
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: MiR-182-5p and miR-96-5p increased hepatocellular carcinoma cell mobility, proliferation and cisplatin resistance partially by targeting RND3
Shiming Yang,
Zhi Chen,
Daguang Fan,
Rui Zhang,
Yuhong Zhang and
Shusheng Wu *
*
Department of General Surgery, Shanxi Provincial People's Hospital, No. 29, Shuangtaisi Street, Taiyuan, Shanxi 030012, China. E-mail: ysm529@126.com; Tel: +86-0351-4960336
First published on 12th October 2018
Abstract
We investigated whether miR-182-5p or miR-96-5p could increase hepatocellular carcinoma (HCC) development by targeting Rho Family GTPase 3 (RND3) gene expression. The expression levels of miR-182-5p, miR-96-5p and mRNA/protein of RND3 in non-HCC liver tissue, HCC tissue and adjacent tissue specimens were evaluated by RT-qPCR and western blot. Patient-derived HCC cell culture was established, and miR-182-5p or miR-96-5p agomir or antagomir treatment was performed to mimic the overexpression or knockdown of the two miRNAs. HCC cell mobility in vitro was monitored by trans-well migration and invasion assay, while HCC cell growth in vitro was evaluated by cell viability, proliferation and apoptosis assay. HCC cell apoptosis was further investigated by caspase-3/-8/-9 activity assay. MiR-182-5p and miR-96-5p were significantly upregulated in HCC tissue specimens compared with non-HCC or adjacent tissue specimens, inversely correlating to RND3 mRNA expression level. Treatment with miR-182-5p or miR-96-5p agomir significantly reduced RND3 mRNA/protein expression level in HCC cells. MiR-182-5p- or miR-96-5p-targeting RND3 mRNA was verified by luciferase reporter assay and AGO2-RNA immunoprecipitation assay. MiR-182-5p or miR-96-5p agomir treatment significantly rescued HCC cell migration and invasion in vitro that were repressed by RND3 overexpression, during which ROCK1 and ROCK2 inhibition were involved. MiR-182-5p or miR-96-5p agomir treatment also increased HCC cell proliferation and cisplatin resistance in vitro, which could be antagonized by RND3 overexpression or ROCK inhibition. Thus, miR-182-5p and miR-96-5p increased HCC cell mobility, proliferation and cisplatin resistance in vitro partially by targeting RND3.
Introduction
Hepatocellular carcinoma (HCC) ranks as the sixth most prevalent neoplasm and the third leading cause of cancer related mortality.1 The oncogenic or tumor suppressive roles as well as clinical values of microRNAs (miRNA) in the pathogenesis and development of (HCC) have been intensively investigated in the past decades.2,3 MiRNA functions most commonly by binding to its target site on a mRNA complementary pairing to its seed sequence, thus blocking the protein translation process and inducing mRNA degradation mediated by the RNA-induced silencing complex.4 MiR-182 and miR-96 are two “oncomirs” that have been found to facilitate HCC progression by repressing the expression of various target genes, such as Forkhead Box O3 (FOXO3), Forkhead Box O1 (FOXO1) and SRY-Box 6 (SOX6).5–7 Notably, Wang et al. reported that miR-182-5p and miR-96-5p could increase HCC cell proliferation and invasion in vitro partially by co-targeting ephrin A5, while Leung et al. reported that beta-catenin could directly activate the transcription of the miR-183-96-182 cluster.8,9During our preliminary research exploring potential target genes of miR-182-5p and miR-96-5p using TargetScanHuman (Ver 7.2) and Diana microT-CDS database,10,11 we found RND3 as a potential target gene of both miRNAs. RND3 (Rho Family GTPase 3, also named RhoE) is a member of the RND protein subgroup belonging to the Ras homologous (Rho) family of GTPases although the RND proteins exhibit no GTPase activity.12,13 Mechanistically, RND3 was found to competitively bind with Rho Associated Coiled-Coil Containing Protein Kinase 1 (ROCK1), preventing the activation of the latter by Ras Homolog Family Member A (RhoA) although the existence of RhoA-ROCK1-independent activity of RND3 has also been anticipated.13 The clinical significance of RND3 as a tumor suppressor gene in HCC was first reported by Luo et al., who documented a significant association between RND3 downregulation and HCC progression, resulting in patients' shortened survival.14 Downregulation of RND3 gene expression in HCC was later confirmed by Grise et al. and Ma et al.; both of them also reported a significant increase in HCC cell mobility in vitro after RND3 knockdown.15,16 In one of their later reports, Ma et al. further demonstrated that RND3 knockdown significantly increased HCC cell chemoresistance to cisplatin or doxorubicin, which seemed to be diminished by RhoA or Rho Associated Coiled-Coil Containing Protein Kinase 2 (ROCK2) silencing rather than by ROCK1 silencing.17 However, Grise et al. postulated that the increase in HCC cell mobility in vitro due to RND3 knockdown could be largely abrogated by Rac Family Small GTPase 1 (RAC1) or Cell Division Cycle 42 (CDC42) knockdown but not by RhoA knockdown, raising the possibility that RND3 might regulate HCC cell mobility and chemoresistance through a distinct mechanism.16 In addition, in HCC, RND3 has also been proposed as either an oncogene or tumor suppressor gene in other cancer types.13,18
In the present research, we first verified that miR-182-5p and miR-96-5p increased HCC cell mobility, proliferation and cisplatin resistance partially by targeting RND3. Using a selective ROCK1/2 kinase inhibitor, YC-S 7001, as well as specific knockdown of ROCK1 and ROCK2, we found that both ROCK paralogs are involved in the HCC cell mobility-promoting effect of RND3 knockdown.
Materials and methods
Collection of tissue specimens
This research was approved by the ethics review committee of Shanxi Provincial People's Hospital. Written informed consent was obtained from each individual whose biospecimens were involved in this research. Non-malignant liver parenchyma tissue specimens were obtained from 15 non-HCC patients (8 male and 7 female, age of 35.13 ± 5.09) who suffered from severe liver injury (grade IV to V according to Moore et al.'s liver injury scale19) and received minimal hepatectomy for debridement (11 cases) or right hepatic resection (4 cases) in our facility during 2012–2016. HCC and paired adjacent tissue specimens were obtained from 25 primary HCC patients (17 male and 8 male, aged 45.84 ± 10.85, HCC stage A to B according to the Barcelona-Clínic liver cancer staging system20) receiving radical hepatectomy before adjuvant chemotherapy in our facility during 2010–2016. After necrotic tissue was removed and briefly rinsed by cold PBS buffer, all excisions were freshly stored in liquid nitrogen before further analysis.HCC cell culture in vitro
HCC primary culture was established from HCC tissue specimens obtained from 3 randomly picked HCC patients (2 female and 1 male, aged 42.92 ± 8.71, HCC stage A to B according to the Barcelona-Clínic liver cancer staging system20) during 2014–2016 in our facility by fine-needle aspiration for diagnosis purposes. Briefly, left-over cells and tissues in the needle and syringe after cytology analysis were resuspended in cold PBS and ground through a 100 μm cell strainer (Solarbio, Beijing, China), followed by centrifugation and resuspension in a complete culture medium composed of DMEM high glucose medium (Solarbio) supplemented with 10% FBS (GIBCO, Thermo Fisher Scientific, Shanghai, China) and 1% penicillin–streptomycin solution (Solarbio). Cells in the culture medium were transferred to a Petri dish (Corning) and cultured overnight in a humidified cell incubator at 37 °C and 5% CO2 atmosphere. After the unattached cells were removed, the attached cells were allowed to grow until about 80% confluence before subculture. Cells at passage 3 were stored in liquid nitrogen and recovered following universal protocols before further analysis. Cells were assayed at a maximum passage 5.Gene manipulation and cell treatment
RND3 overexpression and knockdown, ROCK1 knockdown and ROCK2 knockdown were achieved by lentiviral transfection. Lentiviral particles for gene manipulation were purchased from Cyagen Biosciences (Suzhou, China) and were applied following manufacturer's instructions. For reducing the miR-182-5p or miR-96-5p expression level, cells at 50% confluence on a 24 well plate were treated with antagomir targeting miR-182-5p or miR-96-5p (GenePharma, Shanghai, China) at 50 nM for 24 hours in the presence of 1% (v/v) Lipofectamine 3000 (Thermo Fisher Scientific). To mimic the overexpression of miR-182-5p or miR-96-5p, cells were treated with agomir under the same conditions as that for antagomir treatment. Non-targeting agomir and antagomir controls (agomir NC and antagomir NC, respectively, GenePharma) were used as negative controls for agomir and antagomir treatment, respectively. To inhibit ROCK1/2 kinase activity, cells were treated with TC-S 7001 (Tocris, Bio-techne, Shanghai, China) at 10 nM for 24 hours. Treatment with cisplatin (Tocris) was performed at 30 μg mL−1 together with other treatments, as indicated earlier.Western blot and RT-qPCR
Cell lysis or homogenization of tissue samples was performed with moderate RIPA lysis buffer (Beyotime, Shanghai, China). After refrigerated centrifugation, supernatants from cell lysate or tissue homogenate samples were subjected to western blot or RT-qPCR analysis. The following primary antibodies used for western blotting were purchased from Abcam (Shanghai, China): RND3 (ab171799), MYPT1 (ab32519), ROCK1 (ab97592), ROCK2 (ab228008), PCNA (ab152112), and caspase-3 (ab90437). The following primary antibodies used for western blotting were purchased from Novus Biologicals (Bio-techne): β-actin (AC-15), MYPT1 pT853 (SA19), and active caspase-3 (269518). The protein expression level of each gene was evaluated by gray scale analysis using ImageJ software, and β-actin was used as the housekeeping gene. For RT-qPCR, the total RNA in supernatants from cell lysate or tissue homogenate samples was extracted using a RNA extraction kit (Genepharma) and reverse transcribed to cDNA using a First-Strand cDNA Synthesis Kit (Genecopoeia) following manufacturer's instructions. The expression levels of RND3, ROCK1 and ROCK2 mRNAs as well as miR-96-5p and miR-182-5p were analyzed by RT-qPCR using qPCR mix and primers purchased from Genecopoeia. The primer sequences were as follows: miR-96-5p Fw: 5′-TTTGGCACTAGCACATTTTTGCT-3′; miR-182-5p Fw: 5′-TTTGGCAATGGTAGAACTCACACT-3′; U6 Fw: 5′-GCTTCGGCAGCACATATACTAAAAT-3′; RND3 Fw: 5′-CCTCTCTTACCCTGATTC-3′; RND3 Rv: 5′-TCTTCGCTTTGTCCTTTC-3′; ROCK1 Fw: 5′-AAACTAGCGGCCGCTAGTTGCATTTTTGCCAAGCCATATT-3′; ROCK1 Rv: 5′-CTAGAATATGGCTTGGCAAAAATGCAACTAGCGGCCGCTAGTTT-3′; ROCK2 Fw: 5′-TCAGAGGTCTACAGATGAAGGC-3′; ROCK2 Rv: 5′-CCAGGGGCTATTGGCAAAGG-3′; GAPDH Fw: 5′-ATGGGGAAGGTGAAGGTCG-3′; GAPDH Rv: 5′-ATGGGGAAGGTGAAGGTCG-3′. Semi-quantification was performed by the 2−ΔΔCt method using GAPDH as the reference gene for mRNAs and U6 as that for miRNAs.Luciferase reporter assay
Luciferase reporter plasmids were purchased from Genecopoeia (Fulengen, Guangzhou, China) and were used following manufacturer's instructions. The plasmid-encoded SEAP gene was used as the internal reference and Gaussian Luciferase was used as the reporter gene, which was flanked at 3′ by the cDNA of full length RND3 mRNA 3′ untranslated region (UTR). Cells were transfected with reporter plasmids using Endofectin max transfection reagent (Genecopoeia) following manufacturer's instructions.AGO2-RNA immunoprecipitation
After treatment with agomir, HCC cells were lysed with commercially purchased cell lysis buffer for western and IP (Beyotime, Shanghai) on ice with harsh pipetting. After centrifugation to remove cell debris, the cell lysate samples were incubated with mouse monoclonal anti human Argonaute-2 antibody (Abcam, Shanghai, China) at 15 μg mL−1 for 2 hours with gentle agitation, followed by incubation with protein A/G magnetic beads (Pierce, Thermo Fisher Scientific). After brief washing with cold cell lysis buffer, the beads were isolated from the cell lysate by a magnetic field, and RND3 mRNA co-immunoprecipitated with the beads were analyzed by RT-qPCR.Trans-well migration and invasion assay
Trans-well migration and invasion assays were performed as described by Justus et al. with minor modifications.21 For the invasion assay, matrigel pre-coated cell culture inserts for a 24 well plate (Corning) were used, and for the migration assay, the non-coated inserts were used. Cells were pre-incubated with agomir for 24 hours before the trans-well assay. For the trans-well assay, about 105 cells were seeded in the inserts in the presence of agomir in a serum-free condition, and the inserts were inserted in wells filled with complete cell culture medium with 5% FBS as chemoattractant. After incubation for 24 hours, cells on the top of the insert membrane were removed with a cotton swab and cells on the bottom of the insert membrane were fixed with ethanol and stained with crystal violet before photography.Cell viability, proliferation, apoptosis and caspase-3/-8/-9 activity assay
For cell viability and proliferation assay, about 2000 cells per well were incubated on a 96 well plate with the indicated treatment before assay. For cell apoptosis and caspase-3/-8/-9 activity assay, the initial cell density was 5000 cells per well. Cell viability was assayed using a CCK-8 cell counting kit (Beyotime) following manufacturer's instructions. Cell proliferation was assayed using a Click-iT EdU Microplate Assay kit (Thermo Fisher Scientific) following manufacturer's instructions. Cell apoptosis was assayed using a TiterTACS In Situ Detection Kit (R&D Systems, Bio-techne) following manufacturer's instructions. Caspase-3/-8/-9 activity was assayed using Caspase Multiplex Activity Assay Kit (Abcam) following manufacturer's instructions.Statistical analysis
Data organization and analysis was performed using Graphpad Prism software (Ver. 7). All experiments were performed in three independent biological replicates multiplied by two technical replicates. All data were normalized to the mean value of the control group in the same experimental settings and were presented as fold change ± standard deviance when applicable. For significance tests, Student's t test was used for comparison between two groups, and Tukey's test was used for multiple comparisons. p < 0.05 was considered significant.Results and discussion
miR-182-5p and miR-96-5p reduced RND3 expression in HCC cells in vitro via direct targeting
To investigate the relationship in gene expression level between RND3 and miR-182-5p/miR-96-5p, we analyzed the mRNA and protein expression level of RND3 as well as the expression level of two miRNA in 15 non-malignant liver parenchyma specimens and 25 pairs of HCC-adjacent tissue specimens. RT-qPCR and western blot results showed that RND3 mRNA and protein expression levels in these HCC tissue specimens (HCC) were about 50% compared with those in adjacent counterparts (adjacent) or in non-malignant liver parenchyma (NC). However, the expression of miR-182-5p and miR-96-5p increased by over 1.5-fold in HCC tissue specimens compared with that in adjacent counterparts or in non-malignant liver parenchyma (Fig. 1A–D). Non-malignant liver parenchyma specimens obtained from patients that suffered from mechanical liver injury and adjacent tissue specimens obtained from HCC patients showed no statistically significant difference in the expression level of RND3 or the two miRNAs. We also found a significant inverse-correlation between RND3 mRNA expression level and the expression level of the two miRNAs (Fig. 1E and F).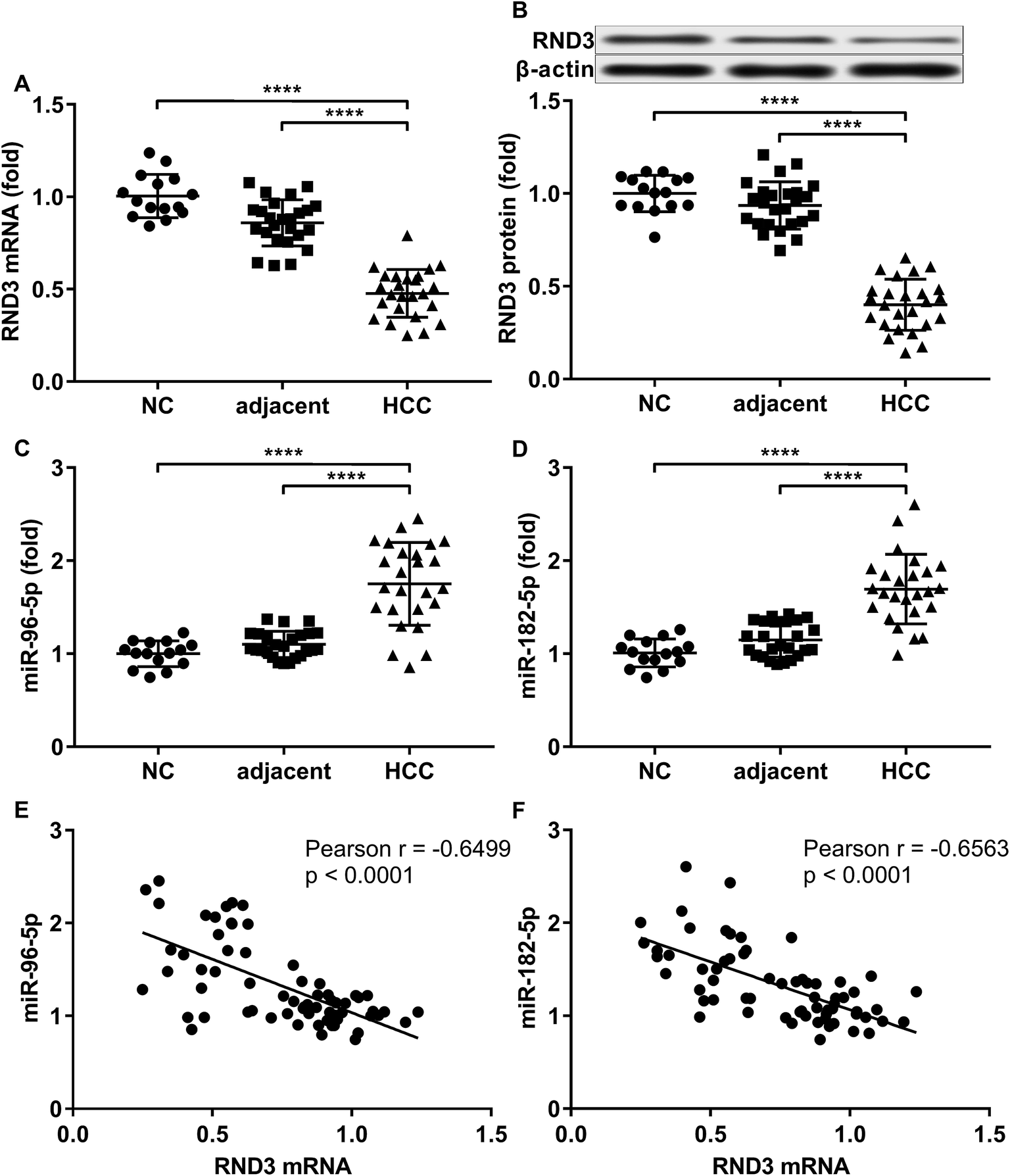 | ||
| Fig. 1 Inverse correlation between RND3 gene expression and the expression level of miR-182-5p or miR-96-5p in liver tissue specimens. (A–D): RT-qPCR and western blotting for evaluating the mRNA/protein expression level of RND3 as well as the expression level of two indicated miRNAs in non-HCC tissue specimens (NC), HCC tissue specimens (HCC) and paired adjacent counterparts (adjacent). (E and F): Pearson correlation analysis evaluating the correlation between the expression level of RND3 mRNA and that of the two indicated miRNAs. ***, p < 0.001; ****, p < 0.0001. | ||
We employed agomir or antagomir treatment to mimic the overexpression or knockdown of the two miRNAs, respectively. As revealed by RT-qPCR and western blot analysis, in patient-derived HCC cells, miR-182-5p or miR-96-5p agomir treatment significantly decreased RND3 mRNA and protein expression level by over 50% compared with the untreated counterparts (NC), which could be significantly increased by treatment with antagomir targeting the two miRNAs (Fig. 2A–E). We next used the luciferase reporter assay and AGO2-RIP assay to validate the binding between RND3 mRNA and the two miRNAs. MiR-182-5p or miR-96-5p agomir treatment significantly reduced the activity of Gaussian luciferase (GLuc) whose gene on the reporter plasmid was flanked at 3′ by the cDNA of RND3 mRNA 3′ untranslated region (Fig. 2F). RT-qPCR analysis revealed that after miR-182-5p or miR-96-5p agomir treatment, the abundance of RND3 mRNA co-immunoprecipitated with AGO2 protein significantly increased compared with that of untranslated counterparts (Fig. 2G). These data suggested that miR-182-5p or miR-96-5p could reduce RND3 mRNA and protein expression in HCC cells in vitro by binding to the 3′ UTR of RND3 mRNA, leading to its degradation.
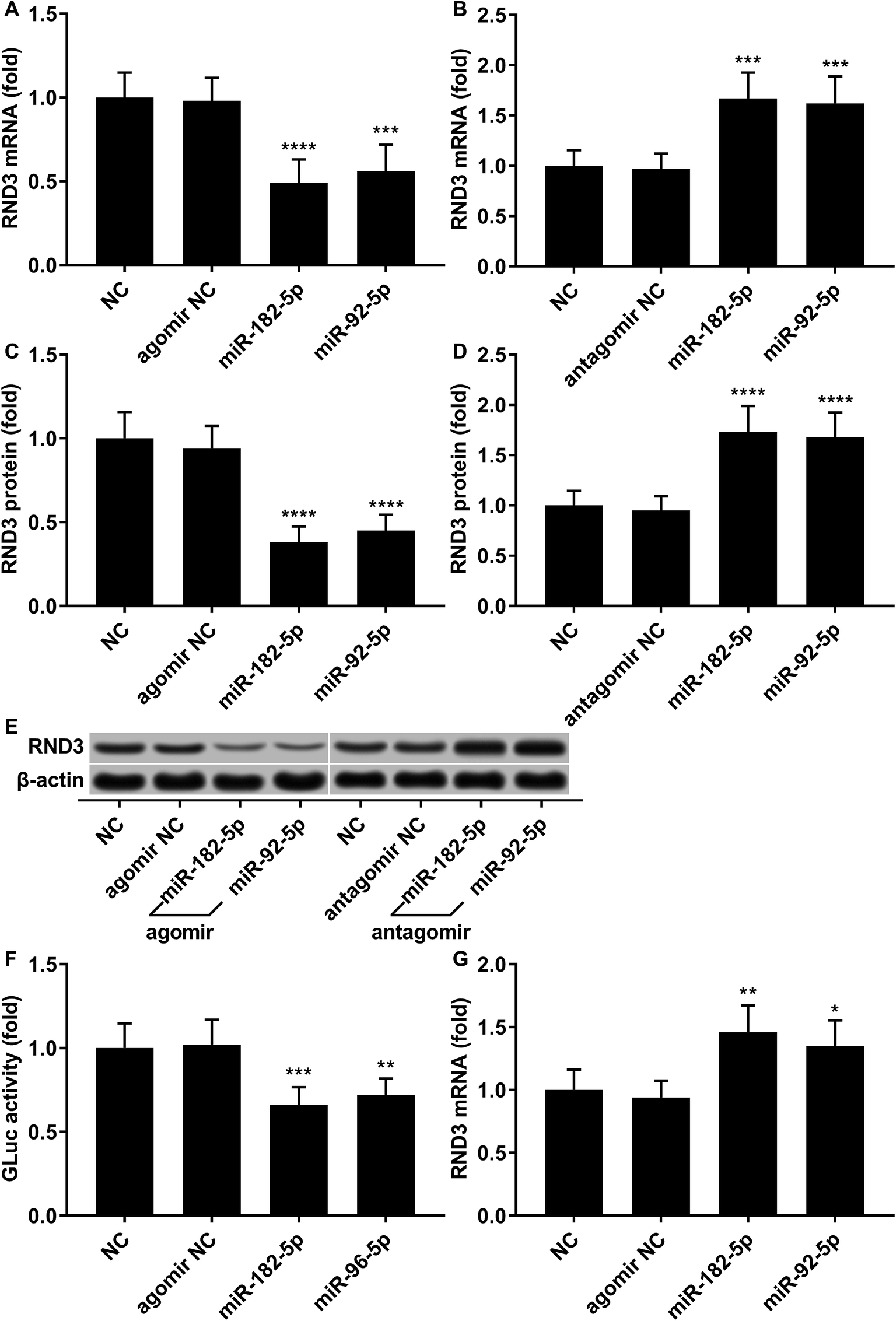 | ||
| Fig. 2 RND3 mRNA is a direct target of miR-182-5p and miR-96-5p in HCC cells. (A–E): RND3 mRNA and protein expression level change in patient-derived HCC cells after treatment with indicated agomir or antagomir. (F): Luciferase reporter assay verifying the targeting potential of the two indicated miRNA agomir to RND3 mRNA 3′ UTR. (G): AGO2 RIP assay verifying the binding potential of the two indicated miRNA agomir to RND3 mRNA. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. | ||
MiR-182-5p or miR-96-5p increased HCC cell mobility in vitro by targeting RND3
To investigate whether RND3-suppression was involved in the prometastatic role of miR-182-5p and miR-96-5p, we constructed RND3-overexpressing HCC cells in vitro using patient-derived HCC cells. As expected, miR-192-5p or miR-96-5p agomir treatment significantly diminished the increase in RND3 mRNA and protein expression induced by RND3 OE (Fig. 3A and B). A trans-well migration and invasion assay revealed that miR-182-5p or miR-96-5p significantly increased while RND3 OE significantly decreased the HCC cell mobility in vitro. Furthermore, miR-192-5p or miR-96-5p agomir treatment significantly rescued cell mobility that was decreased by RND3 OE (Fig. 3C–F). Previous research has suggested the involvement of ROCK1/2 in RND3 knockdown-induced upregulation of HCC cell mobility.15 MYPT1 is a substrate of ROCK1/2 kinase whose pT853 phosphorylation level can be used to monitor ROCK1/2 kinase activity.22 Western blot analysis results showed a significant upregulation in MYPT1 pT853 phosphorylation level in HCC cells induced by miR-192-5p or miR-96-5p agomir treatment and a significant decrease in RND3 OE HCC level compared with that of the WT counterparts, which could be largely restored by miR-192-5p or miR-96-5p agomir treatment (Fig. 3G and H). These data suggested that miR-192-5p or miR-96-5p antagonized with RND3 regulate HCC cell mobility in vitro.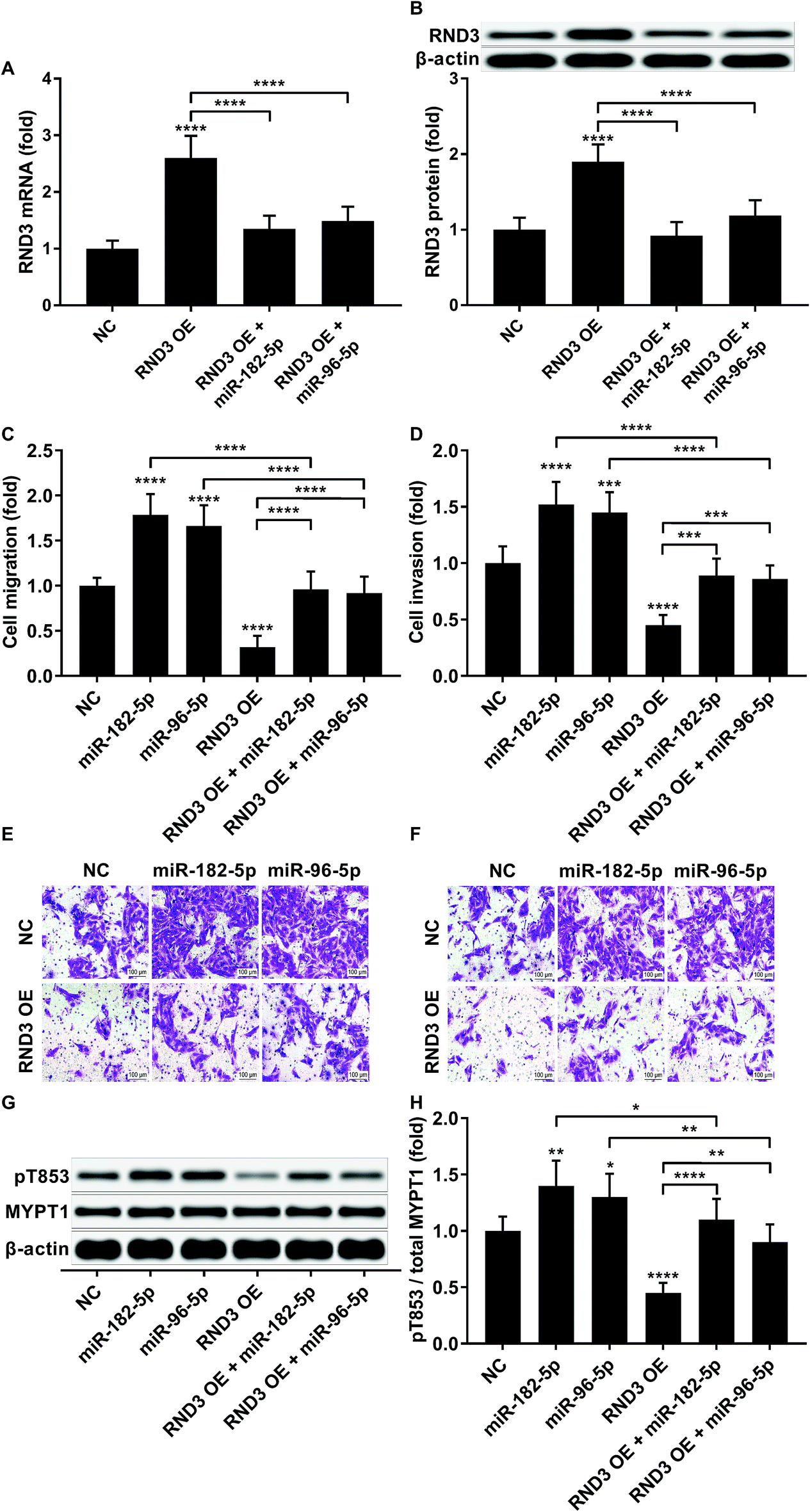 | ||
| Fig. 3 MiR-182-5p or miR-96-5p increased HCC cell mobility in vitro by antagonizing RND3. (A and B): RND3 mRNA and protein expression level in HCC cells after indicated treatment. (C–F): Trans-well migration and invasion of un-modified (NC) or RND3 overexpressing (RND3 OE) HCC cells with or without indicated miRNA agomir treatment. (G and H): MYPT1 protein expression level as well as its Thr853 Phosphorylation (pT853) level in HCC cells in (C and D). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. | ||
To verify that miR-182-5p and miR-96-5p increased HCC cell mobility in vitro by targeting RND3, we combined the agomir treatment with a specific ROCK1/2 inhibitor TC-s 7001 anticipating to inhibit the effect of miR-182-5p or miR-96-5p agomir treatment on HCC cell mobility. However, our trans-well assay results indicated that miR-182-5p or miR-96-5p agomir treatment could increase HCC cell mobility that was repressed by TC-s 7001 treatment, which almost completely abrogated the increase in MYPT1 pT853 phosphorylation level induced by miR-192-5p or miR-96-5p agomir treatment, as revealed by western blot analysis (Fig. 4A–F). These results suggested that miR-182-5p or miR-96-5p could increase HCC cell mobility in vitro through other mechanisms.
 | ||
| Fig. 4 MiR-182-5p or miR-96-5p increased HCC cell mobility in vitro by antagonizing ROCK inhibition. (A–D): Trans-well migration and invasion of HCC cells after the indicated treatment. (E and F): MYPT1 protein expression level as well as its Thr853 phosphorylation (pT853) level in HCC cells after the indicated treatment. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001. | ||
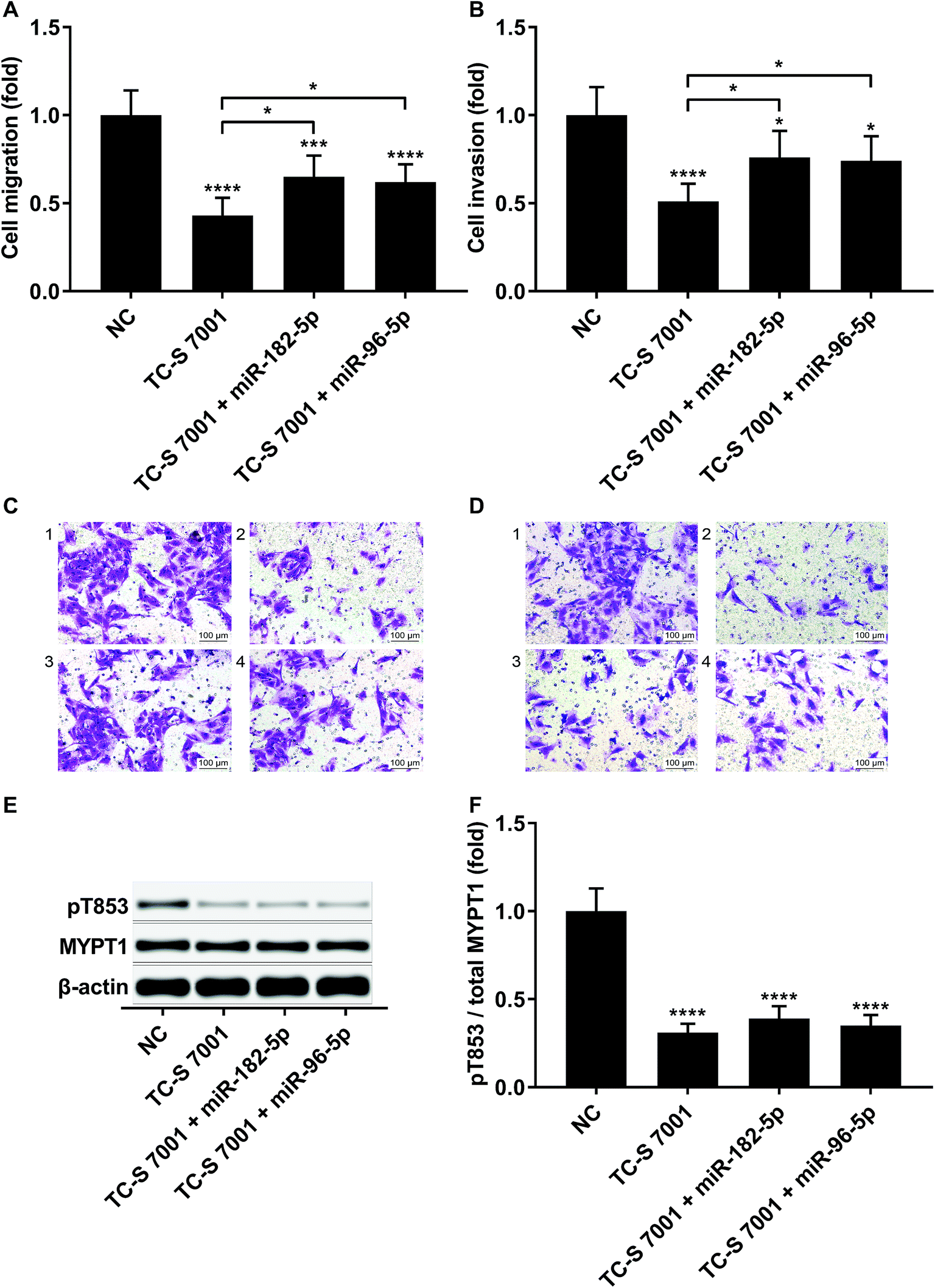
We slightly expanded our research to investigate which ROCK analog was involved in the RND3-mediated suppressive effect on HCC cell mobility as previous research suggested the different roles of ROCK1 and ROCK2 in HCC development.17 Interestingly, our trans-well assay results showed that the upregulation of HCC cell mobility induced by RND3 could be attenuated by both ROCK1 and ROCK2 knockdown or ROCK1/2 inhibitor TC-S 7001 treatment (Fig. 5A–F). Western blot analysis on the MYPT1 pT853 phosphorylation level showed that TC-S 7001 treatment drastically repressed MYPT1 pT853 phosphorylation that was increased by RND3 knockdown, compared to which the effect of ROCK1 or ROCK2 knockdown was less potent (Fig. 5G and H). We inferred from these results that RND3 might regulate HCC cell mobility in vitro through another mechanism aside from ROCK1/2 inhibition.
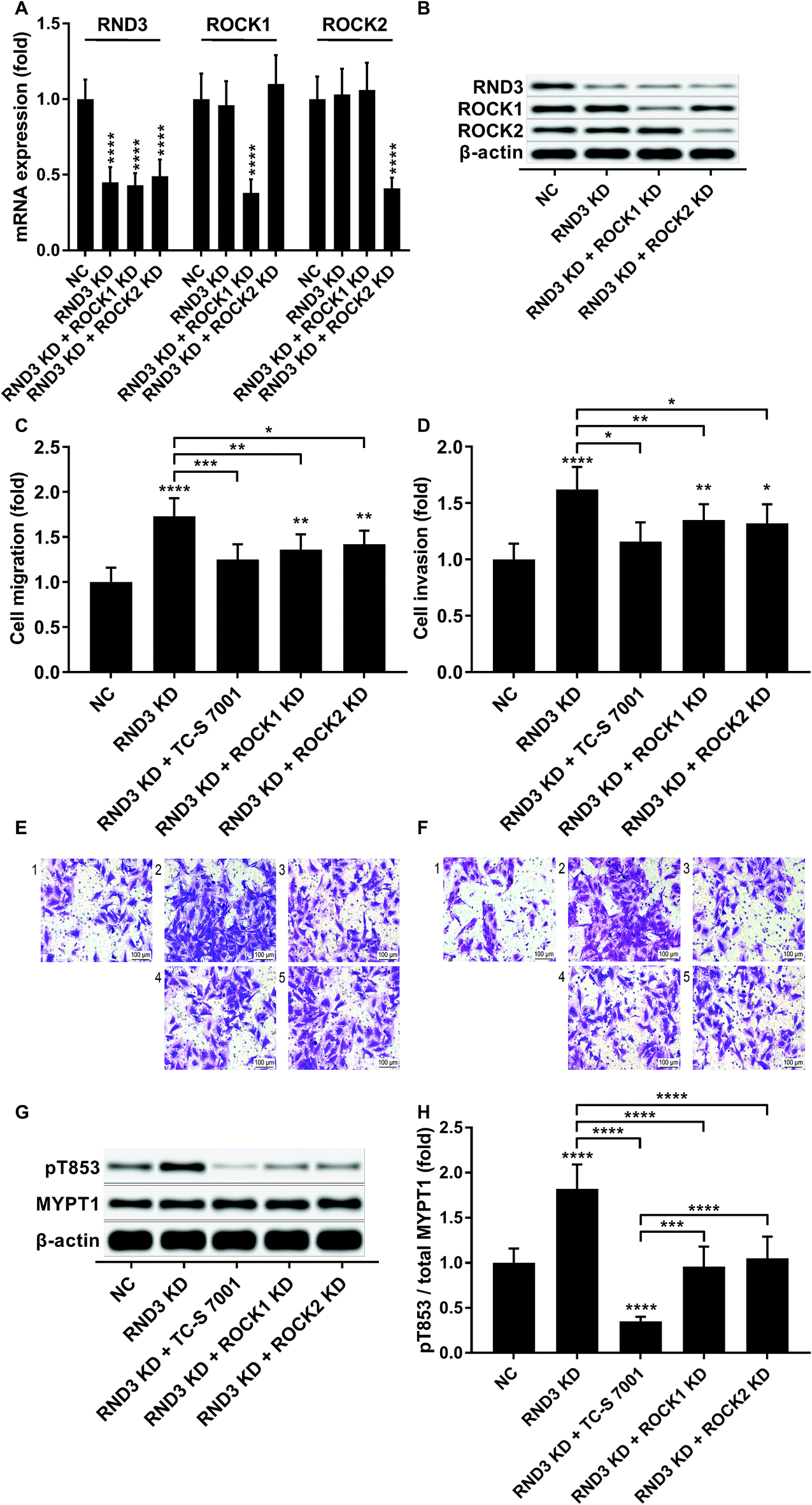 | ||
| Fig. 5 RND3 reduced HCC cell mobility partially by inhibiting ROCK1/2. (A and B): mRNA and protein expression level of indicated genes in HCC cells after RND3 knockdown, ROCK1 knockdown or ROCK2 knockdown. (C–F): Trans-well migration and invasion of HCC cells after different modifications and treatment. (G and H): MYPT1 protein expression level as well as its Thr853 phosphorylation (pT853) level in HCC cells in (C and D). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. | ||
MiR-182-5p or miR-96-5p increased HCC cell proliferation and cisplatin resistance in vitro by targeting RND3
The cell growth-inhibiting effect of RND3 overexpression has been demonstrated in esophageal squamous cell carcinoma cells,23 while RND3 knockdown was found to enhance cell proliferation in glioblastoma cells and neural stem cells.24,25 We therefore investigated whether miR-182-5p and miR-96-5p would influence HCC cell growth in vitro by targeting RND3. Our cell viability and proliferation assay results both suggested that RND3 OE or TC-S 7001 treatment partially attenuated the cell proliferation-promoting effect of miR-182-5p or miR-96-5p agomir treatment in HCC cells in vitro (Fig. 6A and B). Our cell apoptosis assay results revealed no statistically significant influence of miR-182-5p or miR-96-5p agomir treatment or RND3 OE or TC-S 7001 treatment on HCC cell apoptosis in vitro (Fig. 6C). HCC cell proliferation and apoptosis (Fig. 6B and C) were also verified by western blotting by analyzing the expression level of cell proliferation marker protein PCNA and activation of apoptosis marker protein caspase-3 (Fig. 6D). However, when HCC cells were treated with cisplatin, miR-182-5p or miR-96-5p agomir treatment exhibited a significant anti-apoptotic effect, while RND3 OE or TS-C 7002 treatment significantly promoted HCC cell apoptosis induced by cisplatin treatment, which can be partially attenuated by miR-182-5p or miR-96-5p agomir treatment (Fig. 6E). Caspase activity assay results further demonstrated that RND3 OE or TS-C 7002 treatment primarily enhanced the intrinsic apoptosis pathway in cisplatin-treated HCC cells, marked by a significant upregulation of caspase-9 activation compared with the control, along with the extrinsic apoptosis pathway executed by active caspase-8. The effect of RND3 OE or TS-C 7002 treatment on caspase-9 activation was also reduced by miR-182-5p or miR-96-5p agomir treatment (Fig. 6F–H). Collectively, these data indicated that miR-182-5p or miR-96-5p could increase HCC cell proliferation and cisplatin resistance in vitro by targeting RND3.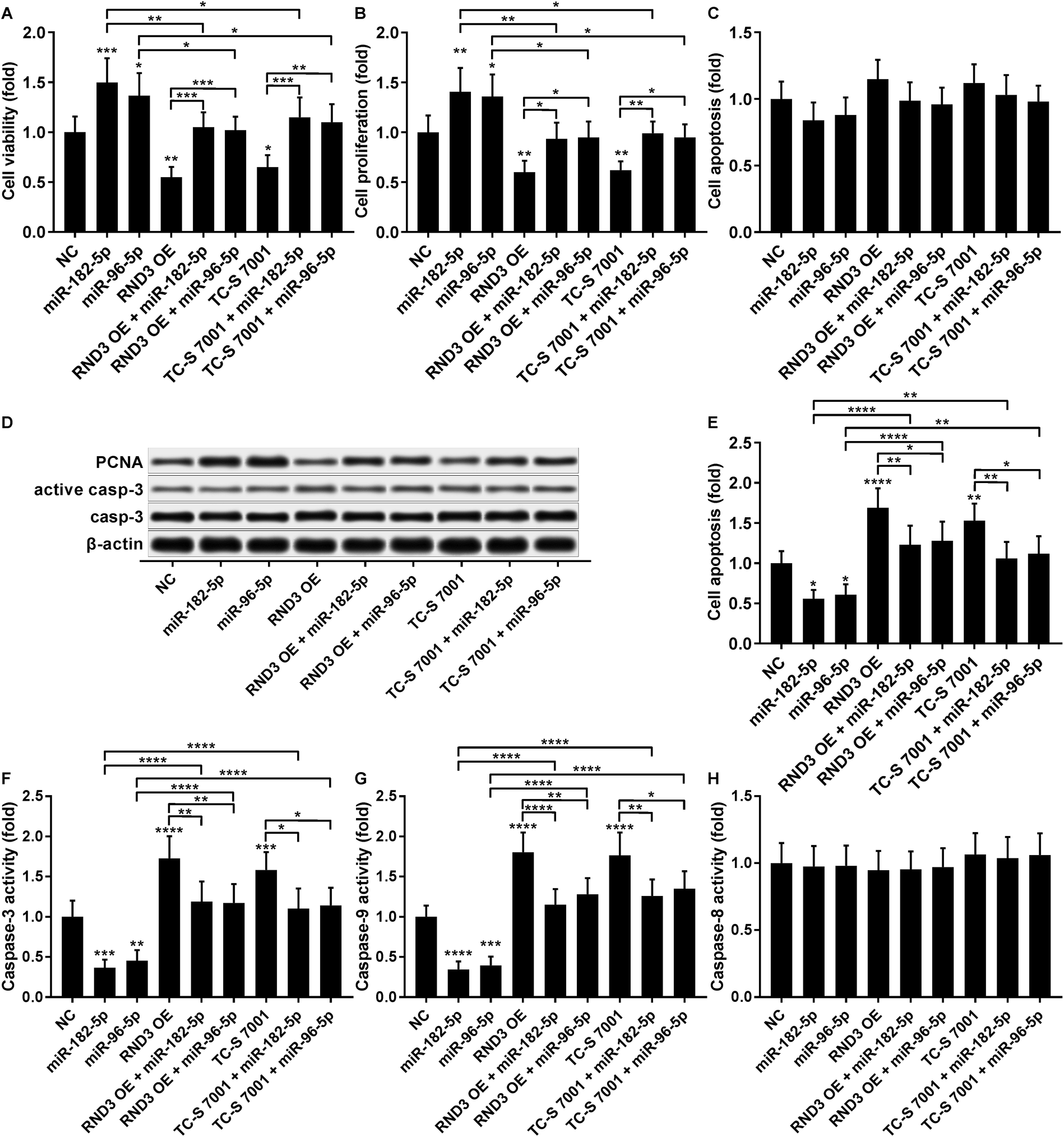 | ||
| Fig. 6 MiR-182-5p or miR-96-5p increased HCC cell proliferation and cisplatin resistance by antagonizing RND3. (A–C): Cell viability, proliferation and apoptosis of HCC cells after indicated modifications and treatment in vitro. (D): Protein expression level of PCNA, caspase-3 and active caspase-3 in HCC treated in (A). (E–H): Cell apoptosis and caspase-3, -8 or -9 activities in HCC cells after indicated modifications and treatment in addition to cisplatin treatment in vitro. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. | ||
Discussions
The present research aimed to discover novel mechanisms underlying the HCC-promoting role of miR-182-5p and miR-96-5p. In our preliminary research using bioinformatic analysis tools, we found RND3 as a potential target of the two miRNAs. Although the HCC-suppressive role of RND3 has been suggested by previous reports,14–16 its mechanism of action in HCC remains largely unresolved. In the present research, we verified that miR-182-5p and miR-96-5p promoted the mobility, proliferation and cisplatin resistance of patient-derived HCC cells in vitro partially by targeting RND3, which exhibited its anti-HCC effect by deactivating ROCK.In the present research, we established a patient-derived HCC cell culture instead of using commercially available HCC cell lines to better simulate real conditions in HCC patients. These cells were assayed at a maximum passage 5 to avoid the accumulation of mutation potentially induced in vitro cell culture. Specifically, we used agomir or antagomir treatment to mimic the overexpression or knockdown of miRNA instead of using miRNA mimic or inhibitor transfection. This is because in our preliminary experiments, we found that the transfection efficiency of miRNA mimic or inhibitor varies greatly among biological replicates. We first examined the expression level of RND3 mRNA/protein as well as that of miR-182-5p and miR-96-5p in different liver tissue samples. We found a significant upregulation of the two miRNAs and significant downregulation of RND3 mRNA/protein expression level in HCC tissue samples compared with those in paired adjacent counterparts or non-HCC liver tissue specimens, which is in accordance with previous reports.26 We also found a significant inverse correlation between RND3 mRNA expression level and the two miRNAs' expression level, implying that these two miRNAs might reduce RND3 expression. This result was also confirmed in vitro by treating HCC cells with miR-182-5p and miR-96-5p agomir or antagomir. We employed the luciferase reporter assay and AGO2-RIP assay to verify that the 3′ UTR of RND3 mRNA is a direct target of miR-182-5p or miR-96-5p. These data suggested that miR-182-5p or miR-96-5p could reduce RND3 gene expression in HCC cells by directly targeting its mRNA.
Previous studies have demonstrated the anti-metastatic and chemo-sensitizing role of RND3 in HCC cells.15–17,27 We therefore investigated whether miR-182-5p and miR-96-5p could increase HCC cell mobility and chemoresistance in vitro by targeting RND3. Our data clearly demonstrated that the pro-metastatic role of the two miRNAs in patient-derived HCC cells could be reduced by RND3 overexpression or ROCK1/2 inhibition. Interestingly, our data also revealed that both ROCK1 and ROCK2 were involved in RND3 knockdown-mediated upregulation of HCC cell mobility in our experimental settings. As mentioned in Jie et al.'s review article, Rnd3 might function in RhoA/ROCK1 in a signaling-dependent or independent manner.13 However, Ma et al. reported that RND3 decreased HCC cell chemoresistance primarily through ROCK2 inhibition.17 We speculated that RND3 might regulate different cancer cell behaviors in different cell types through various mechanisms, which should be further investigated in future research.
In addition to cell migration and invasion, we found that RND3 limited HCC cell growth predominantly by reducing cell proliferation rather than increasing cell apoptosis in vitro, the effect of which could be antagonized by miR-182-5p or miR-96-5p agomir administration. To the best of our knowledge, these results represent the first evidence suggesting that RND3 could regulate HCC cell proliferation in vitro, presumably by inhibiting ROCK1/2 kinas activity. RND3 has been proposed to mediate cell cycle arrest by blocking the S-phase entry in different cell types.28–30 It might be worth verifying whether RND3 could reduce HCC cell proliferation through a similar mechanism in future research. We further revealed that RND3 sensitized HCC cells to cause cisplatin-induced apoptosis by enhancing the intrinsic apoptosis pathway rather than the extrinsic apoptosis pathway, possibly by inhibiting ROCK1/2, which could also be reduced by miR-182-5p or miR-96-5p agomir administration.
A major drawback of the present research is the lack of in vivo investigations. The HCC-promoting role of miR-182-5p and miR-96-5p as well as anti-metastatic role of RND3 in HCC xenograft model has been reported,5,15,31 but whether miR-182-5p and/or miR-96-5p could directly inhibit RND3 expression in vivo remains to be verified. Nevertheless, based on our in vitro experimental results we propose that targeting miR-182-5p and miR-96-5p might be a possible approach for HCC treatment.
Conclusion
Collectively, our data demonstrated that miR-182-5p and miR-96-5p could promote HCC cell proliferation, mobility and cisplatin resistance in vitro at least in part by targeting the tumor suppressive gene RND3, thus adding novel insight into the HCC-promoting mechanism of these two miRNAs. Utilizing antagomirs with enhanced cell membrane penetration compared with that of miRNA inhibitor might serve as a novel therapeutic strategy for HCC management.Ethics approval and consent to participate
The study complied with relevant institutional and national guidelines. The protocol was approved by the ethics review committee of Shanxi Provincial People's Hospital. This research was approved by the ethics review committee of Shanxi Provincial People's Hospital. Written informed consent was obtained from each individual whose biospecimens were involved in this research.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
Not applicable.References
- A. Forner, M. Reig and J. Bruix, Lancet, 2018, 391, 1301–1314 CrossRef.
- C. M. Wong, F. H. Tsang and I. O. Ng, Nat. Rev. Gastroenterol. Hepatol., 2018, 15, 137–151 CrossRef CAS PubMed.
- M. Klingenberg, A. Matsuda, S. Diederichs and T. Patel, J. Hepatol., 2017, 67, 603–618 CrossRef CAS PubMed.
- S. A. Gorski, J. Vogel and J. A. Doudna, Nat. Rev. Mol. Cell Biol., 2017, 18, 215–228 CrossRef CAS PubMed.
- M. Q. Cao, A. B. You, X. D. Zhu, W. Zhang, Y. Y. Zhang, S. Z. Zhang, K. W. Zhang, H. Cai, W. K. Shi, X. L. Li, K. S. Li, D. M. Gao, D. N. Ma, B. G. Ye and C. H. Wang, et al., J. Hematol. Oncol., 2018, 11, 12 CrossRef PubMed.
- Z. Li and Y. Wang, Biochem. Cell Biol., 2018, 96, 365–371 CrossRef CAS PubMed.
- D. Xu, X. He, Y. Chang, C. Xu, X. Jiang, S. Sun and J. Lin, Oncol. Rep., 2013, 29, 653–661 CrossRef CAS PubMed.
- T. H. Wang, C. T. Yeh, J. Y. Ho, K. F. Ng and T. C. Chen, Mol. Carcinog., 2016, 55, 366–375 CrossRef CAS PubMed.
- W. K. Leung, M. He, A. W. Chan, P. T. Law and N. Wong, Cancer Lett., 2015, 362, 97–105 CrossRef CAS PubMed.
- V. Agarwal, G. W. Bell, J. W. Nam and D. P. Bartel, eLife, 2015, 4 DOI:10.7554/eLife.05005.
- M. D. Paraskevopoulou, G. Georgakilas, N. Kostoulas, I. S. Vlachos, T. Vergoulis, M. Reczko, C. Filippidis, T. Dalamagas and A. G. Hatzigeorgiou, Nucleic Acids Res., 2013, 41, W169–W173 CrossRef PubMed.
- P. Chardin, Nat. Rev. Mol. Cell Biol., 2006, 7, 54–62 CrossRef CAS PubMed.
- W. Jie, K. C. Andrade, X. Lin, X. Yang, X. Yue and J. Chang, Compr. Physiol., 2015, 6, 169–186 Search PubMed.
- H. Luo, Z. Dong, J. Zou, Q. Zeng, D. Wu and L. Liu, J. Surg. Oncol., 2012, 105, 699–704 CrossRef CAS PubMed.
- W. Ma, C. C. Wong, E. K. Tung, C. M. Wong and I. O. Ng, Hepatology, 2013, 57, 152–161 CrossRef CAS PubMed.
- F. Grise, S. Sena, A. Bidaud-Meynard, J. Baud, J. B. Hiriart, K. Makki, N. Dugot-Senant, C. Staedel, P. Bioulac-Sage, J. Zucman-Rossi, J. Rosenbaum and V. Moreau, Hepatology, 2012, 55, 1766–1775 CrossRef CAS PubMed.
- W. Ma, K. M. Sze, L. K. Chan, J. M. Lee, L. L. Wei, C. M. Wong, T. K. Lee, C. C. Wong and I. O. Ng, Oncotarget, 2016, 7, 41445–41459 Search PubMed.
- L. Paysan, L. Piquet, F. Saltel and V. Moreau, Mol. Cancer Res., 2016, 14, 1033–1044 CrossRef CAS PubMed.
- E. E. Moore, T. H. Cogbill, G. J. Jurkovich, S. R. Shackford, M. A. Malangoni and H. R. Champion, J. Trauma, 1995, 38, 323–324 CrossRef CAS PubMed.
- J. M. Llovet, C. Bru and J. Bruix, Semin. Liver Dis., 1999, 19, 329–338 CrossRef CAS PubMed.
- C. R. Justus, N. Leffler, M. Ruiz-Echevarria and L. V. Yang, J. Vis. Exp., 2014, e51046 Search PubMed.
- C. A. Hudson, K. J. Heesom and A. Lopez Bernal, Mol. Hum. Reprod., 2012, 18, 265–279 CrossRef CAS PubMed.
- H. Wang, Y. Wang, B. Liang, F. He, Y. Li, J. Che, X. Li, H. Zhao and G. Shi, J. Cancer Res. Ther., 2016, 12, 60–63 CrossRef CAS PubMed.
- B. Liu, X. Lin, X. Yang, H. Dong, X. Yue, K. C. Andrade, Z. Guo, J. Yang, L. Wu, X. Zhu, S. Zhang, D. Tian, J. Wang, Q. Cai and Q. Chen, et al., Cancer Med., 2015, 4, 1404–1416 CrossRef CAS PubMed.
- H. Dong, X. Lin, Y. Li, R. Hu, Y. Xu, X. Guo, Q. La, S. Wang, C. Fang, J. Guo, Q. Li, S. Mao and B. Liu, Oncotarget, 2017, 8, 91112–91122 Search PubMed.
- Q. H. Zhang, H. M. Sun, R. Z. Zheng, Y. C. Li, Q. Zhang, P. Cheng, Z. H. Tang and F. Huang, Gene, 2013, 527, 26–32 CrossRef CAS PubMed.
- D. Wang, N. Zhang, Y. Ye, J. Qian, Y. Zhu and C. Wang, Mol. Med. Rep., 2014, 10, 3268–3274 CrossRef CAS PubMed.
- P. Villalonga, R. M. Guasch, K. Riento and A. J. Ridley, Mol. Cell. Biol., 2004, 24, 7829–7840 CrossRef CAS PubMed.
- W. Xia, J. Li, L. Chen, B. Huang, S. Li, G. Yang, H. Ding, F. Wang, N. Liu, Q. Zhao, T. Fang, T. Song, T. Wang and N. Shao, Mol. Cell. Biochem., 2010, 344, 261–266 CrossRef CAS PubMed.
- M. Lonjedo, E. Poch, E. Mocholi, M. Hernandez-Sanchez, C. Ivorra, T. F. Franke, R. M. Guasch and I. Perez-Roger, J. Biol. Chem., 2013, 288, 30872–30882 CrossRef CAS PubMed.
- S. H. Baik, J. Lee, Y. S. Lee, J. Y. Jang and C. W. Kim, Exp. Mol. Med., 2016, 48, e222 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |