
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtective desilication of highly siliceous H-ZSM-5 by sole tetraethylammonium hydroxide for the methanol to propylene (MTP) reaction†
Yunpeng Zhang,
Minggang Li,
Enhui Xing,
Yibin Luo * and
Xingtian Shu
* and
Xingtian Shu
Research Institute of Petroleum Processing Sinopec, Xueyuan Road No. 18, Beijing, China. E-mail: luoyibin.ripp@sinopec.com
First published on 12th November 2018
Abstract
Protective desilication of highly siliceous H-ZSM-5 was effectively realized by dissolution and recrystallization in tetraethylammonium hydroxide (TEAOH) solution. With better balance between dissolution of OH− and recrystallization of TEA+, intracrystalline mesopores could be generated by selective dissolution of Si by the drilling effects of TEAOH on the micropores, and then Si species in the mother liquor near the external surface could be recrystallized into ZSM-5 shell. With a significantly reduced diffusion length provided by the intracrystalline mesopores, TEAOH-treated samples exhibited longer lifetime and higher propylene selectivity than the parent H-ZSM-5 zeolite. The mediumly-treated T-16 h sample possessed the longest MTP lifetime of 140 h, 5.6 times that of the parent H-ZSM-5 zeolite. Furthermore, the coke content and adsorbed methyl benzene species on the T-16 h sample were heavier than those on the parent H-ZSM-5 sample, which were related to the intracrystalline mesopore structure.
1 Introduction
Propylene, as the raw material for a wide variety of products such as polypropylene, polyacrylonitrile, isopropyl alcohol, acrolein and acrylic acid, is one of important products in the petrochemical industry.1 Propylene was mainly produced from fossil resources via steam cracking and fluid catalytic cracking.2 The methanol-to-propylene (MTP) reaction is one of the most important reactions in C1 chemistry, which has provided an alternative route to fossil-based process for producing propylene.3–5 Zeolites with uniform microporous channels, tunable acidity, and high hydrothermal stability such as ZSM-5,6 ZSM-11,7 ZSM-22,8 and β zeolites9 have been extensively tested for the MTP reaction, among which ZSM-5 has been proven to be the optimum catalyst because of its unique three-dimensional pore structure with straight channels (5.3 × 5.6 Å) and intersecting zigzag channels (5.1 × 5.5 Å).10,11 ZSM-5 catalysts had been commercially available for MTP process by Lurgi.12Better propylene selectivity and longer lifetime are two key challenges for the designing of MTP catalysts.13 In general, acidity and porosity are the two main factors influencing product selectivity and lifetime. However poor diffusion of coke precursors from active centers could lead to coke deposition to block and cover active sites within channels of ZSM-5.14,15 In order to overcome the above disadvantages, ZSM-5 with a secondary network of mesopores and micropores was obtained by dealumination or desilication treatment.16
As for highly siliceous ZSM-5 zeolite, dealumination has poor efficiency in generating enough mesopores. More importantly, framework Al sites are also removed during dealumination. Therefore, base leaching is a simple but effective in generating adequate mesopores, which differs from dealumination, as silicon, rather than aluminum, is effectively extracted from the zeolite matrix, however careful control on desilication depth is desired to avoid the formation of extra-framework Al, equal to, the loss of active centers for MTP reaction.17,18 Thus, selective desilication was adapted to achieve substantial modification on acidity and porosity of zeolites.
Pioneering work by Groen19 showed that ZSM-5 with a Si/Al ratio range of 25–50 was optimal for generating a uniform mesoporous structure because higher amount of Al protected Si from excessive removal resulting in a less mesoporous structure. At higher Si/Al ratio, limited protection by less Al led to excessive Si leaching, thus, larger mesopores and lower product selectivity were obtained, which inevitably deceased shape selectivity effect and amount of framework Al because of severe Si leaching. In most cases, NaOH has been most usually used base to obtain mesoporous ZSM-5 via post treatment,20 however NaOH has difficulty in control on the desilication depth because of its strong dissolution capability and easy diffusion into channels of zeolites.21 Therefore, controllable or protective desilication was designed by incorporating external pore directing agents (Al (OH)4−, Ga (OH)4−, or TPA+) or organic amine in the alkaline medium.22,23 Framework Al should be preserved as much as possible during Si extraction to form sufficient mesopores. For zeolites in alkaline solution, dissolution (desilication) and crystallization (protection on zeolitic frameworks) are two main reactions occurred, which were influenced by OH− and templating capability. Careful consideration on capability between dissolution (NaOH) and crystallization (protecting agent) should be weighted to achieve better balance. Subsequent ion exchange process is usually needed for these zeolites treated in presence of NaOH to act as acid catalysts. In this paper, better balance between dissolution of Si and protecting effects on framework might be perfectly achieved by only one agent without NaOH. Bulky quaternary ammonium hydroxide is a favorable choice to satisfy these requirements with OH− (dissolution of Si) and quaternary ammonium cation (protection on zeolitic framework), however strong tendency of crystallization toward MFI structure might have reverse effects on the formation of mesopores. Therefore, tetraethylammonium hydroxide (TEAOH), which is larger than micro pore sizes of ZSM-5, was intentionally selected as the post treating reagent to achieve better balance between dissolution and crystallization without addition of NaOH.
This paper presented a protective method to introduce mesopores within highly siliceous H-ZSM-5 by sole TEAOH. Various characterizations were utilized to propose the mechanism on the formation of intracrystalline mesopores within H-ZSM-5 zeolite, and to explain corresponding benefits on lifetime, propylene selectivity, and coke species during MTP reaction over TEAOH-treated H-ZSM-5 catalysts.
2 Experimental
2.1 Preparation
Commercially available H-ZSM-5 (SiO2/Al2O3 = 200) was used as provided by Sinopec Catalyst CO., LTD Changling Division. Pseudo-boehmite were firstly dissolved in the TPAOH solution at 140 °C for 2 h to form clear homogenous solution, and then seeds, silica gel, deionized water were mixed to form synthesis gel with batch composition as following: SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3:TPAOH:H2O = 1:0.005:0.06:6. The synthesis was carried out at 120 °C for 24 h with stirring. The product was filtered, washed and dried at 120 °C overnight. Parent H-ZSM-5 was obtained via calcinations of as-made ZSM-5 for 6 h in a muffle furnace to remove organics.
:Al2O3:TPAOH:H2O = 1:0.005:0.06:6. The synthesis was carried out at 120 °C for 24 h with stirring. The product was filtered, washed and dried at 120 °C overnight. Parent H-ZSM-5 was obtained via calcinations of as-made ZSM-5 for 6 h in a muffle furnace to remove organics.
Parent H-ZSM-5 was treated in TEAOH solution with typical batch composition in terms of molar ratio as: TEAOH/SiO2 = 0.04, H2O/SiO2 = 4. The post treatment was carried out in a Teflon-lined autoclave at 170 °C for various time. The product was recovered by filtration, washed and dried in oven at 120 °C overnight, and calcined at 550 °C for 4 h. The final obtained samples were designated as parent ZSM-5 and T-x h, in which T-x h indicated the ZSM-5 zeolite treated by TEAOH for different hours. For comparison, TPAOH-treated H-ZSM-5 (labeled as TPA-16 h) was also characterized and subject to MTP reaction.
2.2 Characterization
The powder X-ray diffraction (XRD) patterns of different zeolite samples were recorded on an EMPYREAN powder diffractometer (PANalytical Corporation, Netherland) using Cu Kα radiation (λ = 0.154 nm) at a scanning rate of 0.02° min−1 from 5° to 35°. The relative crystallinity of the samples were calculated by standard sample in the laboratory. The chemical compositions of the samples were determined on an X-ray fluorescence (XRF) spectrometer ZSX Primus II (Rigaku). X-ray photoelectron spectroscopy data were obtained using an ESCALab 250 electron spectrometer from Thermo Fisher Scientific Corporation with monochromatic 150 W AlKα radiation. Pass energy for the narrow scan is 30 eV. The base pressure was about 6.5 × 10−10 mbar. The binding energies were referenced to the Cls line at 284.8 eV from alkyl or adventitious carbon.Nitrogen adsorption–desorption isotherms were performed on a Micro-meritics ASAP 2420 instrument. Fresh samples were outgassed under vacuum for 15 h prior to the adsorption measurements. The Brunauer–Emmett–Teller (BET) equation was used to calculate the surface area. The total pore volumes were evaluated from the volume adsorbed at p/p0 = 0.99, whereas the micropore surface area and the microspore volume were determined by the t-plot method.
The morphology of samples was examined by scanning electron microscopy (SEM) on a HITACHI S-4800 microscope. Transmission electron microscopy (TEM) images were recorded on a JEM-2100 transmission electron microscope operating at an accelerating voltage of 200 kV.
The 27Al and 29Si magic angle spinning nuclear magnetic resonance (MAS NMR) spectra were recorded on a Bruker Avance III 500 WB spectrometer with the 7 mm ZrO2 rotor. The frequencies of the 27Al and 29Si MAS NMR were 156.4 and 99.3 MHz, respectively. The 27Al MAS NMR spectrum with the single pulse sampling, pulse width was 0.4 μs and the revolution of MAS was 12 kHz. The 29Si MAS NMR spectrum with the single pulse sampling, pulse width was 2.1 μs and the revolution of MAS was 5 kHz.
NH3-TPD experiments were carried out on an Autochem II 2920 unit equipped with a thermal conductivity detector. 50 mg of zeolite was degassed in a He stream at 600 °C for 1 h. After cooling to 150 °C, the sample was put in a mixed gas flow of 10% NH3 and 90% He (40 mL min−1) for the sufficient adsorption of the NH3. After 30 min, the flow was switched to nitrogen, and TPD profiles were recorded from 150 to 600 °C at a constant heating rate of 10 °C min−1 under He flow (40 mL min−1). The desorbed ammonia was monitored by a thermal conductivity detector.
The deactivated samples were characterized using a thermal gravimetric analyzer (STA 449 F5, NETZSCH Corporation). In a typical run, about 15 mg of the spent sample was placed in the crucible and heated to 900 °C in a 60 mL min−1 air stream at a heating rate of 10 °C min−1, while the mass change was recorded.
The soluble coke species were analyzed according to the method described by Guisnet.24 Retained hydrocarbons soluble in CH2Cl2 were extracted by dissolving 20 mg of deactivated catalysts with a given fraction in 1 mL 40% HF for 3 h. After neutralization (pH = 7) with 2 M NaOH solution, 6 mL CH2Cl2 was added to Teflon beaker to extract the organic compounds trapped within deactivated MTP catalysts. Then, organic phase and insoluble coke were separated by separatory funnel. The mixing of organic compounds and insoluble coke were oscillated with ultrasonic for 3 h. Organic compounds were obtained by filtration and concentrated to 0.8–1 mL, and then analyzed by an Agilent 7890 A-5975C equipped with a HP-5 MS column.
2.3 Catalytic tests
MTP reaction was carried out in a fixed-bed continuous-flow reactor with a titanium tube of 12 mm inner diameter and 250 mm in length. In a typical run, 3 g zeolite catalyst (20–40 mesh) was located in the flat-temperature zone of the reactor. The mixture of methanol/water (mass ratio 1:1) was pumped into the reactor by a microtube pump (NS, NP-KX-110) at a methanol WHSV of 4.5 h−1. MTP reaction was conducted at 480 °C under atmospheric pressure. The gas products were sampled and analyzed by an Agilent-7890A gas chromatograph. Lifetime was defined as TOS from initial time to final time at 90% conversion. The deactivation rate around 95% conversion was obtained by linear fitting the data points around 95%.
3 Results and discussion
3.1 Physicochemical properties
Fig. S1† shows the typical powder XRD patterns of the parent, TEAOH-treated H-ZSM-5 and TPA-16 h samples. All samples displayed a characteristic diffraction pattern of MFI topology in ranges of 5–10° and 22.5–25° without any competing phase detected. The relative crystallinity first dropped slightly from 88.3% (parent H-ZSM-5) to 83.5% at 4 h, and then was further reduced to 82.9% at 16 h. After 16 h relative crystallinity was gradually recovered to 85.0% at 24 h and 88.1% at 48 h, respectively. On one hand, OH− of TEAOH could dissolve Si from the framework of parent H-ZSM-5, however bulky TEA+ hindered free diffusion of OH− throughout parent H-ZSM-5. Therefore, there was a slight decrease of relative crystallinity before 16 h. On the other hand, TEAOH was effective structure directing agent for highly siliceous zeolites besides ZSM-5.25 The recrystallization capability of TEAOH toward MFI structure could be proven by the recovery in relative crystallinity at 24 h and 48 h with parent H-ZSM-5 as seeds. No formation of amorphous or other new phase emerge was observed in presence of parent H-ZSM-5, which showed superior stability of ZSM-5 via a dissolution–recrystallization process in TEAOH solution. Moreover, with better recrystallization capability than TEAOH, TPA-16 h showed higher relative crystallinity of 84.5% than that of T-16 h.Table 1 shows the chemical compositions of calcined samples. During post treatment, SiO2/Al2O3 ratio dropped slightly from 197.6 to 193.1 at 16 h, which indicated occurrence of slight desilication. With longer treating time, desilication depth gradually enlarged to decrease SiO2/Al2O3 ratio to 176.6 (24 h) and 174.8 (48 h), respectively. However, the relative crystallinity was even recovered to 85.0% and 88.1% for samples at 24 h and 48 h. This phenomenon could be explained by the dissolution effects and subsequent recrystallization induced by OH− and TEA+ of TEAOH, respectively. These results clearly showed stronger tendency toward MFI structure of TEAOH with parent H-ZSM-5 as seeds than desilication effects of OH− in TEAOH, which contributed to the stable crystalline structure of parent H-ZSM-5. More importantly, there was hardly detection of Al in mother liquor during TEAOH treatment because of suitable alkalinity and protective effects via recrystallization toward MFI structure in presence of H-ZSM-5 seeds. According to the XPS and XRF data, it was clear that parent H-ZSM-5 showed an Al-rich exterior (superficial SiO2/Al2O3 = 115.9 determined by XPS) and Si-rich interior (bulk SiO2/Al2O3 = 197.6 determined by XRF). The reason of discrepancy between bulk (XRF) and surface (XPS) SiO2/Al2O3 molar ratios was decided by the synthesis condition of parent H-ZSM-5. Parent H-ZSM-5 was found from a siliceous core to an Al-rich rim, which resulted to higher bulk SiO2/Al2O3 than superficial SiO2/Al2O3. R. V. Ballmoos first found that Al concentration in the core was below that in the rim of ZSM-5 zeolite in 1981. Moreover, Al was invariably concentrated in the rim of ZSM-5 crystals.26 During TEAOH treatment, superficial SiO2/Al2O3 ratio (115.9, parent H-ZSM-5) was decreased to 93.9 (4 h) and 83.0 (16 h) because desilication occurred on the surface at first. Although further desilication proved by XRF results, superficial SiO2/Al2O3 ratio increased to 84.4 (24 h) and 91.6 (48 h) due to severe exposure of Si-rich interior. During TPAOH treatment, TPA-16 h sample showed bulk SiO2/Al2O3 of 192.9, similar to that of T-16 h (193.1). However, superficial SiO2/Al2O3 ratio of TPA-16 h sample (96.3) was obviously higher than that of T-16 h sample (83.0) which implied that better recrystallization capability of TPAOH transformed more Si species in solution recrystallized into ZSM-5 crystal shell.
| Zeolite | H-ZSM-5 | T-4 h | T-16 h | T-24 h | T-48 h | TPA-16 h |
|---|---|---|---|---|---|---|
| Na2O, w% | 0.03 | 0.07 | 0.05 | 0.04 | 0.03 | 0.04 |
| Al2O3, w% | 0.85 | 0.86 | 0.87 | 0.95 | 0.96 | 0.87 |
| SiO2, w% | 98.8 | 98.8 | 98.8 | 98.7 | 98.7 | 98.7 |
| (SiO2/Al2O3) XRF | 197.6 | 195.3 | 193.1 | 176.6 | 174.8 | 192.9 |
| (SiO2/Al2O3) XPS | 115.9 | 93.9 | 83.0 | 84.4 | 91.6 | 96.3 |
During TEAOH treatment on H-ZSM-5, Si species could be selectively removed from the external surface, and then inner of the H-ZSM-5 crystals, which resulted in causing the formation of mesopores ranging in several nm coupled with a substantial mesopore volume at expense of micropores. Parent H-ZSM-5 possessed a typical I isotherm, indicating microporosity of parent H-ZSM-5. TEAOH-treated H-ZSM-5 samples showed two hysteresis loops in N2 adsorption–desorption isotherms shown in Fig. 1. The first hysteresis loop appearing at p/p0 = 0.1–0.4 was associated with the phenomenon of the fluid-to-crystalline phase transition of adsorbed N2 on the ZSM-5 zeolite with high Si/Al ratio, while the second hysteresis loop appearing at p/p0 = 0.4–1.0 with larger amount of N2 sorption verified the formation of mesopores (Fig. 1a). The efficiency of TEAOH treatment is clearly manifested by the pronounced hysteresis loop of TEAOH-treated samples, which corresponds to a significant mesopore distribution with an obvious peak at 2–3 nm in Fig. 1b. With IV type isotherms, the TPA-16 h sample showed much smaller hysteresis loops and pore distribution than T-16 h. The results about textural properties of parent H-ZSM-5, TEAOH-treated H-ZSM-5 and TPA-16 h samples were listed in Table 2. The BET surface area, micropore and external surface areas of parent H-ZSM-5 were 429 m2 g−1, 399 m2 g−1 and 30 m2 g−1, respectively. The total, micropore and mesopore volumes were 0.29 cm3 g−1, 0.19 cm2 g−1 and 0.10 cm2 g−1, respectively. After TEAOH treatment, the micropore surface area and pore volume obviously decreased, which proved partial destruction of H-ZSM-5 microporous channels by OH− of TEAOH. It seemed that no obvious change in external surface area was observed, however the mesopore volume first increased up to maximum 0.19 cm3 g−1 at 16 h. The increase in mesopore volumes contributed to the enhancement in total pore volume in spite of slight loss of micropore volume. These changes clearly proved that TEAOH transformed micropores to mesopores because of its alkalinity and larger sizes than channels of H-ZSM-5. More importantly, the desilication depth should be carefully controlled to avoid subsequent decrease of formed mesopores due to breakup of H-ZSM-5 crystals. Moreover, T-16 h had the largest average pore diameter of 3.87 nm (Table 2). With treating time longer than 16 h, the formed mesopores decreased because of further desilication, meanwhile the relative crystallinity was recovered by recrystallization. By contrast, after TPAOH treatment, mesopore volume (0.130 cm3 g−1) of TPA-16 h was much smaller than that of T-16 h (0.185 cm3 g−1) because of better recrystallization capability.
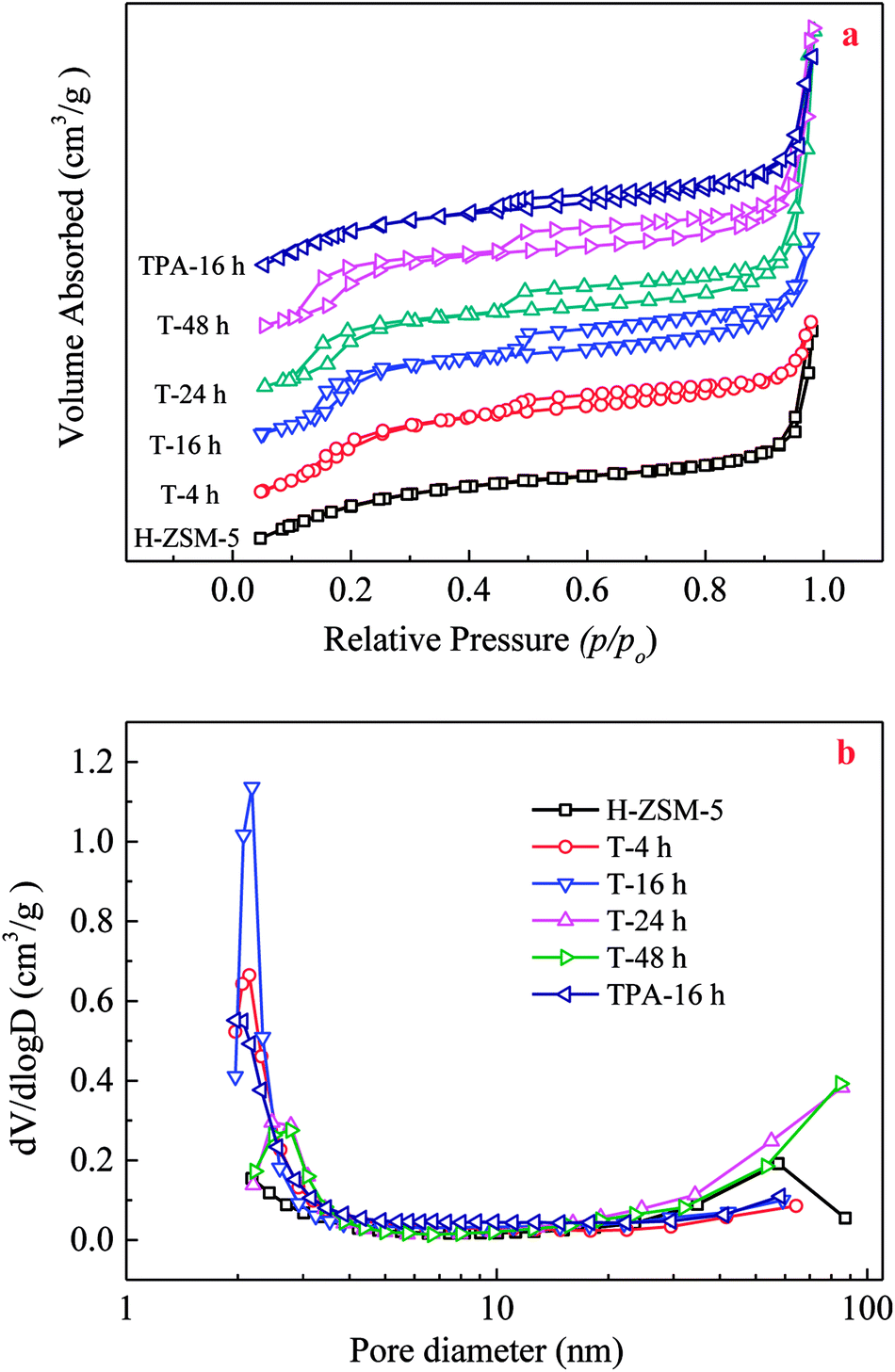 | ||
| Fig. 1 N2 adsorption–desorption isotherms (a) and BJH adsorption pore-size distribution (b) of the parent, TEAOH-treated H-ZSM-5 and TPA-16 h samples. | ||
| Samples | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) | ||||
|---|---|---|---|---|---|---|---|
| SBETa/(m2 g−1) | Sext/(m2 g−1) | Smicrob/(m2 g−1) | Vtotalc/(cm3 g−1) | Vmicrob/(cm3 g−1) | Vmesod/(cm3 g−1) | ||
| a BET surface area.b Micropore surface area evaluated by t-plot method.c Total pore volume at p/p0 = 0.99.d Mesopore volume calculated using Vtotal − Vmicro. | |||||||
| H-ZSM-5 | 429 | 30 | 399 | 0.294 | 0.190 | 0.104 | 3.10 |
| T-4 h | 409 | 40 | 368 | 0.292 | 0.179 | 0.113 | 3.61 |
| T-16 h | 364 | 35 | 339 | 0.356 | 0.171 | 0.185 | 3.87 |
| T-24 h | 396 | 32 | 364 | 0.351 | 0.174 | 0.177 | 3.78 |
| T-48 h | 388 | 33 | 355 | 0.317 | 0.171 | 0.146 | 3.77 |
| TPA-16 h | 387 | 38 | 349 | 0.311 | 0.181 | 0.130 | 3.23 |
SEM and TEM were employed to characterize the evolution of morphology and fine structure of the H-ZSM-5 during TEAOH treatment. As shown in Fig. 2, parent H-ZSM-5 was monodispersed particles with smooth surface in the range from 100 to 300 nm in diameters. After TEAOH treatment, the surface of H-ZSM-5 particles became rough because of desilication by OH− of TEAOH. At 24 h and 48 h, the breakup of ZSM-5 crystals were observed because of further desilication, however the relative crystallinity was well reserved and recovered to 85.0% and 88.1% at 24 h and 48 h, respectively. This also proved the recrystallization effects of TEAOH which could lead to the formation of irregular morphology through the broken crystals. TEM provided clear observation of formed mesopores during TEAOH and TPAOH treatment (Fig. 3). At 4 h, small mesopores could be observed all over the parent H-ZSM-5, while at 16 h the mesopores became larger than those at 4 h. The enlargement of mesopores could be realized by further dissolution of Si by OH− of TEAOH within H-ZSM-5 crystals or merge of several mesopores. Although large amount of mesopores were observed in samples at 4 h and 16 h, it seemed that ZSM-5 shell is completely preserved, which indicated the formation of intracrystalline mesopores. TPA-16 h sample showed rich but smaller bright spots distributing in the crystals (Fig. 3f). During protective desilication in presence of NaOH and TPAOH, the intracrystalline mesopores could also be formed, which is ascribed to the adsorption of TPA+ onto the zeolite, sealing off most of the available external surface, hereby largely inhibiting surface realumination, contrary to what occurs in pure NaOH.27 However, our results supported the fact that higher Si concentration near external surface could be easily recrystallized into ZSM-5 shell to cover the formed mesopores. Meanwhile, without enough Si dissolution within crystals was the major reaction to enlarge intracrystalline mesopores. After 16 h, intracrystalline mesopores became even larger which led to the thin shell of ZSM-5. When the shell was too thin to keep its shape, further desilication led to the breakup of ZSM-5 crystals at 24 h and 48 h (Fig. 2d and e).
 | ||
| Fig. 2 SEM images of the parent and TEAOH-treated H-ZSM-5 samples ((a) parent H-ZSM-5, (b) T-4 h, (c) T-16 h, (d) T-24 h, (e) T-48 h). | ||
 | ||
| Fig. 3 TEM images of the parent, TEAOH-treated H-ZSM-5 and TPA-16 h samples ((a) parent H-ZSM-5, (b) T-4 h, (c) T-16 h, (d) T-24 h, (e) T-48 h, (f) TEA-16 h). | ||
27Al MAS NMR was conducted to characterize the chemical environment of Al of zeolites. As shown in Fig. 4a, a single resonance centered at about 55 ppm was clearly observed for all ZSM-5 samples, which was assigned to four-coordinated framework Al species. No obvious resonance centered at about 0 ppm assigned to six-coordinated extra-framework Al species was observed, which could prove the reservation of framework Al by TEAOH during desilication. Similar results could also be found in the results of 29Si MAS NMR in Fig. 4b. All the 29Si resonances had similar peaks, which could be resolved by deconvolution into three peaks centered at −109, −112, and −116 ppm, respectively. Accordingly, the peaks around −109 ppm corresponded to Si (3Si, 1Al) sites, while the resonances around −112 and −116 ppm were assigned to the Si (4Si, 0Al) sites. After treatment by TEAOH, the resonance of Si (4Si, 0Al) sites was slightly decreased indicating the Si atoms in Si rich surroundings were easy to be selectively dissolved to form mesopores, which moved the resonances at lower field (far from Al) to higher field. With the loss of Si (4Si, 0Al), Si (3Si, 1Al) became slightly increased.
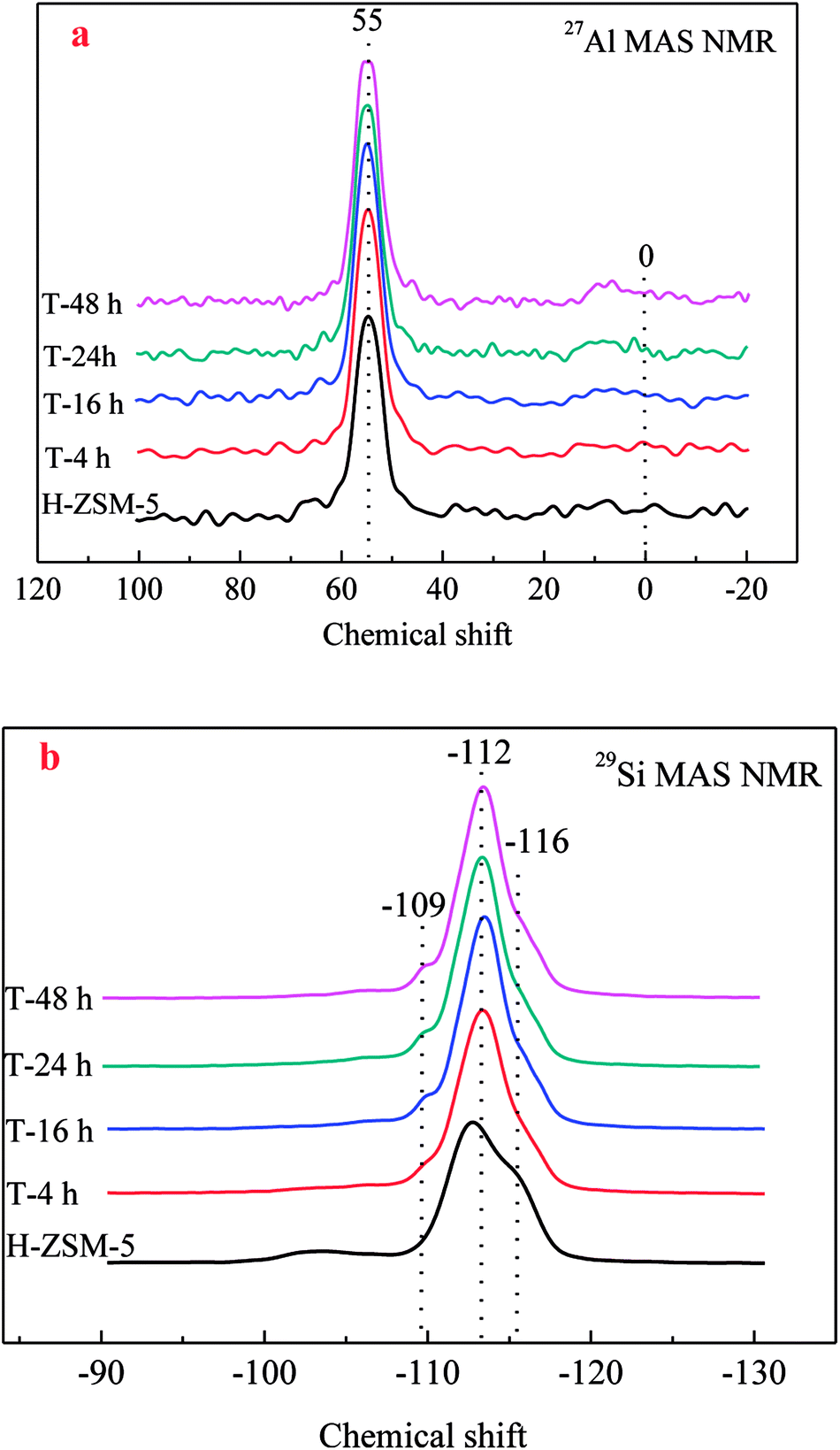 | ||
| Fig. 4 27Al/29Si MAS NMR spectra of the parent and TEAOH-treated H-ZSM-5 samples ((a) 27Al MAS NMR; (b) 29Si MAS NMR). | ||
3.2 Mechanism on the formation of intracrystalline mesopores
Based upon above results of characterization, the mechanism of intracrystalline mesopores in presence of TEAOH was illustrated (Fig. 5). There were two different reactions proceeded when parent H-ZSM-5 was treated by TEAOH solution. The dissolution of framework elements and recrystallization proceeded simultaneously, which showed reverse effects on parent H-ZSM-5 zeolite. Si species was selectively extracted from frameworks in Si rich area by OH− of TEAOH. In general, the dissolution rate and depth mainly depended on the sizes, alkalinity and type of base. As mentioned in literature,21 NaOH was most usually used to generate mesopores of ZSM-5, however easy diffusion of OH− all over the parent zeolites made it difficult to achieve selective desilication of parent zeolites, especially for highly siliceous zeolites. Even worse, framework Al could be transformed to extra-framework Al because of severe desilication. Si species were selectively dissolved with better preservation of crystallinity for effective generation of mesopores within parent H-ZSM-5. TEAOH has lower alkalinity and larger sizes than Na+, therefore it has poorer dissolution capability. However, it might have better capability to form larger mesopores because of bulkier sizes of TEAOH molecules. TPAOH has similar structure to TEAOH, however its strong crystallization capability toward ZSM-5 could lead to lower efficiency during the formation of mesopores within parent H-ZSM-5. For above considerations, TEAOH was selected to be the post treatment agent to generate mesopores within parent H-ZSM-5.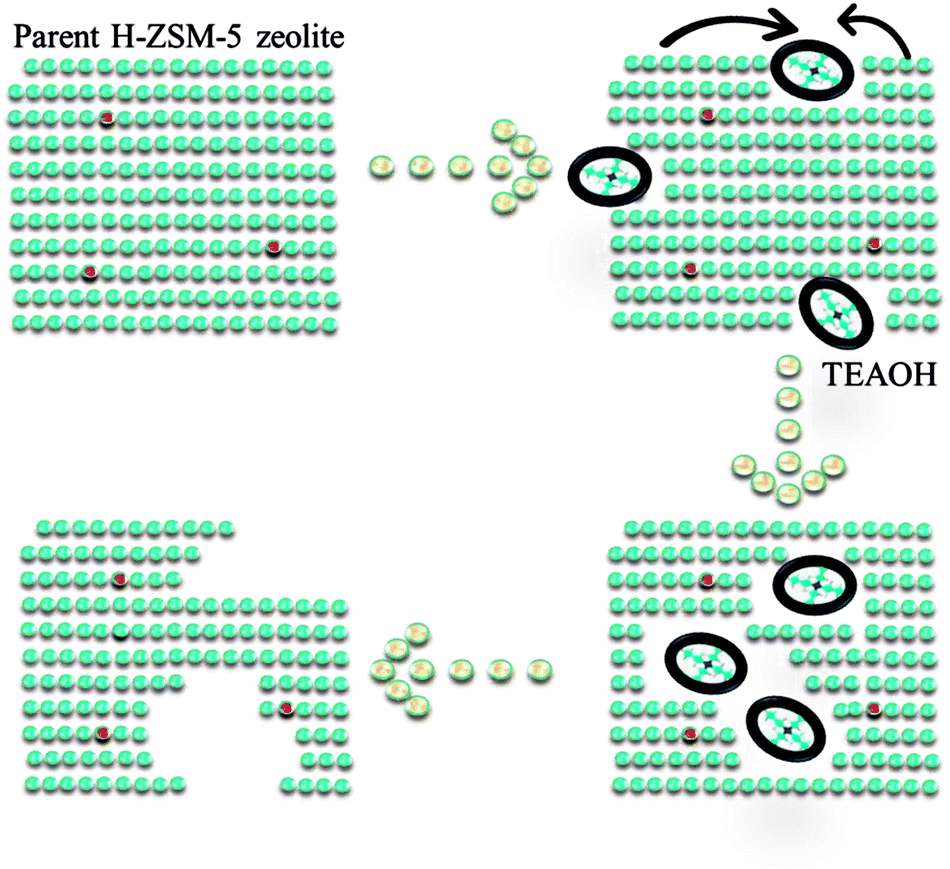 | ||
| Fig. 5 Mechanism diagram on the formation of intracrystalline mesopores. | ||
TEAOH which possesses suitable alkalinity and crystallization capability could make the formation of mesopores more effective. At first, OH− simultaneously dissolved Si to destroy micropores via alkalinity, which led to the decrease of micropore surface area and slight loss of crystallinity (4–16 h). The dissolution of Si led to slight decrease in bulk and superficial SiO2/Al2O3 ratio at 4 h and 16 h. It is well known that sizes of TEAOH were much larger than channels of ZSM-5. Desilication by OH− means the damage on micropores from external surface to inner surface due to diffusion limitation of TEA+. The pore mouth was enlarged by selective dissolution of Si, with enough Si species available on the external surface, ZSM-5 could be recrystallized to form a shell, which is the reason for the formation of intracrystalline mesopores. As a result, mesopores were formed within ZSM-5 crystals, in other words the microporous ZSM-5 shell was preserved at suitable treatment time (Fig. 3b). With prolonged treating time, mesopores became larger and larger to form macropores either via simple desilication or merge of mesopores until the ZSM-5 shell broke up. From TEM and SEM images, broken particles via severe desilication could be found obviously. The framework near Al was reserved as shown in Fig. 4a as a result of protection effect of TEA+ on Al or faster recrystallization rate based on higher Si concentration in mother liquor. A. Galarneau found similar results that the stable mesoporous SBA-15 zeolite could be formed under more severe hydrothermal treatment, or by dissolution/redeposition processes during water treatment.28–30
In all, the relative rate of dissolution and recrystallization during TEAOH-treatment on H-ZSM-5 (SiO2/Al2O3 = 200) was mainly determined by TEAOH and concentration of active Si species. Active Si species in mother liquor could be recrystallized to form ZSM-5 shell to cover generated mesopores and finally balanced between formation of mesopores and growth of ZSM-5 crystal. The formed mesopores might benefit the accessibility of acid sites and diffusion of products off the ZSM-5 channels. The diffusivity was improved because the TEAOH treated H-ZSM-5 at suitable time could be regarded as much shorter diffusion channels connected by intracrystalline mesopores.
3.3 Acidity
NH3-TPD measurements were carried out to investigate the acidity of the parent and TEAOH-treated H-ZSM-5 samples. As shown in Fig. S2,† each of the profile was characterized by two desorption peaks in the NH3-TPD profiles. The low temperature desorption peak (200–240 °C) corresponded to weak acid sites, while the high temperature peak (330–400 °C) to strong acid sites. The weak and strong acid amounts of the parent and TEAOH-treated H-ZSM-5 samples were listed in Table 3. Compared with the parent ZSM-5 of 0.198 mmol g−1, the total acidity of T-4 h and T-16 h decreased to 0.200 mmol g−1 while T-24 h and T-48 h increased to 0.220 and 0.205 mmol g−1, respectively. Such enhancement in strong acid sites is accordingly attributed to the increased Al concentration after desilication (Table 1). Moreover, with longer treating time, the desorption peak of strong acid sites also shifted to higher temperature. As shown in Table 3, T-16 h has fewer strong acid sites than parent and TEAOH-treated H-ZSM-5 samples which was expected to improve the catalytic performance in the MTP reaction.| Samples | Acidity/(mmol g−1) | ||
|---|---|---|---|
| Total acidity | Strong acidity | Weak acidity | |
| H-ZSM-5 | 0.198 | 0.021 | 0.183 |
| T-4 h | 0.200 | 0.026 | 0.174 |
| T-16 h | 0.200 | 0.030 | 0.170 |
| T-24 h | 0.220 | 0.022 | 0.198 |
| T-48 h | 0.205 | 0.018 | 0.187 |
3.4 Catalytic performance of MTP reaction
Methanol conversion versus time on stream (TOS) over H-ZSM-5 zeolites was shown in Fig. 6. The lifetime for methanol conversion higher than 90% varied among the samples. All catalysts showed high initial activity with complete conversion of methanol. For parent H-ZSM-5, methanol was completely converted within 20 h, and then quick deactivation occurred. The activity fell below 90% after 25 h. In contrast, TEAOH treatment obviously promoted the catalytic longevity. The complete conversion of methanol over TEAOH-treated H-ZSM-5 lasted for 30 h (T-4 h), 48 h (T-16 h), 36 h (T-24 h), and 37 h (T-48 h), while the longevity increased to 106 h (T-4 h), 140 h (T-16 h), 120 h (T-24 h), and 96 h (T-48 h), respectively. With suitable treatment time, T-16 h sample had the longest lifetime. The conversion remained above 99% for 79 h, and the activity fell below 90% after 140 h on stream. The whole lifetime was 5.6 times longer than parent H-ZSM-5. However, the catalyst lifetime of T-24 h and T-48 h was slightly shorter than that of T-16 h, which were 120 h and 96 h, respectively. TPA-16 h sample possessed the same trend of TOS as the TEAOH-treated samples (Fig. S3†). However, the complete conversion of methanol over TPA-16 h sample lasted for 31 h, which was shorter than T-16 h sample of 48 h. Moreover, the catalytic longevity of TPA-16 h sample was 100 h compared with T-16 h sample of 140 h. The excessive protective effect of TPA+ resulted in formation of mesopores, however better recrystallization capability limited the increase in mesopores. Table 4 compares methanol treatment capacity and average propylene selectivity under similar conditions in the literature.31–35 Under the condition of 480 °C, 4.5 h−1, the methanol treatment capacity of T-16 h sample was 648 g methanol per g catalyst which was superior among results reported in the MTP reaction.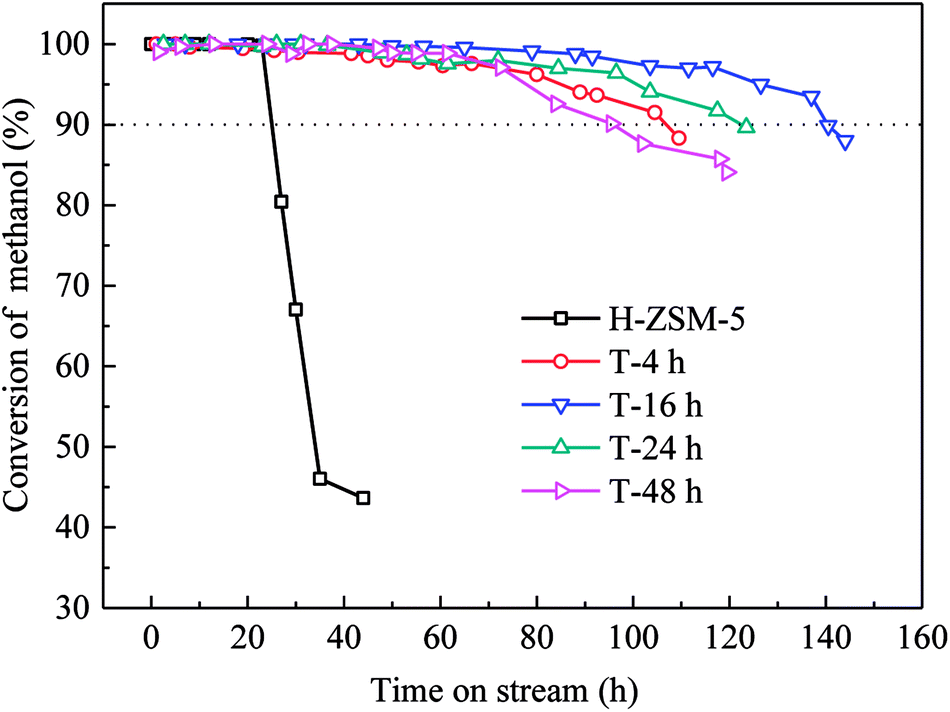 | ||
| Fig. 6 Variation of methanol conversion with the time-on-stream (TOS) over the parent and TEAOH-treated H-ZSM-5 samples. (Reaction conditions: T = 480 °C, WHSV = 4.5 h−1, P = 0.1 MP, m(H2O):m(CH3OH) = 1:1). | ||
Different deactivation rates were observed over parent and TEAOH-treated samples. The instantaneous deactivation rate of methanol conversion at 95% over ZSM-5 catalysts was 4.69% h−1 (parent H-ZSM-5), 0.21% h−1 (T-4 h), 0.18% h−1 (T-16 h), 0.22% h−1 (T-24 h), and 0.28% h−1 (T-48 h), respectively. Nevertheless, all the TEAOH-treated H-ZSM-5 samples showed much slower deactivation rates than parent H-ZSM-5. Obviously, post treatment of H-ZSM-5 by TEAOH was effective to decrease deactivation rates during MTP reaction. More importantly, this method worked in a rather wide range. Even short treatment time could significantly improve the diffusion and catalytic performance of H-ZSM-5 catalysts.
According to results of characterization and proposed mechanism of intracrystalline mesopores formation, mesopores formed within ZSM-5 crystals significantly decreased diffusion length of ZSM-5 channels. The overall TEAOH-treated H-ZSM-5 could be described as much shorter micro channels of ZSM-5 connected by intracrystalline mesopores. With prolonged treating time, the intracrystalline mesopores became larger either via simple desilication or emerge of adjacent mesopores, which led to the further decrease in diffusion length within micropores but increase in mesopore volumes. With the longer time (24 h and 48 h), the broken ZSM-5 crystals led to disappearance of intracrystalline mesopores. With the recovery of relative crystallinity, the micropore channels could be recovered to increase diffusion length. In general, the improvement in lifetime was achieved by significantly decrease length of micropore channels, which were connected by formed intracrystalline mesopores.
Similar to lifetime and deactivation rates of ZSM-5 catalysts, intracrystalline mesopores showed important effects on the coke deposits of deactivated catalysts. In this section, the deactivated samples (parent H-ZSM-5 and T-16 h) were characterized to verify the reason for lifetime improvement. As shown in Fig. 7, the weight loss between 300 °C and 700 °C was due to coke burn-off, which could be regarded as total coke content. The deactivated parent H-ZSM-5 has a coke content of around 3.59%, whereas deactivated T-16 h sample showed a higher coke content of 5.27%. The coke content of the deactivated T-16 h sample was obviously higher than the deactivated parent ZSM-5, however the rate of deactivation of T-16 h sample (0.18% h−1) was much slower than parent H-ZSM-5 (4.69% h−1). The higher content was due to better capacity for coke deposits in mesopores, the mesopore volumes of which was 0.185 cm3 g−1, much larger than 0.104 cm3 g−1 of parent H-ZSM-5. Moreover, the better capacity for coke deposits of T-16 h sample might explain the longer lifetime of T-16 h sample. It is also reported previously36,37 that the coke content of mesoporous zeolite was higher than parent zeolite, because of mesopore formation for more coke deposit holding. Generally, higher coke content means serious deactivation, however, T-16 h sample showed much slower deactivation rate than parent H-ZSM-5 because of much shorter diffusion length due to intracrystalline mesopores. Not only T-16 h sample showed a higher coke content than parent H-ZSM-5 zeolite, but also the soluble carbonaceous residuals were different. Fig. 8 shows the GC-MS for the retained species of deactivated parent H-ZSM-5 and T-16 h samples. Aromatic molecules such as xylenes, trimethylbenzenes and tetramethyl benzenes were detected within these two samples. For the deactivated parent H-ZSM-5, the content of xylenes, trimethylbenzenes and tetramethyl benzenes was 43.9%, 26.3%, and 7.0%, respectively. By contrast, the situation was quite different for deactivated T-16 h sample with intracrystalline mesopores. The content of xylenes of the deactivated T-16 h decreased to 31.4%, while tetramethyl benzenes rose to 11.3%. Moreover, there existed a small peak of pentamethyl benzenes reaching to 1.0%, which supported the formation of heavier compounds in deactivated T-16 h sample. The T-16 h sample had larger space, which had the chance to accommodate polymethyl benzenes. Interestingly, K. Góra-Marek et al.38 also found that the desilication procedure could lead to catalyst modifications which allow larger molecules to be formed within the zeolite crystals. The presence of larger aromatic molecules over desilicated sample was straightforwardly associated with improved lifetime because of larger mesopores. With formation of intracrystalline mesopores, also much shorter diffusion length contributed to slower deactivation rate.
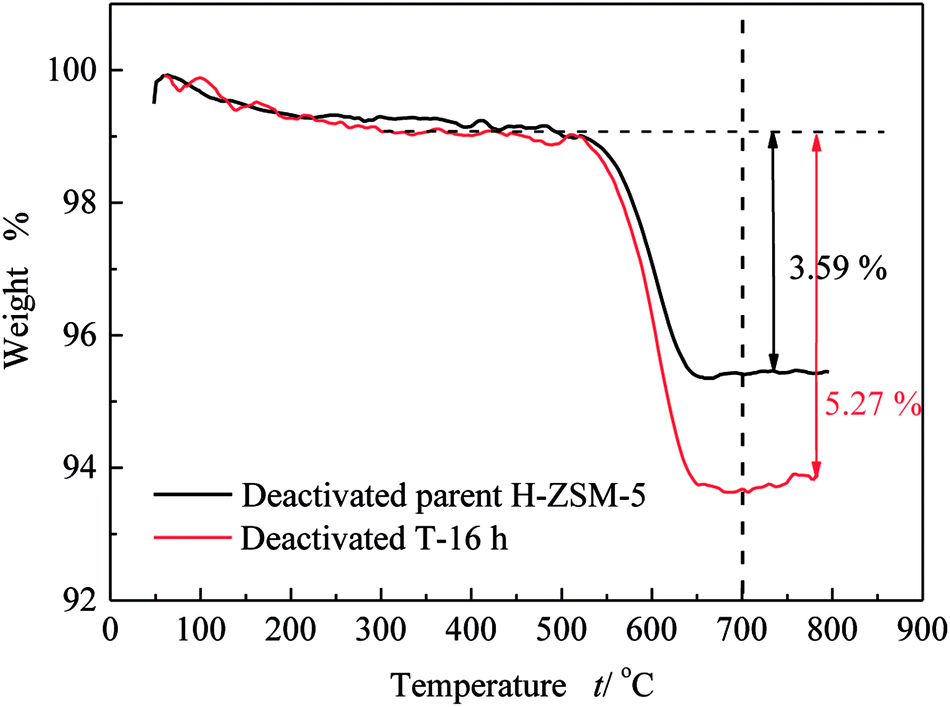 | ||
| Fig. 7 TG results of deactivated parent H-ZSM-5 and T-16 h samples. | ||
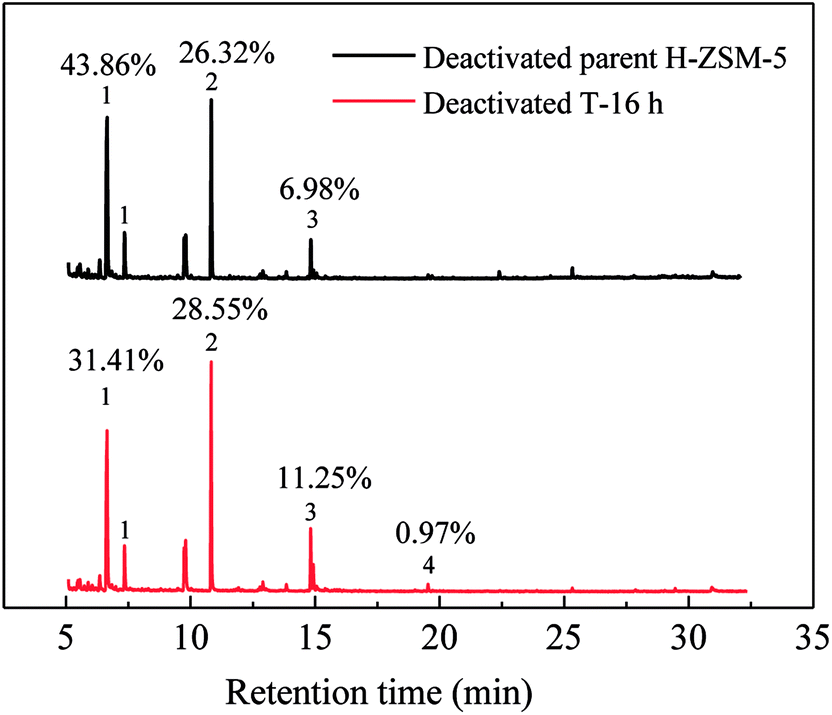 | ||
| Fig. 8 GC-MS analyses of retained hydrocarbons of deactivated parent H-ZSM-5 and T-16 h samples. Chromatograms from the dissolution–extraction experiments for deactivated samples. (1) Xylenes, (2) trimethylbenzenes, (3) tetramethyl benzenes, (4) pentamethyl benzenes. | ||
The propylene selectivity, P/E ratio (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 3/C2, Spropylene/Sethylene) and C3 hydrogen transfer index (C3-HTI, Spropane/Spropylene) were important factors to evaluate the performance of catalyst. Propylene selectivity, C3/C2 and C3-HTI of the parent and TEAOH-treated H-ZSM-5 samples versus TOS are shown in Fig. 9. As seen from Fig. 9a, when methanol was completely converted from initial time to 24 h, the propylene selectivity of parent H-ZSM-5 sample varied between 32.6% and 34.5%, and then decreased sharply along TOS because of fast deactivation. Ethylene selectivity decreased from 11.2% to 9.6%, which resulted in higher P/E ratio from 2.9 to 3.6 (Fig. 9b and e). Parent H-ZSM-5 showed the highest methane selectivity as it grew from 0.9% to 2.9% during the whole lifetime (Fig. 9d). C3 hydrogen transfer index (C3-HTI) is a convenient assessment of the hydrogen transfer activity of a catalyst, and often used to evaluate the extent of secondary reaction in MTH reaction. Obviously, the parent H-ZSM-5 zeolite had the highest initial C3-HTI of 0.047 (Fig. 9f), which indicated secondary reaction of propylene was extremely high. By contrast, the TEAOH-treated H-ZSM-5 samples with mesoporosity were prominent for their higher propylene selectivity and lower methane selectivity. When TOS increased, the TEAOH-treated samples showed little change in propylene selectivity, decreasing ethylene selectivity, and increasing methane selectivity. Propylene selectivity of T-4 h varied between 34.5% and 37.8%. Ethylene selectivity decreased from 7.7% to 5.7%, meanwhile methane selectivity increased from 0.1% to 0.5%. The trends of propylene and ethylene selectivity resulted in the higher P/E ratio from 4.8 to 6.1 (Fig. 9e). T-16 h, T-24 h and T-48 h samples had higher propylene and much lower methane selectivity (below 0.3%) than T-4 h sample. Moreover, the T-16 h sample with the largest mesopores volume also showed lowest methane selectivity (below 0.3%) (Fig. 9d), and the lowest initial C3-HTI of 0.027 (Fig. 9f) presumably due to the shortened diffusion lengths. By contrast, the TPA-16 h sample (Fig. S4†) with rich but smaller mesopores volume showed propylene selectivity decreased slightly from 39.5% to 35.6%, meanwhile ethylene selectivity decreased from 9.5% to 5.3%. The trends of propylene and ethylene selectivity resulted in P/E ratio changed from 4.1 to 6.8. Moreover, TPA-16 h sample also had higher methane selectivity (from 0.3% to 0.8%) and initial C3-HTI of 0.034 due to the different diffusion performance compared with T-16 h sample.
3/C2, Spropylene/Sethylene) and C3 hydrogen transfer index (C3-HTI, Spropane/Spropylene) were important factors to evaluate the performance of catalyst. Propylene selectivity, C3/C2 and C3-HTI of the parent and TEAOH-treated H-ZSM-5 samples versus TOS are shown in Fig. 9. As seen from Fig. 9a, when methanol was completely converted from initial time to 24 h, the propylene selectivity of parent H-ZSM-5 sample varied between 32.6% and 34.5%, and then decreased sharply along TOS because of fast deactivation. Ethylene selectivity decreased from 11.2% to 9.6%, which resulted in higher P/E ratio from 2.9 to 3.6 (Fig. 9b and e). Parent H-ZSM-5 showed the highest methane selectivity as it grew from 0.9% to 2.9% during the whole lifetime (Fig. 9d). C3 hydrogen transfer index (C3-HTI) is a convenient assessment of the hydrogen transfer activity of a catalyst, and often used to evaluate the extent of secondary reaction in MTH reaction. Obviously, the parent H-ZSM-5 zeolite had the highest initial C3-HTI of 0.047 (Fig. 9f), which indicated secondary reaction of propylene was extremely high. By contrast, the TEAOH-treated H-ZSM-5 samples with mesoporosity were prominent for their higher propylene selectivity and lower methane selectivity. When TOS increased, the TEAOH-treated samples showed little change in propylene selectivity, decreasing ethylene selectivity, and increasing methane selectivity. Propylene selectivity of T-4 h varied between 34.5% and 37.8%. Ethylene selectivity decreased from 7.7% to 5.7%, meanwhile methane selectivity increased from 0.1% to 0.5%. The trends of propylene and ethylene selectivity resulted in the higher P/E ratio from 4.8 to 6.1 (Fig. 9e). T-16 h, T-24 h and T-48 h samples had higher propylene and much lower methane selectivity (below 0.3%) than T-4 h sample. Moreover, the T-16 h sample with the largest mesopores volume also showed lowest methane selectivity (below 0.3%) (Fig. 9d), and the lowest initial C3-HTI of 0.027 (Fig. 9f) presumably due to the shortened diffusion lengths. By contrast, the TPA-16 h sample (Fig. S4†) with rich but smaller mesopores volume showed propylene selectivity decreased slightly from 39.5% to 35.6%, meanwhile ethylene selectivity decreased from 9.5% to 5.3%. The trends of propylene and ethylene selectivity resulted in P/E ratio changed from 4.1 to 6.8. Moreover, TPA-16 h sample also had higher methane selectivity (from 0.3% to 0.8%) and initial C3-HTI of 0.034 due to the different diffusion performance compared with T-16 h sample.
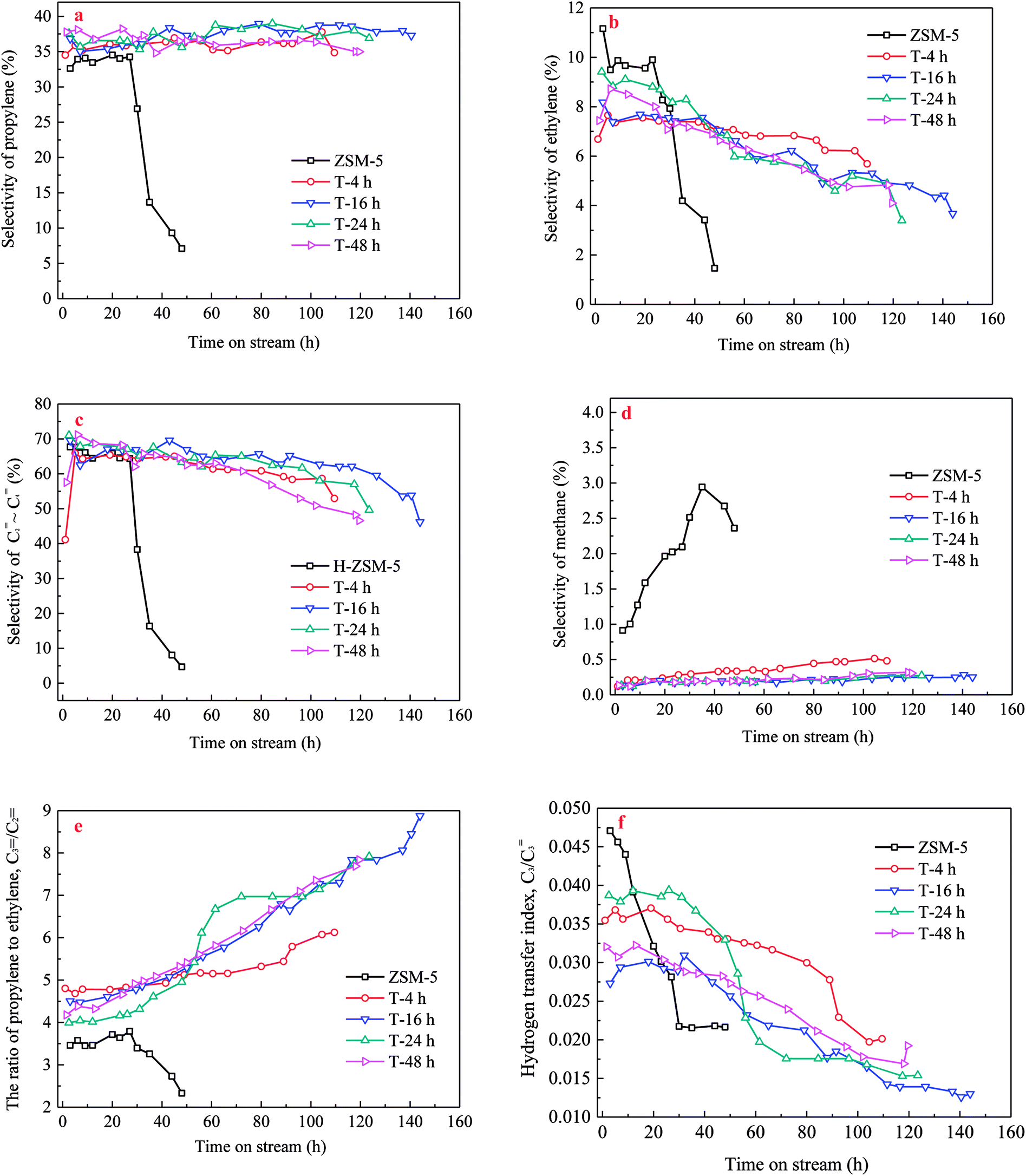 | ||
| Fig. 9 Product selectivity ((a) propylene, (b) ethylene, (c) C2 ∼ C4, (d) methane), C3/C2 (e) and C3 hydrogen transfer index (f) of the parent and TEAOH-treated H-ZSM-5 samples for MTP reaction as a function of time. Reaction conditions: T = 480 °C, WHSV = 4.5 h−1, P = 0.1 MP, m(H2O):m(CH3OH) = 1:1. | ||
Recently, “dual cycle” mechanism proposed in pioneering work provided a good explanation to the formation of ethylene and propylene, which showed two reaction cycles running synchronously in the methanol-to-hydrocarbon process: an aromatic-based alkylation–dealkylation cycle and an olefin-based methylation-cracking cycle.39,40 The scheme of dual-cycle concept is summarized in Fig. 10, propylene and ethylene could be generated by the aromatic-based cycle, whereas C2+ olefins mainly propylene was produced by the olefin-based cycle without formation of ethylene. The dual cycle mechanism was rather a closely connected two circles with mutual influence than simple paralleling reactions. The olefins generated in the aromatic-based cycle were active intermediates for olefin-based cycles, while higher alkenes formed in olefins-based cycles could be transformed into aromatics via dimerization, cyclization and dehydrogenation. As for post treatment of H-ZSM-5 by TEAOH, the main difference lied in the diffusion properties of catalyst. It has been extensively proved that better diffusion properties and shorter contact time could restrict alkylation–dealkylation reactions, which led to lower ethylene selectivity.41,42 It is also well documented in the literature that reduction of crystal sizes could shorten the resident time of aromatics which would restrict the aromatic-based alkylation–dealkylation cycle, thus led to much lower ethylene selectivity.43 R. Khare et al. also found that self-pillared ZSM-5 with higher diffusion performance showed lower ethylene selectivity.44 Thus, methods for improving diffusion performance such as reduction of crystal sizes and creation of mesopores were considered effective to enhance mass transfer and to improve catalytic performance. Introduction of intracrystalline mesopores into H-ZSM-5 remarkably decreased the length of channels with more opening mouth, which could significantly promote the diffusion of reactant/product into/off to acid sites for longer lifetime and better propylene selectivity. More importantly, the micro pores could be gradually blocked because of coke deposition. Olefins and aromatics were active intermediates for the olefin-based and the aromatic-based cycles respectively, which differed in concentrations, sizes, reaction activity and so on. In general, more olefins with much higher activity than aromatics were generated during MTP reaction, more importantly as active intermediates olefins in the olefin-based cycle needed much smaller space for the methylation-cracking route than aromatic in the aromatic-based cycle for the alkylation–dealkylation. The free space size within zeolite channels gradually decreased along TOS, which had fewer restricting effects on olefin-based cycle. However, space restriction was more influential to the aromatic-based cycle, which led to the gradual decrease in ethylene selectivity. With further blocking of channels of H-ZSM-5, there was no enough free space for the formation of transitional states of methylation of aromatics, which resulted in a decrease in methanol conversion. More importantly, methylation-cracking in the olefin-based cycle could also be decreased with severe blockage of channels.
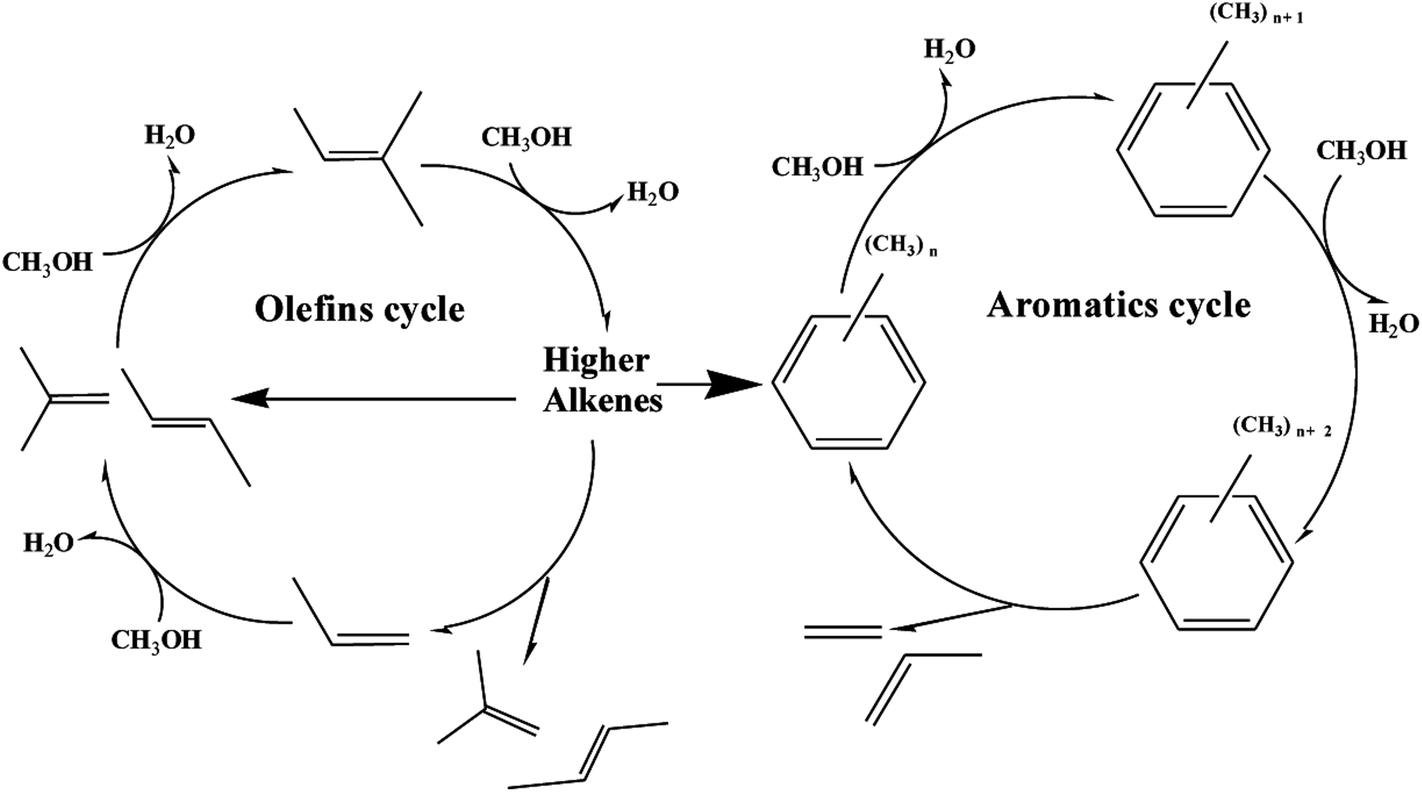 | ||
| Fig. 10 Scheme of dual-cycle concept (olefins cycle and aromatics cycle) during MTP reaction over H-ZSM-5 zeolite. | ||
It was proposed that methane was formed via reduction of a methyl group by hydride transfer39 and decomposition of methanol. On one hand, methanol could dehydrate to form dimethyl ether on acid sites and was further converted to hydrocarbons. On the other hand, methanol decomposition occurred simultaneously. Parent H-ZSM-5 sample had an especially high methane selectivity, which might be related to the poor diffusion. Poor accessibility of acid site means methanol decomposition a certain probability occurs. However, the TEAOH-treated samples showed much lower amount of methane compared to parent H-ZSM-5 sample. This may be ascribed to the hollow structure, introducing a mesoporous structure improved the accessibility of acid site which could suppress the content of methanol decomposition. Thus, methanol decomposition occurred at a much slower rate than methanol conversion to olefins which results to much lower methane selectivity. Moreover, the methane selectivity of parent H-ZSM-5 and TEAOH-treated samples both increased along TOS. This may be ascribed to the decreasing of acidity due to coke deposition.
In all, the diffusion length of H-ZSM-5 could be remarkably decreased by intracrystalline mesopores in zeolite via protective desilication in TEAOH, which could significantly enhance lifetime, product selectivity and coke tolerance with much lower methane selectivity and deactivation rates.
4 Conclusions
In summary, this study presented a protective method to introduce intracrystalline mesopores within highly siliceous H-ZSM-5 by TEAOH treatment, during which there were two different reactions: dissolution of Si by OH− and recrystallization of dissolved Si into ZSM-5 by TEA+ in presence of seeds. At first, the external crystal Si was selectively dissolved to form mesopores at cost of micropores, and then active Si species in mother liquor could be recrystallized to form ZSM-5 shell to cover generated intracrystalline mesopores. With prolonged treating time, intracrystalline mesopores became larger and larger either via simple desilication or merge of adjacent mesopores. With intracrystalline mesopores, original channels of parent H-ZSM-5 was cut into much shorter ones by formed mesopores. The formed mesopores within ZSM-5 was broken from inner via further desilication to restore relative crystallinity and micropore surface.Because of much shorter diffusion length, the TEAOH-treated H-ZSM-5 samples were demonstrated to be an excellent MTP catalyst with longer lifetime and higher propylene selectivity. A good correlation between the mesoporosity and the catalytic longevity was found. Lifetime and high propylene selectivity have a closely relationship with pore structure. T-16 h sample, with the largest mesopore volumes, exhibited the longest lifetime (140 h) and higher propylene selectivity (36.6%) relative to the parent H-ZSM-5 sample.
This method proved a simple method for intracrystalline mesopore formation with better balance between dissolution (OH−) and recrystallization (TEA+) in TEAOH only for better design on MTP catalyst with high stability and propylene selectivity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Careful characterization of samples from the Department of Analysis in Research Institute of Petroleum Processing, Sinopec was greatly acknowledged.Notes and references
- M. Firoozi, M. Baghalha and M. Asadi, Catal. Commun., 2009, 10, 1582–1585 CrossRef CAS.
- A. Muntasar, R. L. V. Mao and H. Yan, Ind. Eng. Chem. Res., 2010, 49, 3611–3616 CrossRef CAS.
- C. Sun, J. M. Du, J. Liu, Y. Yang, N. Ren, W. Shen, H. L. Xu and Y. Tang, Chem. Commun., 2010, 46, 2671–2673 RSC.
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- S. Hu, J. Shan, Q. Zhang, Y. Wang, Y. Liu, Y. Gong and T. Dou, Appl. Catal., A, 2012, 445, 215–220 CrossRef.
- B. Jiang, F. Xiang, L. Yan, Y. Jiang, Z. Liao, J. Wang and Y. Yang, Ind. Eng. Chem. Res., 2014, 53, 4623–4632 CrossRef CAS.
- X. Meng, Q. Yu, Y. Gao, Q. Zhang, C. Li and Q. Cui, Catal. Commun., 2015, 61, 67–71 CrossRef CAS.
- J. Wang, Y. Wei, J. Li, S. Xu, W. Zhang, Y. He, J. Chen, M. Zhang, A. Zheng, F. Deng, X. Guo and Z. Liu, Catal. Sci. Technol., 2016, 6, 89–97 RSC.
- Z. Liu, X. Dong, Y. Zhu, A. H. Emwas, D. Zhang, Q. Tian and Y. Han, ACS Catal., 2015, 5, 5837–5845 CrossRef CAS.
- S. Ilias and A. Bhan, ACS Catal., 2012, 3, 18–31 CrossRef.
- G. T. Kokotailo, S. L. Lawton, D. H. Olson and W. M. Meier, Nature, 1978, 272, 437–438 CrossRef CAS.
- H. Koempel and W. Liebner, Stud. Surf. Sci. Catal., 2007, 167, 261–267 CrossRef.
- M. Khanmohammadi, S. Amani, A. B. Garmarudi and A. Niaei, Chin. J. Catal., 2016, 37, 325–339 CrossRef.
- K. Barbera, F. Bonino, S. Bordiga, T. V. W. Janssens and P. Beato, J. Catal., 2011, 280, 196–205 CrossRef CAS.
- S. Al-Khattaf, C. D'Agostino, M. N. Akhtar, N. Al-Yassir, N. Y. Tan and L. F. Gladden, Catal. Sci. Technol., 2014, 4, 1017–1027 RSC.
- C. Dai, A. Zhang, L. Li, K. Hou, F. Ding, J. Li, D. Mu, C. Song, M. Liu and X. Guo, Chem. Mater., 2013, 25, 4197–4205 CrossRef CAS.
- S. Abello, A. Bonilla and J. Perez-Ramirez, Appl. Catal., A, 2009, 364, 191–198 CrossRef CAS.
- D. Verboekend and J. Pérez-Ramírez, Catal. Sci. Technol., 2011, 1, 879–890 RSC.
- J. C. Groen, L. A. A. Peffer, J. A. Moulijn and J. Pérezramírez, Microporous Mesoporous Mater., 2004, 69, 29–34 CrossRef CAS.
- J. Ahmadpour and M. Taghizadeh, C. R. Chim., 2015, 18, 834–847 CrossRef CAS.
- M. Bjørgen, F. Joensen, M. S. Holm, U. Olsbye, K. Lillerud and S. Svelle, Appl. Catal., A, 2008, 345, 43–50 CrossRef.
- H. Liu, S. Xie, W. Xin, S. Liu and L. Xu, Catal. Sci. Technol., 2016, 6, 1328–1342 RSC.
- D. Wang, L. Zhang, L. Chen, H. Wu and P. Wu, J. Mater. Chem. A, 2014, 3, 3511–3521 RSC.
- P. Magnoux, P. Roger, C. Canaff, V. Fouche, N. S. Gnep and M. Guisnet, Stud. Surf. Sci. Catal., 1987, 34, 317–330 CrossRef CAS.
- P. R. H. P Rao and M. Matsukata, Chem. Commun., 1996, 12, 1441–1442 RSC.
- R. V. Ballmoos and W. M. Meier, Nature, 1981, 289, 782–783 CrossRef.
- D. Verboekend, G. Vile and J. Pérez-Ramírez, Cryst. Growth Des., 2012, 12, 3123–3132 CrossRef CAS.
- A. Galarneau, M. Nader, F. Guenneau, F. D. Renzo and A. Gedeon, J. Phys. Chem. C, 2007, 111, 8268–8277 CrossRef CAS.
- A. Galarneau, H. Cambon, F. D. Renzo and F. Fajula, Langmuir, 2001, 17, 8328–8335 CrossRef CAS.
- A. Galarneau, H. Cambon, F. D. Renzo, R. Ryoo, M. Choi and F. Fajula, New J. Chem., 2003, 27, 73–79 RSC.
- M. Rostamizadeh and A. Taeb, Synth. React. Inorg. Met.-Org. Chem., 2016, 46, 665–671 CrossRef CAS.
- H. Chen, Y. Wang, F. Meng, C. Sun, H. Li, Z. Wang, F. Gao, X. Wang and S. Wang, Microporous Mesoporous Mater., 2017, 244, 301–309 CrossRef CAS.
- H. Chen, Y. Wang, C. Sun, F. Gao, L. Sun, C. Wang, Z. Wang and X. Wang, Catal. Commun., 2017, 100, 107–111 CrossRef CAS.
- M. Rahmani and M. Taghizadeh, React. Kinet., Mech. Catal., 2017, 122, 409–432 CrossRef CAS.
- H. Li, Y. Wang, C. Fan, C. Sun, X. Wang, C. Wang, X. Zhang and S. Wang, Appl. Catal., A, 2018, 551, 34–48 CrossRef CAS.
- Z. Wan, G. Li, C. Wang, H. Yang and D. Zhang, Appl. Catal., A, 2017, 549, 141–151 CrossRef.
- F. L. Bleken, K. Barbera, F. Bonino, U. Olsbye, K. P. Lillerud, S. Bordiga, P. Beato, T. V. W. Janssens and S. Svelle, J. Catal., 2013, 307, 62–73 CrossRef CAS.
- K. Sadowska, K. Góra-Marek, M. Drozdek, P. Kuśtrowski, J. Datka, J. M. Triguero and F. Rey, Microporous Mesoporous Mater., 2013, 168, 195–205 CrossRef CAS.
- X. Sun, S. Mueller, Y. Liu, H. Shi, G. L. Haller, M. Sanchez-Sanchez, A. C. V. Veen and J. A. Lercher, J. Catal., 2014, 317, 185–197 CrossRef CAS.
- J. Li, M. Liu, X. Guo, S. Xu, Y. Wei, Z. Liu and C. Song, ACS Appl. Mater. Interfaces, 2017, 9, 26096–26106 CrossRef CAS PubMed.
- R. Khare, D. Millar and A. Bhan, J. Catal., 2015, 321, 23–31 CrossRef CAS.
- M. Zhang, S. Xu, Y. Wei, J. Li, J. Wang, W. Zhang, S. Gao and Z. Liu, Chin. J. Catal., 2016, 37, 1413–1422 CrossRef CAS.
- N. L. Michels, S. Mitchell and J. Perez-Ramirez, ACS Catal., 2014, 4, 2409–2417 CrossRef CAS.
- R. Khare and A. Bhan, J. Catal., 2015, 329, 218–228 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD, NH3-TPD, methanol conversion and products selectivity over TPA-16 h sample in MTP reaction. See DOI: 10.1039/c8ra06786d |
| This journal is © The Royal Society of Chemistry 2018 |