ACE inhibition rate versus the sample concentration.
2.16. Statistical analysis
All results were expressed as the mean standard deviation (SD). Data were analyzed using the SPSS statistics program (SPSS 10.0 for Windows, SPSS Inc., Chicago, IL). Statistical differences between sample treatments were determined by one-way analysis of variance (ANOVA), and p < 0.05 was considered statistically significant.3. Results and discussion
3.1. Extraction, purification and chemical composition of sulfated glycosaminoglycans
GAGs were extracted for the first time from the skin of Atlantic bluefin tuna (SGAT). After proteolytic treatment, SGAT was purified on an anion-exchange resin. Based on the wet weight, a sulfated GAGs extraction yield of about 7.78% was obtained. This yield was similar to those previously reported for skins of grey triggerfish and smooth hound.5 The approximate composition of the purified SGAT is shown in Table 1. The total sugars of SGAT determined by the phenol/sulfuric acid method was about 80.12%. The sugar content was similar to those reported for sulfated polysaccharides from squid (85.06%)33 and red seaweed (85.6%).34 Furthermore, quantitative analyses by the carbazole method showed that SGAT contained 69% uronic acid. This is similar to the uronic acid amount previously described for CS/DS from grey triggerfish skins (70%).12skin (SGAT). Values are given as mean ± SD from triplicate determinations. The physico-chemical composition was calculated based on dry matter
| SGAT | |
|---|---|
| Total sugar (%) | 80.12 ± 1.29 |
| Uronic acid (%) | 69 ± 0.14 |
| Sulfate (%) | 25.43 ± 1.6 |
|
|
| Colour | |
| L* | 80.74 ± 0.3 |
| a* | 15.71 ± 0.12 |
| b* | 1.7 ± 0.06 |
| Yield (%) | 7.78 ± 0.7 |
The values recorded for the sulfate content in SGAT were 25.43 ± 1.6%. These values are similar to the sulfate amounts previously described by Ben Mansour et al.35 for sulfated GAGs extracted from ray skins (25%). However, they are higher than those previously described for polysaccharides extracted from grey triggerfish (8.13%) and smooth hound (8.77%).5 It may therefore be concluded that the amount of sulfate in GAGs depends on their origin, nature of raw material, and type of extraction. Duarte et al.36 indicated that the bioactivities of polysaccharides depend on the degree and position of sulfation.
Generally, color influences the overall acceptability of products. In this respect, the analysis revealed that the colour of SGAT was light (80.74). The results also showed that SGAT displayed a relatively high degree of redness (a* = 15.71) but showed low yellowness (b* = 1.7).
3.2. Purity identification and acetate cellulose electrophoresis
The ultraviolet-visible spectrum of SGAT in the range of 190 to 700 nm is presented in Fig. 1A. The SGAT sample showed a stronger absorption peak at 200 to 205 nm, which is characteristic of polysaccharides. However, SGAT had no absorbance at 260 or 280 nm, indicating the absence of nucleic acids and proteins. The UV spectrum of sulfated GAGs extracted from Norway lobster showed an extremely weak absorbance at 280 nm, indicating that the sample contains very small amounts of proteins.4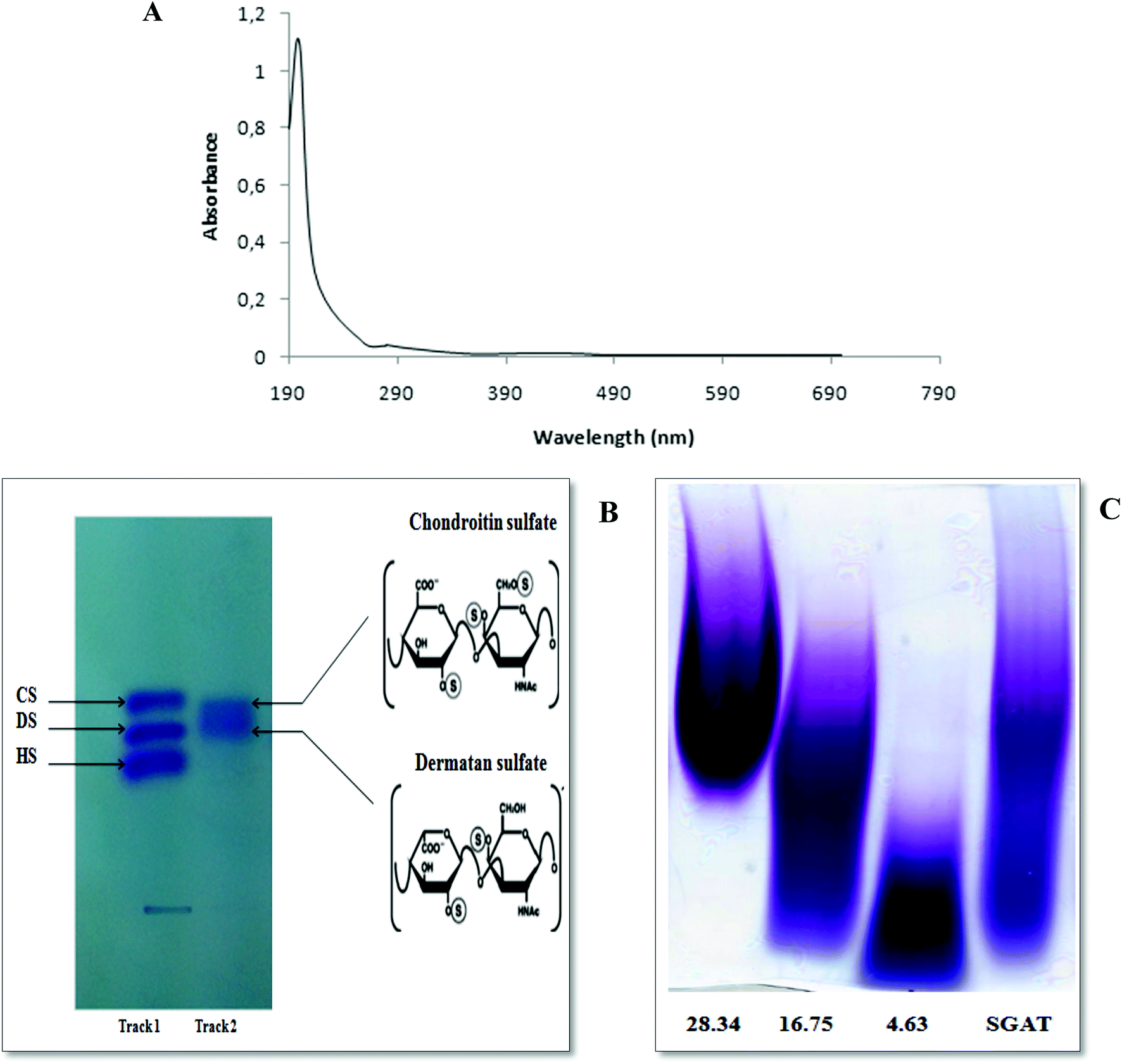 | ||
| Fig. 1 (A) UV spectrum of the sulfated glycosaminoglycans purified from Atlantic bluefin tuna skin (SGAT) in the wavelength range from 190 to 700 nm obtained using a UV-vis spectrophotometer. (B) Acetate cellulose electrophoresis of sulfated glycosaminoglycans from Atlantic bluefin tuna skin. Standard GAGs (dermatan sulfate (DS), chondroitin sulfate (CS) and heparan sulfate (HS)) (Track 1), purified GAGs from Atlantic bluefin tuna skin (Track 2). (C) PAGE analysis of CS/DS purified from Atlantic bluefin tuna skin (SGAT). The calibration curve was constructed using standards prepared from chondroitin sulfate with known molecular masses of 4.63 kDa, 16.75 kDa and 28.34 kDa. | ||
The analysis of SGAT by cellulose acetate electrophoresis revealed two single bands visualized with alcian blue and migration; these bands were chondroitin sulfate (CS) and dermatan sulfate (DS) (Fig. 1B). In the same context, Ben Mansour et al.35 reported the isolation of hyaluronic acid and dermatan sulfate from ray skin. Furthermore, Krichen et al.20 reported that sulfated GAGs from the skins of smooth hound and grey triggerfish revealed the presence of hyaluronic acid (HA), chondroitin sulfate (CS) and dermatan sulfate (DS).
3.3. Fish CS/DS molecular mass determination
It is well known that GAGs from different sources have variable structures and properties. In particular, they can have repeating disaccharides with various numbers and positions of sulfate groups as well as various amounts. Moreover, GAGs possess various molecular masses and polydispersities depending on the source. The PAGE analysis of SGAT was calculated based on a calibration curve of CS fractions of known molecular masses (Fig. 1C). The results showed that SGAT had a molecular mass of around 24.07 kDa. This molecular mass was similar to that reported for CS/DS from grey triggerfish (23.8 kDa).12 In the same context, Chatziioannidis et al.17 and Ben Mansour et al.13 isolated DS with molecular weights of 31.2 kDa and 33 kDa from skins of the rays Raja clavata and Raja radula, respectively. Moreover, Maccari et al.6 confirmed that the molecular mass values of CS from bony fishes ranged from 13.46 to 48.68 kDa.3.4. Infra-red spectroscopic analysis
FTIR spectroscopy was performed in the 4000 to 450 cm−1 region to further characterize and corroborate the data obtained for SGAT. As shown in Fig. 2A, the spectrum showed strong OH stretches at 3394 cm−1 and 3150 cm−1. The small band observed at around 2354 cm−1 was attributed to C–H stretching.37 Furthermore, the presence of uronic acid was evidenced by a strong absorbance band at 1650 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching).37 The most intense band was observed at 1390 cm−1, corresponding to ester sulfate groups (SO). The presence of sulfate (C–O–S) was also confirmed by the absorption at 792 cm−1.38
O stretching).37 The most intense band was observed at 1390 cm−1, corresponding to ester sulfate groups (SO). The presence of sulfate (C–O–S) was also confirmed by the absorption at 792 cm−1.38
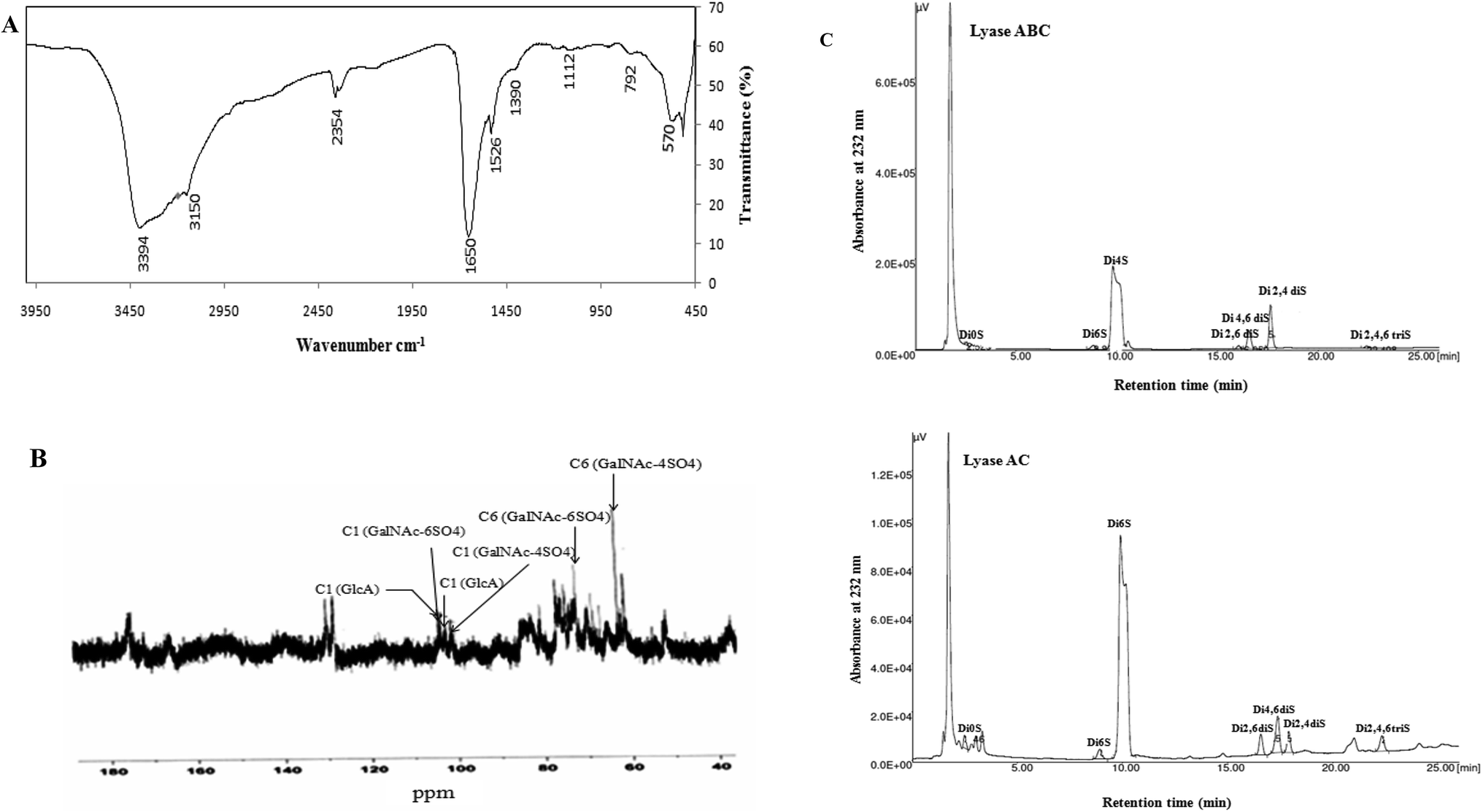 | ||
| Fig. 2 Structural characterization of SGAT. (A) FT-IR spectrometry of purified CS/DS from Atlantic bluefin tuna skin (SGAT). (B) 13C NMR spectrum of the purified CS/DS from Atlantic bluefin tuna skin. (C) SAX-HPLC separation of the unsaturated disaccharides produced by CS/DS purified from Atlantic bluefin tuna skin and treated with chondroitinase ABC or AC. ΔDi0S (ΔUA-GalNAc), ΔDi6S (ΔUA-GalNAc6S), ΔDi4S (ΔUA-GalNAc4S), ΔDi2,6diS (ΔUA2S-GalNAc6S) ΔDi4,6diS (ΔUA GalNAc4, 6diS), ΔDi2,4diS (ΔUA2S-GalNAc4S), ΔDi2,4,6triS (ΔUA2S-GalNAc4,6 diS). The identities of the disaccharide species were determined by co-elution with purified standards (Seikagaku Co./Sigma-Aldrich). All measurements were performed in triplicate. | ||
3.5. NMR analysis
The 13C NMR spectrum of SGAT is shown in Fig. 2B. The inspection of the 50 to 70 and 100 to 110 ppm regions revealed a relatively high amount of CS/DS sulfated in position 4 or 6 of the GalNAc.7,39 In the 13C NMR spectra, two anomeric carbon signals at 104.1 and 102.08 ppm were attributed to GlcA and GalNAc, respectively. Indeed, the signals at 104.1 and 102.08 were assigned to C1 of GlcA and to C1 of GalNAc-6SO4, respectively, and the signals at 104.03 and 101.11 were assigned to C1 of GlcA and to C1 of GalNAc-4SO4, respectively. These results were similar to those described for sturgeon CS.7 Additionally, Mou et al.40 reported that the signal at 103.8 can be attributed to C1 of GlcA in the spectrum of fucosylated CS from sea cucumber. The signals between 50 and 85 ppm were attributed to the chemical shifts of carbon in the glycosylic rings of different monosaccharide residues. Furthermore, the signal at 72.06 was related to C6 of GalNAc-6SO4, and an additional signal at 62.5 indicated C6 of GalNAc-4SO4; this peak may shift to a higher magnetic field because of the substitution of sugar.Coppa et al.41 reported that the signals at 70.5 and 64.1 in the spectrum of CS from the Italian cheese Parmigiano-Reggiano indicated that C6 of GalNAc residues was sulfated in positions 6 and 4, respectively. Therefore, the 13C NMR spectrum of SGAT confirmed that this polymer contains a high amount of CS/DS sulfated in positions 4 and 6 of the GalNAc.
3.6. Enzymatic treatments and disaccharide evaluation of CS/DS
To characterize the structures of GAGs from fish skins, the purified CS/DS were subjected to treatment with two lyases, chondroitinase ABC and AC, while the unsaturated disaccharides were analyzed by SAX-HPLC (Fig. 2C).Chondroitinase ABC is an endolytic lyase that can totally degrade DS and CS.42 GAG lyase enzymes act through an eliminative mechanism to cleave a HexNAc-HexA bond, leaving a (4,5)-unsaturated (termed Δ unsaturated) HexA residue at the new non-reducing terminus.
As shown in Table 2, chondroitinase ABC produced various unsaturated disaccharides in different percentages from GAGs purified from Atlantic bluefin tuna skin. The percentage of nonsulfated disaccharide Di0S of SGAT was trace (0.671%), while the percentages of monosulfated disaccharides ΔDi6S and ΔDi4S were evaluated to be 2.138 and 74.376, respectively. Additionally, Krichen et al.12 reported that the percentages of monosulfated disaccharides ΔDi6S and ΔDi4S recorded in grey triggerfish skins were 18.2 and 59.0, respectively. Furthermore, Maccari et al.6 reported that CS extracted from tuna cartilage contained 28.3% ΔDi4S and 63.2% ΔDi6S. In contrast, Hashiguchi et al.43 reported that CS extracted from ray fish cartilage contained 61.9% ΔDi6S and 27.0% ΔDi4S. Interestingly, disulfated disaccharides were observed in SGAT at different rates. SGAT contains a low percentage of disulfated disaccharide ΔDi2,6diS (1.016%), while the disulfated disaccharide ΔDi2,4diS was found in elevated amounts (14.135%). However, this amount was lower than that previously described by Krichen et al.12 for CS/DS from smooth hound skin (20.1%). Additionally, it is higher than the amount of disaccharide sulfated in positions 2 and 4 in CS/DS from grey triggerfish skin (7.03%). Furthermore, the results revealed that SGAT had little trisaccharide content (0.75%). The presence of disulfated disaccharides produced a high overall charge density of SGAT, which was 1.23. This value is similar to that reported for CS/DS from grey triggerfish and smooth hound skins.12
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SGAT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Chondroitinase AC | Chondroitinase ABC | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a The scheme illustrates the CS/DS unsaturated disaccharides produced by the action of chondroitinase lyases. CS, chondroitin sulfate; DS, dermatan sulfate, Δ UA, 4,5-unsaturated uronic acid; GalNAc, N-acetyl-galactosamine; S, sulfate group. The percentage of each identified disaccharide was determined by purified standards (Seikagaku Co./Sigma-Aldrich) and reported as the weight percentage. Charge density was calculated by considering the number of sulfated groups per disaccharide unit. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ΔDi0S (ΔUA-GalNAc) (%) | 1.566 | 0.671 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ΔDi6S (ΔUA-GalNAc 6S) (%) | 2.075 | 2.138 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ΔDi4S (ΔUA-GalNAc 4S) (%) | 79.286 | 74.376 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ΔDi2, 6S (ΔUA2S-GalNAc 6S) (%) | 3.555 | 1.016 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ΔDi4, 6S (ΔUA 4S-GalNAc 6S) (%) | 7.049 | 6.912 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ΔDi2, 4S (ΔUA2S-GalNAc 4S) (%) | 3.292 | 14.135 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ΔDi2, 4,6 Tris (ΔUA2S-GalNAc 4,6 diS) (%) | 3.180 | 0.753 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Charge density | 1.19 | 1.23 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
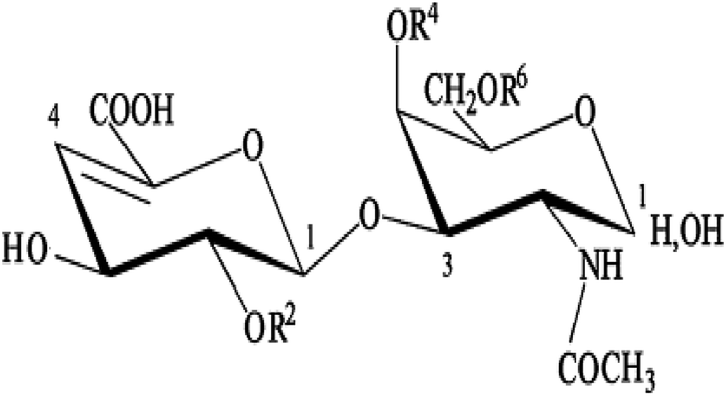
In order to obtain more accurate structure information for SGAT, it was subjected to treatment with chondroitinase AC. This enzyme cleaves GalNAc-GlcA bonds where GalNAc is 4- or 6-O-sulfated but is inactive against GalNAc-IdoA bonds. Therefore, it is only able to cleave chondroitin sulfate. As shown in Fig. 2C and Table 2, chondroitinase AC produced various unsaturated disaccharides in different percentages from SGAT. The analysis by SAX-HPLC chromatography showed that the two chromatograms distinguished disulfated disaccharides in positions 2 and 4 and disulfated disaccharides in positions 2 and 6. By treatment with chondroitinase AC, SGAT was found to be very particular because of the presence inside the polysaccharide backbone of a high percentage of disaccharides with two sulfate groups in positions 2 and 6 of N-acetyl-galactosamine (3.55%). Furthermore, more of the disulfated disaccharide in positions 2 and 4 (Di2, 4 dis) was produced by chondroitinase ABC than AC (14.13% and 3.29%, respectively); this confirms increased concentrations of DS, considering that Di4S and Di2,4diS mainly belong to DS as opposed to the Di6S and Di2,6diS species, which belong to CS.44
The percentages of CS and DS were determined by calculating the proportion of the total area of the chromatogram of the AC-samples and the total area of the ABC-samples. In fact, our results suggest that the percentages of CS and DS recovered in SGAT were 39.5% and 60.5%, respectively.
3.7. Biological functions of purified CS/DS
On the basis of the numbers and positions of sulfate on the disaccharides, the biological functions of GAGs are highly diversified. Accordingly, in the present study, we investigated the anticoagulant and antihypertensive activities of SGAT.Numerous reports have revealed that the aPTT test provides information about bleeding risk in preoperative evaluation, monitoring of antithrombotic treatment with heparin and screening of thrombosis risk factors. The anticoagulant activity of SGAT determined with the aPTT test is shown in Fig. 3A. The purified SGAT prolonged the anticoagulant activity in a concentration-dependent manner. Further, the addition of 500 μg mL−1 of SGAT in the reaction medium caused a significant (p < 0.05) prolongation of the clotting time. The ratio, which reflects the increase in the clotting time of plasma compared to the clotting time of control plasma, is equal to 2.08 at 500 μg mL−1. In this context, Sayari et al.4 reported that sulfated GAGs from Norway lobster shell prolonged anticoagulant activity 3 times more than the control at a concentration of 100 μg mL−1. Furthermore, Ben Mansour et al.9 reported concentration-dependent anticoagulant activity as measured by aPTT which was 2 to 3-fold lower than that of heparin for the skin of the ray Raja radula. The anticoagulant activity of SGAT, revealed by aPTT assay prolongation, was expected because sulfate groups are necessary to provide anticoagulant effects of polysaccharides. These are not only dependent on the sulfate content but also on the position of the sulfate groups and the molecular weight.47,48 Previous studies have indicated that the aPTT assay results of the low molecular weight fraction of GAGs from sea cucumber (Mw = 24.755 kDa) are similar to those of heparin, with significantly fewer side effects.48 In the present work, the in vitro anticoagulant activity of SGAT showed relationships with the concentration, the position and amount of sulfate groups and the molecular mass (MM = 24.07 kDa).
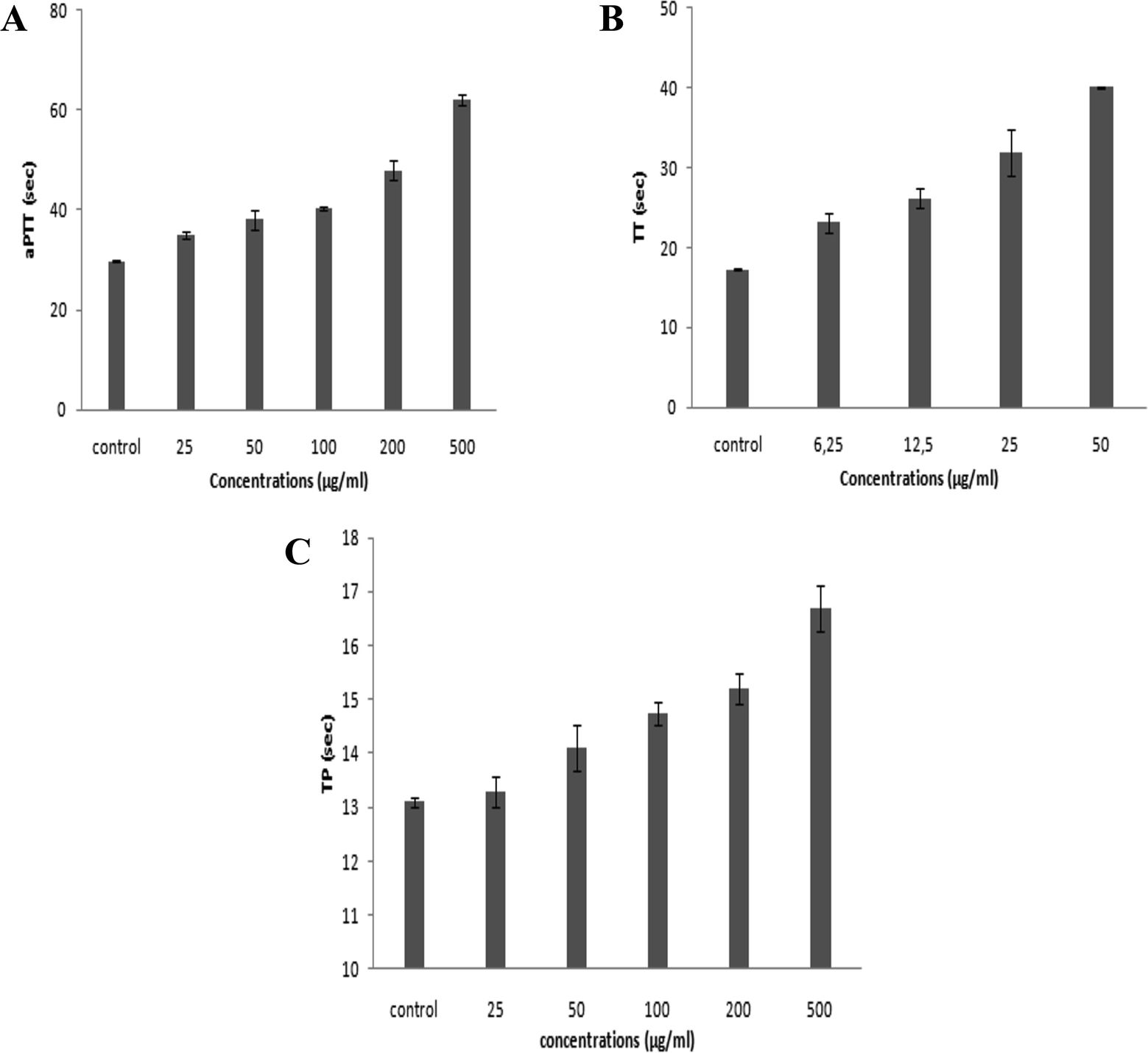 | ||
| Fig. 3 (A) Anticoagulant activity of the purified CS/DS from Atlantic bluefin tuna skin (SGAT) at different concentrations evaluated by measurement of the activated partial thromboplastin time (aPTT). (B) Anticoagulant activity of SGAT at different concentrations evaluated by measurement of the thrombin time (TT). (C) Anticoagulant activity of SGAT at different concentrations evaluated by measurement of the prothrombin time (PT). All results are expressed as mean ± SD (n = 3) and all measurements were performed in triplicate. | ||
Furthermore, it is highly important to compare the effects of SGAT and commercial heparin used as a positive control on clotting times. Heparin caused a significant prolongation of aPTT, and its clotting times were higher than those of SGAT. Indeed, the use of 7 μg mL−1 of heparin prolonged the coagulation time, which reached a value greater than 120 s. This clotting time value indicates that heparin can induce high risk of bleeding. As reported in Table 3, the aPTT anticoagulant activity of SGAT was evaluated using a parallel standard curve based on the International Heparin Standard. The aPTT anticoagulant activity of SGAT required 44.5 IU mg−1 at 500 μg mL−1. These obtained results were lower compared to the anticoagulant effect of sulfated GAGs from A. pleuronectus.49
| aPTT | TT | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μg mL−1 | IU mg−1 | μg mL−1 | IU mg−1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a The activity was expressed as international units per mg using a parallel standard curve based on the International Heparin Standard. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SGAT | 25 | 14.39 | 6.25 | 10.8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 50 | 17.57 | 12.5 | 13 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 100 | 19.88 | 25 | 18.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 200 | 27.79 | 50 | 20.86 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 500 | 44.5 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The anticoagulant effect of purified CS/DS was also evaluated via TT assay. This test is a measure of the conversion of fibrinogen to fibrin by high doses of thrombin, and an increase in TT suggests either thrombin inhibition or impaired fibrin polymerization.50 The TT results of SGAT at different concentrations are presented in Fig. 3B, which demonstrates that TT was prolonged in a concentration-dependent manner. Interestingly, SGAT significatively prolonged TT about 2.33 (40.1 s ± 0.14 s) times more than the control at a concentration of 50 μg mL−1. In this respect, Yang et al.48 studied the anticoagulant activity of GAGs from sea cucumber (Apostichopus japonicas) using the TT assay; they reported that the activities of GAGs were slightly higher than that of heparin at similar concentrations. Furthermore, the TT of SGAT was more prolonged than that previously described for the polysaccharides from Gentiana scabra Bunge roots (1.1 fold at the concentration of 1.5 mg mL−1).51 Thus, the anticoagulant effects of SGAT can be explained by inhibition of fibrin formation, which is associated with intrinsic pathway inhibition.
Compared with the positive control, the TT of SGAT was lower than that of heparin. Indeed, the use of 7 μg mL−1 of heparin prolonged the coagulation time by more than 60 s. Following this, the TT assay of SGAT reported a clotting time of 20.86 IU mg−1 at 50 μg mL−1 (Table 3).
The PT test is used to characterize extrinsic coagulation factors. An abnormal or prolonged PT generally designates a deficiency in one or more factors in the extrinsic or common pathway of blood coagulation. This deficiency may be caused by vitamin K deficiency, hereditary coagulation disorders, liver disease or drug administration.
The anticoagulant activity of SGAT was also evaluated by its ability to extend the prothrombin time (PT). As shown in Fig. 3C, SGAT prolonged the anticoagulant activity in a concentration-dependent manner. Furthermore, SGAT showed significantly prolonged PT about 1.28 fold greater than that of the control at a concentration of 500 μg mL−1. Indeed, SGAT showed a more prolonged PT than that previously described for sulfated GAGs from grey triggerfish (1.1 fold at the same concentration); however, the PT was shorter than that previously described for sulfated GAGs from smooth hound (1.55 fold at 500 μg mL−1).20 These results indicate that SGAT has important anticoagulant activity in the extrinsic pathway of blood clots. In this assay, it is impossible to evaluate the PT of heparin because the reagent used (neoplastin) contains a heparin-neutralizing agent.
To conclude, these results suggest that the purified CS/DS recovered from Atlantic bluefin tuna skin may be utilized as an anticoagulant agent both in the inhibition of the intrinsic and extrinsic or common blood coagulation pathways, probably with fewer bleeding side effects than heparin. It has been reported that there is a relationship between the anticoagulant activity and structure of GAGs, such as the degree of sulfation, sulfation position, molecular weight, type of sugar, glycosidic branching and three-dimensional structure of the sulfated polysaccharide.52 In this context, Pereira et al.53 reported that the disaccharide unit of GAGs monosulfated at the 4-position (Di4S) and/or disulfated at the 2- and 4-positions (Di2,4diS) is necessary for thrombin inhibition by activating heparin cofactor II and antithrombin, respectively. This is in line with the results of our SAX-HPLC analysis.
The purified SGAT was evaluated in terms of its ACE-inhibitory activity. As shown in Fig. 4, SGAT exhibited concentration-dependent ACE inhibitory activity. SGAT was also noted to exhibit strong ACE-inhibitory activity, with the highest levels (70.81%) being observed at a concentration of 0.8 mg mL−1. The IC50 value, defined as the concentration of inhibitor required to prevent 50% of ACE activity, was calculated to be 0.25 mg mL−1. This IC50 value is higher than the IC50 of sulfated polysaccharides from grey triggerfish and smooth hound.18
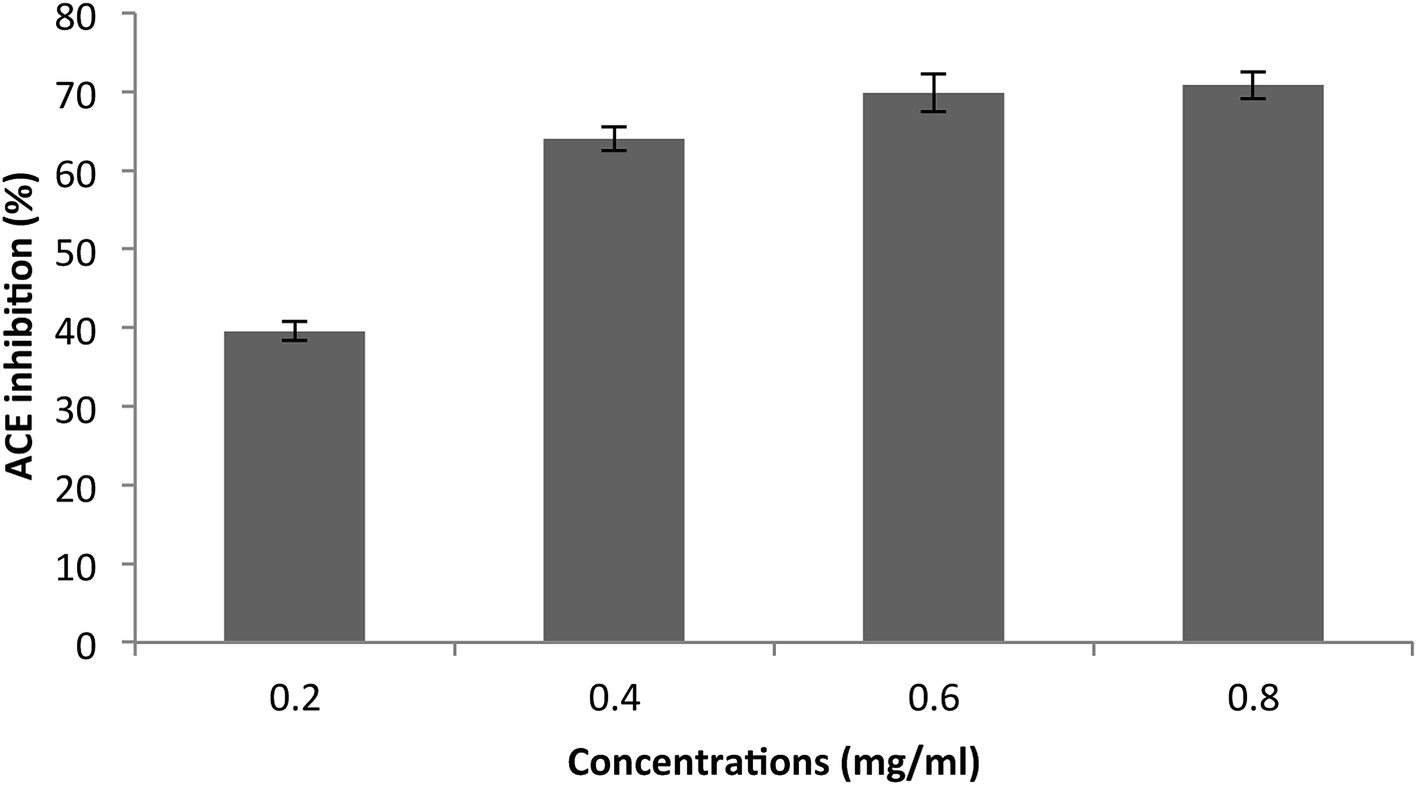 | ||
| Fig. 4 Angiotensin I-converting enzyme inhibitory activity of SGAT. The values are the means of three replications ± SD. | ||
However, Tan and Gan54 demonstrated that the ACE inhibitory activity of polysaccharides may be due to the presence of uronic acid, which can be ionized. This process creates an acidic environment, which is not suitable for ACE because its optimum pH is 8.3. Under acidic conditions, ACE would be denatured and therefore lose its activity.
4. Conclusion
CS/DS were extracted and purified for the first time from the skin of Atlantic bluefin tuna, and their structural characterization and properties are described in the present study. Furthermore, on the basis of the collected data, CS/DS from tuna skin showed high percentages of monosulfated and disulfated disaccharides produced by chondroitinase ABC and AC. SGAT had effective in vitro anticoagulant activity; it prolonged blood clotting time based on activated partial thromboplastin time (aPTT), thrombin time (TT) and prothrombin time (PT) tests and exhibited strong ACE-inhibitory activity. Finally, these new CS/DS are potentially useful for pharmacological applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Tunisian Ministry of Higher Education and Scientific Research.References
- U. Lindahl, Heparin: Chemical and Biological Properties, Clinical Applications, Edward Arnold, London, 1989, vol. 159 Search PubMed.
- L. Kjellen and U. Lindahl, Annu. Rev. Biochem., 1991, 60, 443 CrossRef CAS PubMed.
- V. H. Pomin, Carbohydr. Res., 2015, 413, 41–50 CrossRef CAS PubMed.
- N. Sayari, R. Balti, M. Ben Mansour, I. Ben Amor, I. Graiet, J. Gargouri and A. Bougatef, Biomed. Pharmacother., 2016, 80, 322–330 CrossRef CAS PubMed.
- F. Krichen, W. Karoud, A. Sila, B. E. Abdelmalek, R. Ghorbel, S. Ellouz-Chaabouni and A. Bougatef, Int. J. Biol. Macromol., 2015, 75, 283–289 CrossRef CAS PubMed.
- F. Maccari, F. Galeotti and N. Volpi, Carbohydr. Polym., 2015, 129, 143–147 CrossRef CAS PubMed.
- F. Maccari, F. Ferrarini and N. Volpi, Carbohydr. Res., 2010, 345, 1575–1580 CrossRef CAS PubMed.
- T. Zhao, Y. Zhou, G. Mao, Y. Zou, J. Zhao, S. Bai, L. Yang and X. Wu, J. Sci. Food Agric., 2013, 93, 1633–1640 CrossRef CAS PubMed.
- M. Ben Mansour, H. Majdoub, I. Bataille, M. S. Roudesli, M. Hassine, N. Ajzenberg, F. Chaubet and R. M. Maaroufi, Thromb. Res., 2009a, 123, 671–678 Search PubMed.
- K. Sugahara, S. Nadanaka, K. Takeda and T. Kojima, Eur. J. Biochem., 1996, 239, 871–880 CrossRef CAS PubMed.
- H. Bougatef, F. Krichen, F. Capitani, I. Ben Amor, F. Maccari, V. Mantovani, F. Galeotti, N. Volpi, A. Bougatef and A. Sila, Carbohydr. Polym., 2018, 196, 272–278 CrossRef CAS PubMed.
- F. Krichen, N. Volpi, A. Sila, F. Maccari, V. Mantovani, F. Galeotti, S. Ellouz-Chaabouni and A. Bougatef, Int. J. Biol. Macromol., 2017, 95, 32–39 CrossRef CAS PubMed.
- M. Ben Mansour, M. Dhahri, I. Bertholon, V. Ollivier, I. Bataille, N. Ajzenberg, M. Hassine, M. Jandrot-Perrus, F. Chaubet and R. M. Maaroufi, Thromb. Res., 2009b, 123, 887–894 Search PubMed.
- T. Tsegenidis, Comp. Biochem. Physiol. B, 1992, 103, 275–279 CrossRef.
- N. Volpi, J. Pharm. Sci., 2007, 96, 3168–3180 CrossRef CAS PubMed.
- N. Volpi, J. Pharm. Pharmacol., 2009b, 61, 1271–1280 Search PubMed.
- C. C. Chatziioannidis, N. K. Karamanos, S. T. Anagnostides and T. Tsegenidis, Biochimie, 1999a, 81, 187–196 Search PubMed.
- F. Krichen, W. Karaoud, N. Sayari, A. Sila, F. Kallel, S. Ellouz-Chaabouni and A. Bougatef, J. Polym. Environ., 2016, 24(2), 166–175 CrossRef CAS.
- S. F. Chavante, A. S. Brito, M. Lima, E. Yates, H. Nader, M. Guerrini, G. Torri and A. Bisio, Carbohydr. Res., 2014, 390, 59–66 CrossRef CAS PubMed.
- F. Krichen, Z. Ghlissi, I. Ben Amor, N. Sayari, R. Kallel, J. Gargouri, Z. Sahnoun, T. Boudawara, S. Ellouz-Chaabouni and A. Bougatef, Exp. Toxicol. Pathol., 2017b, 69, 45–53 Search PubMed.
- A. S. Brito, D. S. Arimatéia, L. R. Souza, M. A. Lima, V. O. Santos, V. P. Medeiros, C. V. Ferreira, G. Z. Justo, E. L. Leite, G. P. Andrade, F. W. Oliveira, H. B. Nader and S. F. Chavante, Bioorg. Med. Chem., 2008, 16, 9588–9595 CrossRef CAS PubMed.
- J. A. Marcum, J. B. McKenney, S. J. Galli, R. W. Jackman and R. D. Rosenberg, Am. J. Physiol., 1986, 250(5), H879–H888 CAS.
- K. Majumder and J. Wu, J. Agric. Food Chem., 2009, 57, 471–477 CrossRef CAS PubMed.
- L. Vercruysse, J. V. Camp and G. Smagghe, J. Agric. Food Chem., 2005, 53, 8106–8115 CrossRef CAS PubMed.
- C. P. Alderman, Ann. Pharmacother., 1996, 30, 55–61 CrossRef CAS PubMed.
- M. Cesaretti, E. Luppi, F. Maccari and N. Volpi, Carbohydr. Polym., 2003, 54, 59–61 CrossRef CAS.
- M. Dubois, K. A. Gilles, J. K. Hamilton, P. A. Rebers and F. Smith, Anal. Chem., 1956, 28, 350–356 CrossRef CAS.
- Y. Wegrowski and F. X. Maquart, Methods Mol. Biol., 2001, 171, 175–179 CAS.
- R. E. Edens, A. al-Hakim, J. M. Weiler, D. G. Rethwisch, J. Fareed and R. J. Linhardt, J. Pharm. Sci., 1982, 81, 823–827 CrossRef.
- D. Buzzega, F. Maccari and N. Volpi, J. Pharm. Biomed. Anal., 2010, 51, 969–972 CrossRef CAS PubMed.
- M. Dathe, M. Schumann, T. Wieprecht, A. Winkler, M. Beyermann, E. Krause, K. Matsuzaki, O. Murase and M. Bienert, Biochemistry, 1996, 35, 12612–12622 CrossRef CAS PubMed.
- Y. Nakamura, N. Yamamoto, K. Sakai, A. Okubo, S. Yamazaki and T. Takano, J. Dairy Sci., 1995, 78, 777 CrossRef CAS PubMed.
- B. E. Abdelmalek, A. Sila, F. Krichen, W. Karoud, O. Martinez-Alvarez, S. Ellouz-Chaabouni, M. A. Ayadi and A. Bougatef, Int. J. Biol. Macromol., 2015, 72, 1143–1151 CrossRef CAS PubMed.
- B. W. SSouza, M. A. Cerqueira, A. I. Bourbon, A. C. Pinheiro, J. T. Martins, J. A. Teixeira, M. A. Coimbra and A. A. Vicente, Food Hydrocolloids, 2012, 27, 287–292 CrossRef.
- M. Ben Mansour, M. Dhahri, M. Hassine, N. Ajzenberg, L. Venisse, V. Ollivier, F. Chaubet, M. Jandrot-Perrus and R. M. Maaroufi, Comp. Biochem. Physiol. B, 2010, 156, 206–215 CrossRef PubMed.
- M. E. R. Duarte, M. A. Cardoso, M. D. Noseda and A. S. Cerezo, Carbohydr. Res., 2003, 333, 281–293 CrossRef.
- D. Santhiya, S. Subramanian and K. A. Natarajan, J. Colloid Interface Sci., 2002, 256, 237–248 CrossRef CAS PubMed.
- S. S. Bandyopadhyay, M. H. Navid, T. Ghosh, P. Schnitzler and B. Ray, Phytochemistry, 2011, 72, 276–283 CrossRef CAS PubMed.
- Y. Kariya, S. Watabe, K. Hashimoto and K. Yoshida, J. Biol. Chem., 1990, 265, 5081–5085 CAS.
- J. Mou, Q. Li, X. Qi and J. Yang, Carbohydr. Polym., 2018, 185, 41–47 CrossRef CAS PubMed.
- G. V. Coppa, F. Maccari, L. Zampini, L. Santoro, T. Galeazzi, O. Gabrielli and N. Volpi, Food Chem., 2012, 134, 195–199 CrossRef CAS.
- K. A. Jandik, K. Gu and R. J. Linhardt, Glycobiology, 1994, 4, 289–296 CrossRef CAS PubMed.
- T. Hashiguchi, T. Kobayashi, D. Fongmoon, A. K. Shetty, S. Mizumoto, N. Miyamoto, T. Nakamura, S. Yamada and K. Sugahara, Biochim. Biophys. Acta, 2011, 1810, 406–413 CrossRef CAS PubMed.
- C. Malavaki, S. Mizumoto, N. Karamanos and K. Sugahara, Connect. Tissue Res., 2008, 49, 133–139 CrossRef CAS PubMed.
- R. Branislav, M. Marijana and S. Slobodan, World J. Microbiol. Biotechnol., 2008, 24, 1239–1242 CrossRef.
- E. R. Carbonero, F. R. Smiderle, A. H. P. Gracher, C. G. Mellinger, G. Torri and T. Ahti, Carbohydr. Polym., 2006, 63, 13–18 CrossRef CAS.
- J. A. G. Rodrigues, I. N. L. Queiroz, A. L. G. Quindere, B. C. Vairo, P. A. S. Mourao and N. M. B. Benevides, Cienc. Rural, 2011, 41, 634–639 CrossRef CAS.
- J. Yang, Y. Wang, T. Jiang and Z. Lv, Int. J. Biol. Macromol., 2015, 72, 911–918 CrossRef CAS PubMed.
- R. Saravanan and A. Shanmugam, Carbohydr. Polym., 2011, 86, 1082–1084 CrossRef CAS.
- L. Ye, L. Xu and J. Li, Carbohydr. Polym., 2012, 87(3), 2052–2057 CrossRef CAS.
- W. Cai, H. Xu, L. Xie, J. Sun, T. Sun, X. Wu and Q. Fu, Carbohydr. Polym., 2016, 140, 308–313 CrossRef CAS PubMed.
- S. T. Olson, I. Bjork and S. C. Bock, Trends Cardiovasc. Med., 2002, 12, 198–205 CrossRef CAS PubMed.
- M. S. Pereira, F. R. Melo and P. A. S. Mourao, Glycobiology, 2002, 12, 573–580 CrossRef CAS PubMed.
- H. F. Tan and C. Y. Gan, Int. J. Biol. Macromol., 2016, 85, 487–496 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |