
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkylhalovinylboranes: a new class of Diels–Alder dienophiles†
Pablo L. Pisano and
Silvina C. Pellegrinet*
and
Silvina C. Pellegrinet*
Instituto de Química Rosario (CONICET), Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Suipacha 531, Rosario (2000), Argentina. E-mail: pellegrinet@iquir-conicet.gov.ar
First published on 2nd October 2018
Abstract
The Diels–Alder reactions of alkylhalovinylboranes have been investigated theoretically and experimentally. Alkylhalovinylboranes presented higher reactivity than the corresponding dialkylvinylboranes. Although endo/exo selectivities were high for the reactions with cyclopentadiene, facial selectivities for the chiral analogues were low. Our results demonstrate that the replacement of an alkyl group on the boron atom by a halogen increases the dienophilicity considerably.
Introduction
In the past decades, Singleton and others have extensively explored the synthetic utility of alkenyl-, alkynyl- and dienylboranes in Diels–Alder reactions.1–18 Given that many variations of the Diels–Alder reaction of vinylboranes have been developed, it is surprising that there is only one precedent in the use of chiral vinylboranes in asymmetric [4 + 2] cycloaddition reactions. This Diels–Alder reaction was based on the use of dialkylvinylborane 1, derived from (+)-camphene, with 2-phenyl-1,3-butadiene (Scheme 1).8 However, although the yield of the oxidized products and the para regioselectivity were high, the enantiomeric excess of the major regioisomer was lower than 10%.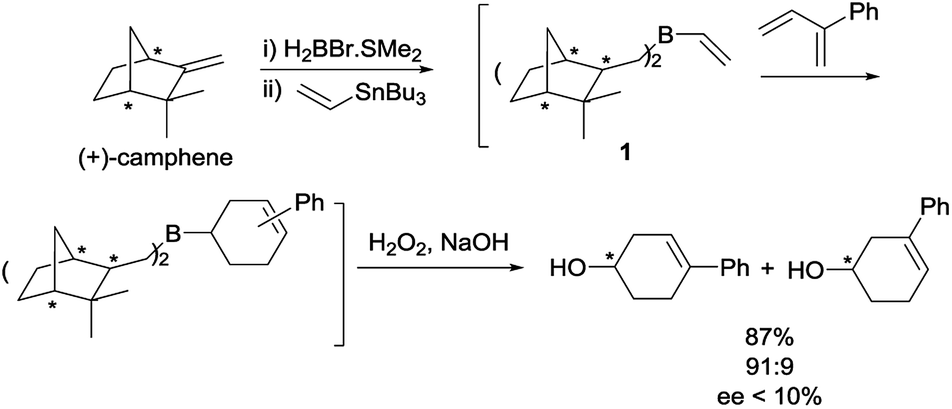 | ||
| Scheme 1 Synthesis and Diels–Alder reaction of chiral dialkylvinylborane 1. | ||
One of the main goals of our group is to develop new chiral unsaturated boron compounds that exhibit high reactivity and selectivity as dienophiles in Diels–Alder reactions. Such compounds have a tremendous potential as precursors of chiral building blocks in asymmetric synthesis.
Initially, we thought that dialkylvinylboranes 2–6 shown in Scheme 2 would be good candidates because the stereocenters are directly bonded to the boron atom, and therefore closer to the prochiral olefin than in the derivative of (+)-camphene (1).19 Furthermore, compounds 2–4 could be readily synthesized from commercial optically pure terpenes (+)-α-pinene, (+)-2-carene and (+)-3-carene.20,21 For that purpose, we tested the reaction sequence shown in Scheme 3 for (+)-α-pinene, analogous to that developed previously by Singleton for (+)-camphene (Scheme 1),8 consisting of hydroboration, transmetallation, Diels–Alder reaction and oxidation. In situ preparation of intermediates 7, 2 and 8 under inert atmosphere would avoid the manipulation of the labile boranes. Unfortunately, although numerous reactions were tested for the three chiral terpenes with different reagents and reaction conditions, we failed to detect the presence of the desired products.22 By monitoring the reaction by 1H NMR under inert atmosphere we observed that, although the hydroboration step occurred efficiently, transmetallation of the dialkylhaloboranes did not go to completion. We managed to avoid this difficulty by generating the monohaloborane free of dimethylsulfide in situ with trihaloborane and triethylsilane.23 Nevertheless, when using this method we could not detect the formation of product 9 either, possibly due to the competitive dimerization of the vinylboranes6 and the protodeboronation of the cycloadducts.
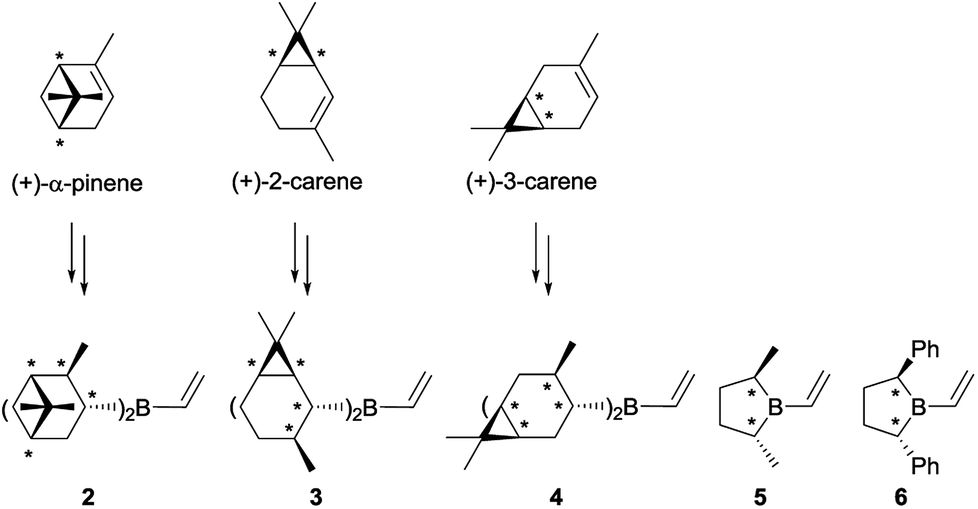 | ||
| Scheme 2 Chiral dialkylvinylboranes with secondary carbons attached to boron. | ||
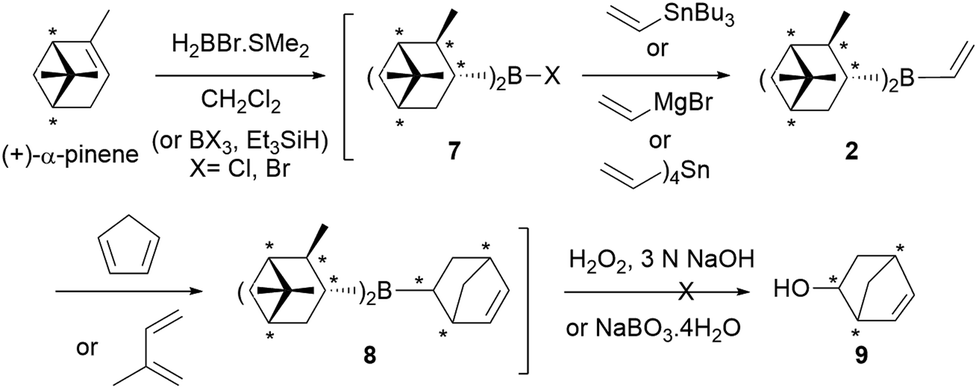 | ||
| Scheme 3 One-pot synthesis of chiral dialkylvinylborane 2, followed by Diels–Alder reaction and oxidation. The same procedure was tested for (+)-2-carene and (+)-3-carene and cyclohexene. | ||
Alternative use of cyclohexene as an achiral model of starting alkene, to generate dicyclohexylvinylborane (10), also gave negative results. These initial results demonstrated that dialkylvinylboranes with secondary carbons attached to the boron atom, such as 2–4 and 10, are highly congested, which complicates their synthesis and the course of their Diels–Alder reactions.19
To circumvent these problems, it occurred to us that alkylhalovinylboranes would represent an interesting alternative to dialkylvinylboranes because the boron atom would be less sterically hindered and more electron deficient, which should ultimately increase the reactivity in Diels–Alder reactions (Fig. 1).24 In this paper, we present the results of a theoretical study of the Diels–Alder reactions of a number of alkylhalovinylboranes with cyclopentadiene, together with the experimental development of a one-pot procedure that includes the synthesis of the dienophiles, the Diels–Alder reaction and the oxidation of the products.
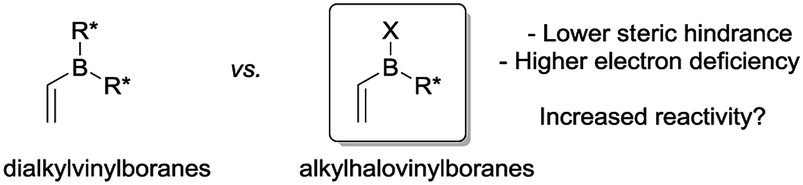 | ||
| Fig. 1 Dialkylvinylboranes and alkylhalovinylboranes. | ||
Results and discussion
Model studies with cyclohexene vinylborane derivatives
To validate our hypothesis, we first performed a comparative study of the reactivity of dicyclohexylvinylborane (10), chlorocyclohexylvinylborane (11a) and bromocyclohexylvinylborane (11b), derived from cyclohexene as achiral models of the dienophiles. Initially, we investigated the Diels–Alder reactions of vinylboranes 10, 11a and 11b with cyclopentadiene, a highly reactive and easily accessible diene, with theoretical methods. Due to the absence of chiral centres in the alkyl moiety, only endo and exo attacks should be considered, which, in turn, would give rise to diastereomeric products as racemic mixtures. After oxidation, racemic endo and exo alcohols 9 might arise.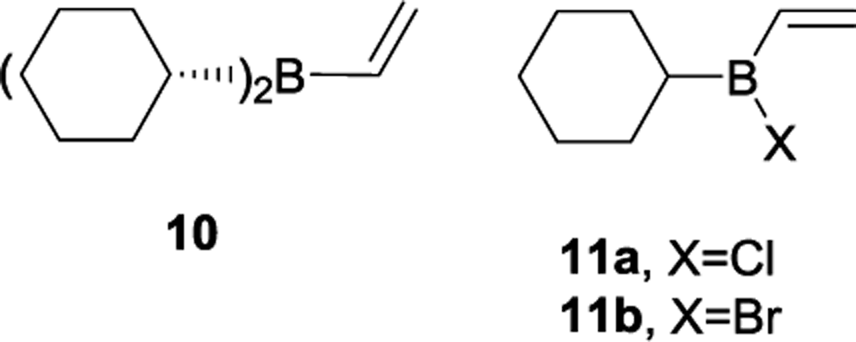
All calculations were performed with the Gaussian 09 package.25 We carried out thorough conformational analyses to locate the lowest energy geometry for all the structures under study. Final geometry optimizations were carried out using the B3LYP hybrid functional together with the 6-31G* basis set. The selection of this level of theory was based on existing records in the literature for the modelling of Diels–Alder reactions.19,26–32 Additionally, solvent effects in dichloromethane (ε = 8.93) and heptane (ε = 1.91) were calculated for the most stable geometries of reagents, transition structures (TSs), and products using the Polarized Continuum Model of Self-Consistent Reaction Field method (PCM method).33,34 Furthermore, the molecular orbitals (MOs) of reagents were calculated to analyse the Frontier molecular orbital interactions and Natural Bond Orbitals (NBOs) analysis of TSs were performed using Wiberg bond indices for interpreting most relevant electronic interactions.35–38 Finally, we performed Intrinsic Reaction Coordinate calculations (IRC) to verify if the TSs were directly connected to the reactants and the products.
Fig. 2 shows the correlation diagrams for the Diels–Alder reactions of vinylboranes 10, 11a, and 11b with cyclopentadiene. Analysis of MOs of the reagents shows that all Diels–Alder reactions under study are of normal electron-demand dominated by the HOMOdiene–LUMOdienophile interaction. As expected, the LUMOs of the vinylboranes have large coefficients on the two carbon atoms of the carbon–carbon double bond of the vinyl group, and also on the boron atoms. This feature promotes the non-classical [4 + 3] secondary orbital interaction (SOI) between the boron atom of vinylboranes and C1 of the diene, resulting in an increase in the observed endo-selectivity. Both FMOs of haloalkylvinylboranes 11a and 11b also show important coefficients in the halogen atoms too. The presence of the halogen attached to the boron atom lowers the LUMO energy significantly (ca. 0.5 eV) and, therefore, the energy gap for the HOMOdiene–LUMOdienophile interaction of 11a and 11b is lower than the value obtained for 10, which suggests greater reactivity for alkylhalovinylboranes with respect to the corresponding dialkylvinylborane.39
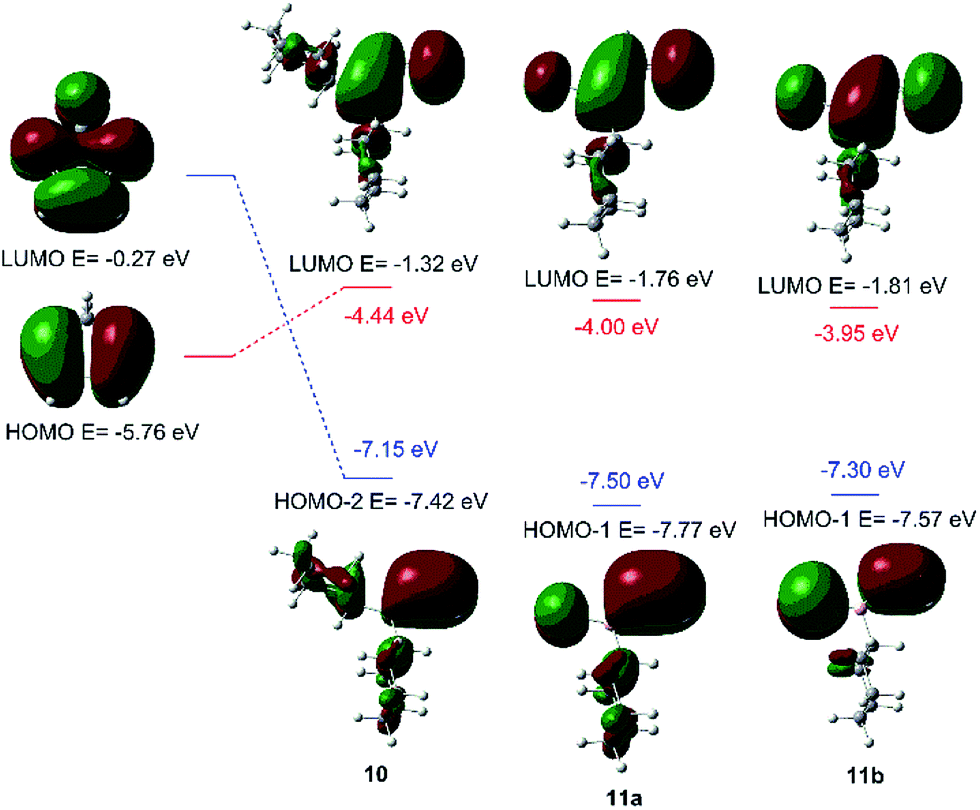 | ||
| Fig. 2 Correlation diagrams for the Diels–Alder reaction of vinylboranes 10, 11a, and 11b with cyclopentadiene. Energy gaps are shown in red and blue. | ||
Fig. 3 shows the optimized geometries of the most stable conformers for the TSs for the Diels–Alder reaction of vinylboranes 10, 11a, and 11b with cyclopentadiene, with the carbon–carbon and C6–B bond distances (in Å) and Wiberg bond indices in parentheses. C6–B distances are approximately 3.1 Å and NBOs of 0.08 and 0.06 indicating that the SOI [4 + 3] are weak. In the IRC studies no intermediate structures between reagents and TSs and between TSs and products were found, confirming that the Diels–Alder reactions under study are concerted. Although in these reactions all bonds are formed and broken in a single stage, all TSs are asynchronous with carbon–carbon distances approximately of 2.0 Å for C2–C3 and 2.6 Å for C1–C6.40,41 As shown in Fig. 3, the dienophile portion of the structures corresponding to 11a and 11b adopts the same conformation in all TSs, the halogen attached to the boron atom is anti relative to the adjacent axial hydrogen atom of the cyclohexyl group and the double bond is eclipsed with the boron–halogen bond. Table 1 shows the activation free energies (ΔG‡) calculated in the gas phase and heptane of the three systems derived from cyclohexene. As expected, the relative energies of the TSs in the gas phase are considerable higher than in solution. However, in both cases the reactions of 11a and 11b have lower activation free energies than those for 10, which in principle indicates higher reactivity for the alkylhalovinylborane compounds relative to the dialkylvinylborane counterpart. Also, endo TSs were computed to be more stable than the exo isomers for all systems. Selectivity calculations were performed at 298 K using the Boltzmann equation with the activation free energies calculated for the TSs. The selectivity values obtained for 10, 11a and 11b are shown in Table 1. For alkylhalovinylboranes 11a and 11b, the endo/exo selectivity is significantly higher in solution than the gas phase. The computed energies of the products also suggested that the reactions under study are exergonic (see the ESI†).
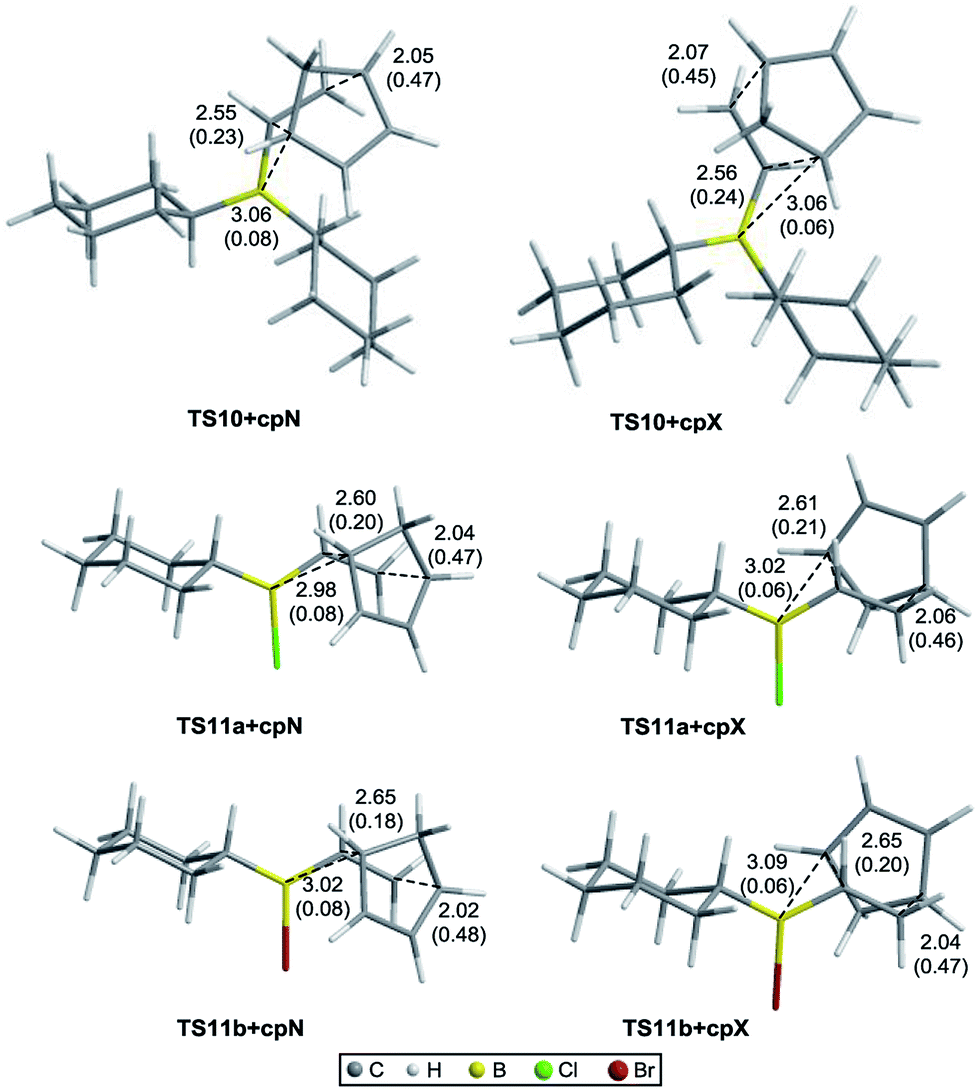 | ||
| Fig. 3 Optimized geometries of the TSs for the Diels–Alder reaction of vinylboranes 10, 11a, and 11b with cyclopentadiene, with carbon–carbon and C6–B bond distances (in Å) and Wiberg bond indices in parentheses. | ||
| Vinylborane | ΔG‡, kcal mol−1 (ΔG‡rel) | endo/exo | ||
|---|---|---|---|---|
| endo | exo | |||
| Gas phase | 10 | 32.28 (0.00) | 32.39 (0.12) | 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45 :45 |
| 11a | 30.10 (0.00) | 30.22 (0.12) | 55:45 |
|
| 11b | 27.22 (0.00) | 27.34 (0.12) | 55:45 |
|
| Heptane | 10 | 16.52 (0.00) | 16.73 (0.21) | 59:41 |
| 11a | 12.71 (0.00) | 13.25 (0.54) | 71:29 |
|
| 11b | 10.62 (0.00) | 11.01 (0.38) | 65:35 |
|
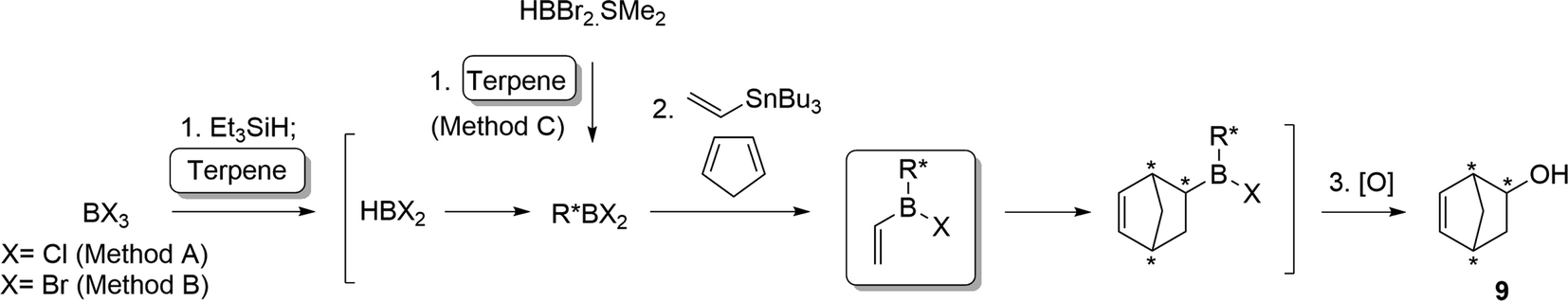
Encouraged by these promising results, we then studied and optimised the reaction sequence shown in Scheme 4. Many experiments were performed using different reaction times and temperatures, as well as number of equivalents and modes of addition of reagents. Fortunately, our hypothesis, which was supported by the calculations, proved to be correct and we managed to obtain the expected products. Initial reaction of trichloroborane with one equivalent of triethylsilane in the presence of 1.1 equivalents of cyclohexene readily gave dichloroborane, which subsequently hydroborated the starting alkene, giving rise to dichlorocyclohexylborane (12). Simultaneous addition of one equivalent of tributylvinyltin and three equivalents of cyclopentadiene successively generated the desired dienophile 11a and cycloadduct (11a+cp). Final oxidation of the reaction mixture conducted to 5-norbornen-2-ol (9) in 48% global yield with 80:20 endo/exo selectivity. The consecutive steps of the synthetic sequence were monitored by 11B NMR under inert atmosphere (Fig. 4), which allowed the optimization of the reaction conditions.
 | ||
| Scheme 4 One-pot synthesis of chlorocyclohexylvinylborane (11a), followed by Diels–Alder reaction and oxidation. | ||
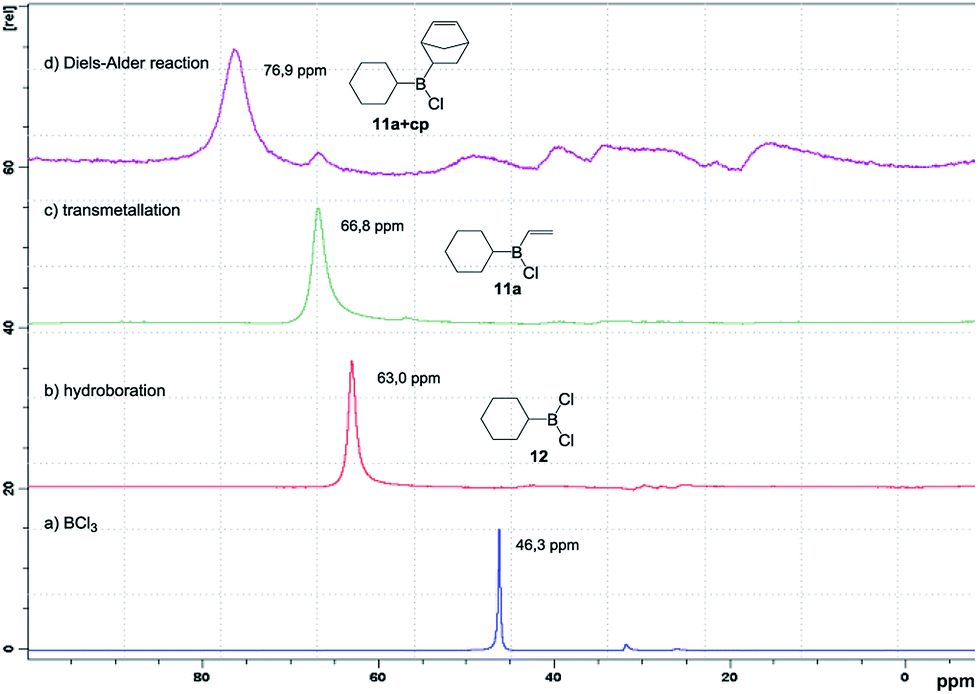 | ||
| Fig. 4 11B NMR of the consecutive steps of the synthetic sequence. | ||
Studies with chiral terpene vinylborane derivatives
Having accomplished the proof of concept with the achiral models, we then turned our attention to the Diels–Alder reactions of chiral alkylhalovinylboranes derived from (−)-α-pinene, (+)-2-carene, (+)-3-carene as starting alkenes. To increase the structural diversity of the ligands, we also included (−)-myrtenol, which has the same carbon skeleton as (−)-α-pinene and allows the preparation synthetic derivatives 13–15 due to the presence of the hydroxymethyl group (for synthetic procedures see the ESI†). We figured that the presence of a heteroatom or an aromatic ring in the side chain of the dienophile would introduce further steric and electronic features, which in turn might contribute to reduce the conformational flexibility and, therefore, increase the stereoselectivity of the cycloaddition.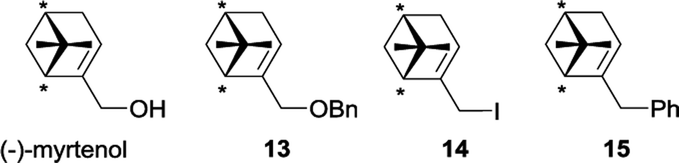
The Diels–Alder reactions of the resulting alkylhalovinylboranes 16–22 with cyclopentadiene were first studied theoretically (Scheme 5). In these cases, four diasteromeric Diels–Alder cycloadducts might arise from the endo and exo attacks of the diene to both faces of the double bond of the chiral alkylhalovinylborane (Re and Si). Upon oxidation such compounds would be converted into two diasteromeric pairs of enantiomers.
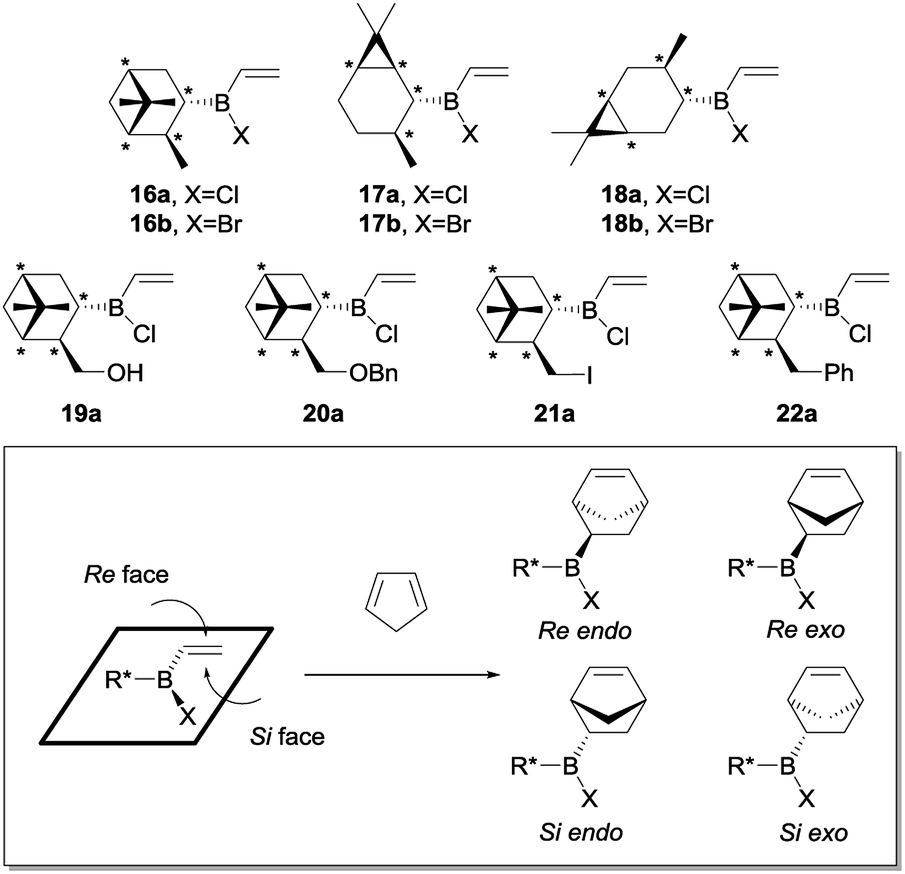 | ||
| Scheme 5 Diels–Alder reactions of chiral alkylhalovinylboranes 16–22 with cyclopentadiene. | ||
As an example, the optimized geometries of the most stable conformers for the TSs for the Diels–Alder reaction of vinylborane 16a with cyclopentadiene are depicted in Fig. 5. Tables 2 and 3 present the activation free energies (ΔG‡) and the stereoselectivities calculated in the gas phase and heptane for the Diels–Alder reactions of chiral alkylhalovinylboranes. In general terms, all [4 + 2] cycloadditions of chiral alkylhalovinylboranes present the same features as the reactions of cyclohexylhaloboranes 11a and 11b:
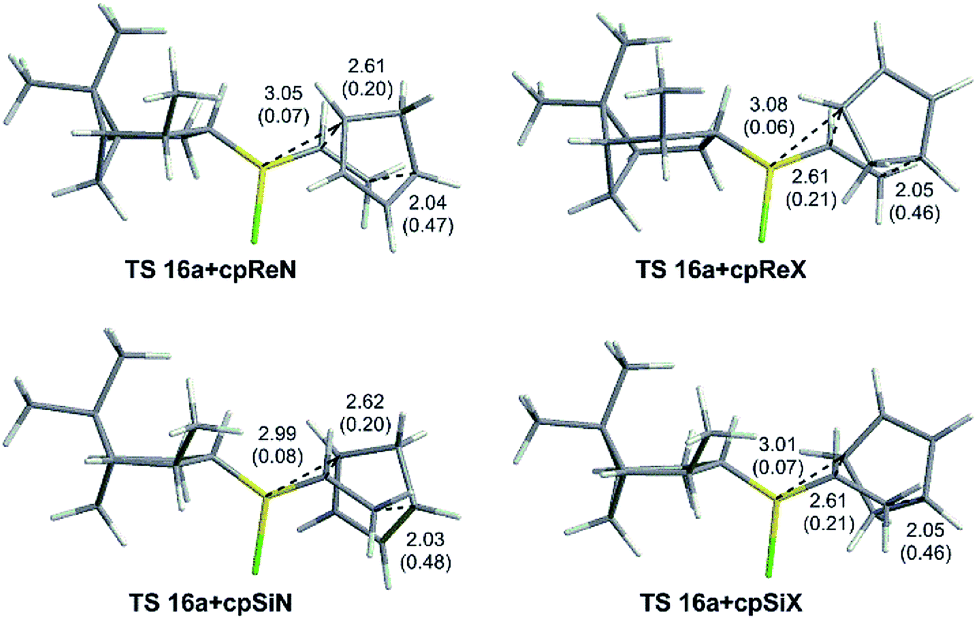 | ||
| Fig. 5 Optimized geometries of the TSs for the Diels–Alder reaction of vinylboranes 16a with cyclopentadiene with the carbon–carbon and C6–B bond distances (in Å) and Wiberg bond indices in parentheses. | ||
| Vinylborane | ΔG‡, kcal mol−1 (ΔG‡rel) | ||||
|---|---|---|---|---|---|
| Re endo | Re exo | Si endo | Si exo | ||
| Gas phase | 16a | 29.89(0.78) | 29.79(0.68) | 29.23(0.12) | 29.11(0.00) |
| 16b | 27.32(0.42) | 27.43(0.52) | 26.90(0.00) | 26.91(0.01) | |
| 17a | 30.09(0.51) | 30.37(0.78) | 29.58(0.00) | 29.68(0.10) | |
| 17b | 28.40(1.04) | 27.72(0.35) | 27.50(0.13) | 27.37(0.00) | |
| 18a | 29.18(0.00) | 29.44(0.26) | 30.36(1.18) | 30.05(0.87) | |
| 18b | 27.31(0.30) | 27.02(0.00) | 27.71(0.69) | 27.54(0.53) | |
| 19a | 30.80(2.03) | 28.76(0.00) | 30.25(1.49) | 29.78(1.02) | |
| 20a | 29.97(0.00) | 30.64(0.67) | 30.58(0.61) | 30.78(0.80) | |
| 21a | 31.64(0.69) | 31.56(0.60) | 31.17(0.22) | 30.95(0.00) | |
| 22a | 29.30(0.74) | 29.37(0.81) | 28.56(0.00) | 28.87(0.31) | |
| Heptane | 16a | 13.43(0.46) | 14.07(1.11) | 12.96(0.00) | 13.48(0.52) |
| 16b | 11.46(0.50) | 11.67(0.71) | 10.96(0.00) | 11.20(0.24) | |
| 17a | 13.56(0.23) | 13.97(0.63) | 13.34(0.00) | 13.72(0.38) | |
| 17b | 11.65(0.27) | 11.53(0.15) | 11.40(0.02) | 11.38(0.00) | |
| 18a | 12.99(0.00) | 13.50(0.51) | 13.16(0.16) | 13.57(0.58) | |
| 18b | 10.86(0.00) | 11.14(0.28) | 11.26(0.40) | 11.06(0.21) | |
| 19a | 13.71(0.83) | 12.88(0.00) | 13.79(0.91) | 13.82(0.93) | |
| 20a | 15.40(1.38) | 14.02(0.00) | 14.02(0.00) | 14.48(0.46) | |
| 21a | 16.00(0.92) | 16.54(1.47) | 15.08(0.00) | 15.69(0.61) | |
| 22a | 14.67(1.10) | 15.15(1.59) | 13.56(0.00) | 14.07(0.51) | |
| Vinylborane | endo/exo | endo Re/Si | exo Re/Si | Re/Si | |
|---|---|---|---|---|---|
| Gas phase | 16a | 45:55 |
25:75 |
24:76 |
25:75 |
| 16b | 51:49 |
33:67 |
29:71 |
31:69 |
|
| 17a | 56:44 |
30:70 |
24:76 |
27:73 |
|
| 17b | 38:62 |
18:82 |
36:64 |
29:71 |
|
| 18a | 57:43 |
88:12 |
73:27 |
82:18 |
|
| 18b | 39:61 |
66:34 |
71:29 |
69:31 |
|
| 19a | 9:91 |
29:71 |
85:15 |
80:20 |
|
| 20a | 70:30 |
74:26 |
56:44 |
68:32 |
|
| 21a | 42:58 |
31:69 |
27:73 |
29:71 |
|
| 22a | 60:40 |
23:77 |
30:70 |
26:74 |
|
| Heptane | 16a | 72:28 |
31:69 |
27:73 |
30:70 |
| 16b | 59:41 |
30:70 |
31:69 |
31:69 |
|
| 17a | 66:34 |
41:59 |
40:60 |
40:60 |
|
| 17b | 47:53 |
40:60 |
44:56 |
42:58 |
|
| 18a | 68:32 |
57:43 |
53:47 |
56:44 |
|
| 18b | 53:47 |
66:34 |
47:53 |
57:43 |
|
| 19a | 28:72 |
53:47 |
83:17 |
74:26 |
|
| 20a | 43:57 |
9:91 |
68:32 |
43:57 |
|
| 21a | 73:27 |
18:82 |
19:81 |
18:82 |
|
| 22a | 70:30 |
14:86 |
14:86 |
14:86 |
- Normal electron demand Diels–Alder reactions (HOMOdiene–LUMOdienophile ∼4 eV).
- LUMOs of the dienophiles show high coefficients on the C–C double bond, the boron and the halogen atoms.
- Concerted reactions with asynchronous TSs with classical [4 + 2] character and weak C–B [4 + 3] SOI (C2–C3, C1–C6 and C6–B distances 2.0, 2.6, and 3.0 Å respectively).
- ΔG‡ in heptane 13–15 kcal mol−1 for chloroalkylboranes and 11 kcal mol−1 for bromoalkylboranes.
Computed endo/exo stereoselectivities are higher for the chloro analogues than for the bromo counterparts and vary considerably (from 28:72 to 72:28 in heptane). Likewise, Re/Si facial selectivities are variable, ranging from 74:26 for 19a to 14:86 for 22a. Since the free energies of the four TSs are within 1 kcal mol−1 for most systems, none of the approximations would be clearly favoured and all cycloadducts would be formed, albeit in different amounts. Both electronic, steric and stereoelectronic effects seem to contribute to give the computed reactivities and selectivities. As found before, in all TSs the vinylborane portion adopts the same conformation in which the halogen attached to the boron atom and the hydrogen of the vicinal carbon adopt a antiperiplanar relationship and the double bond is eclipsed with the boron–halogen bond.
As a consequence the substituents of the carbon backbone (for instance, methyls for 16–18) block preferentially one face of the dienophile and the attack of the diene occurs from the other face.
As an example, in the TSs of the vinylborane derived from (−)-α-pinene (16a), the methyl shields the Re face, making the Si endo approximation more favourable (Fig. 5). The same situation is observed for the derivatives of (+)-2-carene (17), while for the (+)-3-carene counterparts (18) the facial selectivity is reversed because the methyl group is on the other side of the molecule and, therefore, steric clashes would make the attack of the Si face more difficult. The synthetic analogues derived from (−)-myrtenol 21a and 22a were predicted to give the highest endo/exo and Re/Si facial stereoselectivities.
Since the reactions conditions had already been optimised using cyclohexene as the starting alkene (Scheme 4), we next investigated the use of the chiral terpenes and synthetic derivatives as staring materials. The reaction sequences were first investigated using (+)-3-carene and different reagents as the boron source: BCl3 (1 M in hexanes), BBr3 and HBBr2·SMe2 (1 M in CH2Cl2). The reoptimised reaction conditions for the consecutive steps of the synthetic sequences were then used with the chiral terpenes and derivatives (Table 4).
|
||||||
|---|---|---|---|---|---|---|
| Terpene/vinylborane | Methoda | Global yield (%) | endo/exo | endo Re/Si (R/S) | exo Re/Si (R/S) | Re/Si |
| a Method A: (1) BCl3 (1 M in hexanes, 1 mL), Et3SiH (1 equiv.), alkene (1.1 equiv.), −10 °C to RT, 1 h; (2) tributilvinylstannane (1 equiv.), cyclopentadiene (5 equiv.), 0 °C to RT, 3 h; (3) THF (3 mL), Et3N (1 mL), NaOH 3 N (3 mL), H2O2 30% (3 mL), 0 °C to RT, 15 h. Method B: (1) CH2Cl2 (1 mL), BBr3 (1 mmol), Et3SiH (1 equiv.), alkene (1.1 equiv.), −40 °C to RT, 2 h; (2) same as Method A, 5 h; (3) same as Method A. Method C: (1) HBBr2·SMe2 (1 M in CH2Cl2, 1 mL), alkene (1.1 equiv.), −10 °C to reflux, 2 h; (2) tributilvinylstannane (1 equiv.), cyclopentadiene (5 equiv.), 0 °C to reflux, 5 h, then RT, 15 h; (3) same as Method A, 4 h.b Compounds 16′a and 16′b are the enantiomers of 16a and 16b, respectively. | ||||||
| (+)-α-Pinene/16′ab | A | 51 | 80:20 |
53:47 |
50:50 |
51:49 |
| (+)-(2)-Carene/17a | A | 34 | 76:24 |
30:70 |
48:52 |
39:61 |
| (+)-(3)-Carene/18a | A | 54 | 79:21 |
62:38 |
48:52 |
55:45 |
| (−)-Myrtenol/19a | A | 21 | 76:24 |
48:52 |
50:50 |
49:51 |
| 13/20a | A | 27 | 78:22 |
47:53 |
44:56 |
46:54 |
| 14/21a | A | 24 | 79:21 |
45:55 |
45:55 |
45:55 |
| 15/22a | A | 26 | 78:22 |
47:53 |
50:50 |
49:51 |
| (+)-Longifolene/23a | A | 42 | 79:21 |
44:56 |
50:50 |
47:53 |
| (−)-Camphene/24a | A | 41 | 78:22 |
48:52 |
52:48 |
50:50 |
| (+)-α-Pinene/16′bb | B | 36 | 90:10 |
50:50 |
50:50 |
50:50 |
| (+)-(2)-Carene/17b | B | 35 | 91:9 |
49:51 |
50:50 |
50:50 |
| (+)-(3)-Carene/18b | B | 32 | 93:7 |
48:52 |
50:50 |
49:51 |
| 13/20b | B | 27 | 86:14 |
48:52 |
50:50 |
49:51 |
| 15/22b | B | 30 | 96:4 |
45:55 |
50:50 |
48:52 |
| (+)-Longifolene/23b | B | 32 | 91:9 |
47:53 |
50:50 |
49:51 |
| (−)-Camphene/24b | B | 28 | 93:7 |
50:50 |
50:50 |
50:50 |
| (+)-(2)-Carene/17b | C | 10 | 60:40 |
45:55 |
50:50 |
48:52 |
| (+)-(3)-Carene/18b | C | 23 | 74:26 |
50:50 |
49:51 |
50:50 |
(+)-Longifolene and (−)-camphene with exocyclic double bonds, which generate primary alkylhalovinylboranes 23 and 24 respectively, were also included in the experimental study (Scheme 6).
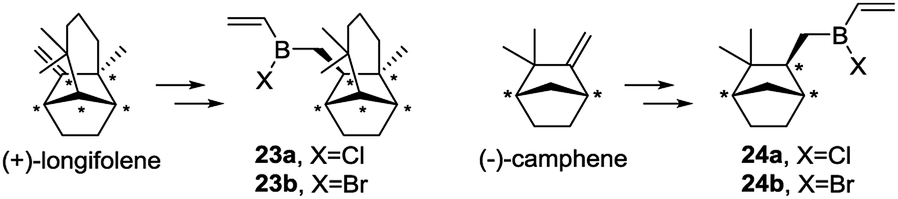 | ||
| Scheme 6 (+)-Longifolene and (−)-camphene and derived alkylhalovinylboranes 23 and 24. | ||
Based on the literature, and the results of our theoretical calculations, we assumed that different reactivities would be observed for boron trichloride and tribromide (Methods A and B). However, only differences in reactivity were observed in the initial steps, which had to be initiated at −40 °C for boron tribromide to avoid that temperatures increased and secondary reactions took place. 11B NMR indicated that transmetallation and Diels–Alder reactions occurred at similar rates. On the other hand, when HBBr2·SMe2 was used (Method C), much lower reactivity was observed, probably due to the lower electron deficiency on boron of the dimethyl sulfide complexes of the reacting species in the studied sequence. As a consequence, reaction times and temperatures had to be increased and yields dropped considerably perhaps as a result of secondary reactions.
In general, global yields for chiral terpenes and synthetic derivatives were comparable to those previously obtained with achiral cyclohexene, although much lower numbers were obtained for HBBr2·SMe2 and consequently only (+)-(2)- and (+)-(3)-carene were tested with Method C.
Facial selectivities (Re/Si) and resulting enantiomeric ratios and absolute configurations (R/S) of the products of oxidation (endo- and exo-5-norbornen-2-ol, 9) were determined by 1H NMR using a methodology developed in our group that involves the use of (S)-(+)-O-acetylmandelic acid as chiral derivatising agent.42,43 Endo-selectivities were good to excellent. In particular, bromovinylboranes obtained with BBr3 (Method B) gave endo/exo ratios higher than 86:14, with vinylborane 22b derived from synthetic analogue 15 displaying the best endo-selectivity (endo/exo 96:4). When BCl3 was used, (+)-α-pinene-derived vinylborane 16′a showed the highest endo-selectivity (endo/exo 80:20). Disappointingly, facial selectivities were low. The chlorovinylborane derived from (+)-2-carene 17a exhibited the highest chiral induction (Re/Si 30/70 for the major endo diastereoisomer). Although we hoped to get better enantiomeric ratios, such value is considerably higher than those obtained for other Diels–Alder reactions of boron-substituted dienophiles (ee < 10%), such as the one of dialkylvinylborane 1, derived from (+)-camphene, with 2-phenyl-1,3-butadiene (Scheme 1).8,44,45
As can be observed in Tables 3 and 4, experimental endo-selectivities for the Diels–Alder reactions of chlorovinylboranes were correctly predicted by the theoretical calculations. In contrast, computed endo-selectivities for bromovinylboranes were lower than those for chlorovinylboranes and than the experimental values. In general, experimental facial selectivities were in accordance to the ones computed in the theoretical study. In addition, the calculations predicted that activation free energies for bromovinylboranes would be 2 kcal mol−1 lower than those for the chloro counterparts. However, experimental yields were comparable, and in some cases lower to those obtained with chlorovinylboranes. We believe that this is caused by the great reactivity of BBr3, which decomposes rapidly during manipulation and generates experimental difficulties.
To determine the scope of the studied reactions, we performed preliminary experiments for the Diels–Alder reactions of alkylchlorovinylboranes 16′a–18a with other dienes (1,3-cyclohexene, isoprene and trans,trans-1,4-diphenyl-1,3-butadiene) (Scheme 7).
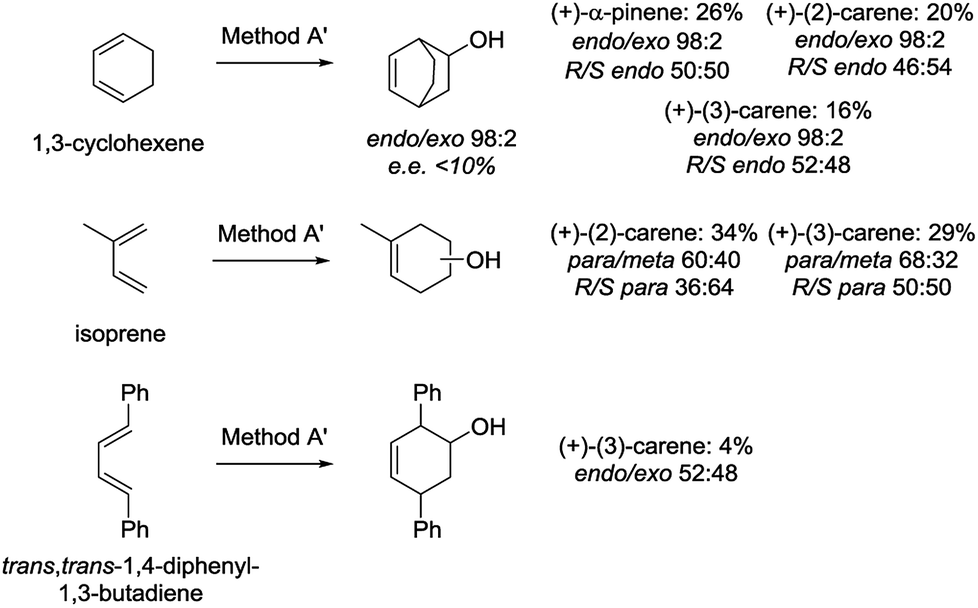 | ||
| Scheme 7 Diels–Alder reactions of alkylchlorovinylboranes 16′a–18a with other dienes. | ||
Reaction conditions were the same as for cyclopentadiene (Method A, Table 4), except that the transmetallation-Diels–Alder reaction step was carried out at reflux for 5 h (Method A′). Global yields for 1,3-cyclohexene and isoprene were lower than those obtained with cyclopentadiene (16–34%) and poor for trans,trans-1,4-diphenyl-1,3-butadiene. Furthermore, the endo/exo selectivity for 1,3-cyclohexene was excellent (endo/exo 98:2), while the para/meta regioselectivity for isoprene above 60:40. In general, enantiomeric excesses for both dienes were <10%, except for the reaction of isoprene with the chlorovinylborane derived form (+)-(2)-carene 17a (R/S para 36:64). This indicates that more experiments should be performed to optimise the reaction conditions.
Conclusions
In this investigation we explored the synthesis and the Diels–Alder reactivity of alkylhalovinylboranes with computational and experimental methods. We verified the viability of our initial postulate that the replacement of an alkyl group in dialkylvinylboranes by a halogen on the boron atom of the dienophile increases the reactivity of the vinylborane significantly. Overall, experimental results were in line with those predicted by theoretical calculations. Alkylhalovinylboranes displayed high endo/exo selectivities, in particular for bromovinylboranes. Unfortunately, in general enantioselectivities obtained for the oxidized products were low. However, the highest value obtained with (+)-2-carene-derived chlorovinylborane (17a) was much higher than those described in the literature for Diels–Alder reactions of other boron-substituted dienophiles. This work contributes to a little-explored area of study, as is the use of chiral vinylboranes in asymmetric cycloaddition reactions. Finally, this novel example of the use of alkylhalovinylboranes in synthesis highlights the practical potential of such organoboron species, which may be applied to other transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank CONICET, ANPCyT and Universidad Nacional de Rosario for financial support.Notes and references
- G. Hilt and P. Bolze, Synthesis, 2005, 2091–2115 CrossRef CAS.
- D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, Wiley-VCH Verlag GmbH, Weinheim, 2005 Search PubMed.
- D. A. Singleton and J. P. Martinez, J. Am. Chem. Soc., 1990, 112, 7423–7424 CrossRef CAS.
- D. A. Singleton and J. P. Martinez, Tetrahedron Lett., 1991, 32, 7365–7368 CrossRef CAS.
- D. A. Singleton, J. P. Martinez and J. V. Watson, Tetrahedron Lett., 1992, 33, 1017–1020 CrossRef CAS.
- D. A. Singleton, J. P. Martinez, J. V. Watson and G. M. Ndip, Tetrahedron, 1992, 48, 5831–5838 CrossRef CAS.
- D. A. Singleton and S. W. Leung, J. Org. Chem., 1992, 57, 4796–4797 CrossRef CAS.
- D. A. Singleton, J. P. Martinez and G. M. Ndip, J. Org. Chem., 1992, 57, 5768–5771 CrossRef CAS.
- D. A. Singleton, K. Kim and J. P. Martinez, Tetrahedron Lett., 1993, 34, 3071–3074 CrossRef CAS.
- J.-M. Jego, B. Carboni, A. Youssofi and M. Vaultier, Synlett, 1993, 595, 597 Search PubMed.
- D. A. Singleton and A. M. Redman, Tetrahedron Lett., 1994, 35, 509–512 CrossRef CAS.
- D. A. Singleton and Y. K. Lee, Tetrahedron Lett., 1995, 36, 3473–3476 CrossRef CAS.
- D. A. Singleton, S. W. Leung, J. P. Martinez and Y. K. Lee, Tetrahedron Lett., 1997, 38, 3163–3166 CrossRef CAS.
- D. A. Singleton and S. W. Leung, J. Organomet. Chem., 1997, 544, 157–161 CrossRef CAS.
- Y.-K. Lee and D. A. Singleton, J. Org. Chem., 1997, 62, 2255–2258 CrossRef CAS PubMed.
- N. Noiret, A. Youssofi, B. Carboni and M. Vaultier, J. Chem. Soc., Chem. Commun., 1992, 1105–1107 RSC.
- R. A. Batey, D. Lin, A. Wong and C. L. S. Hayhoe, Tetrahedron Lett., 1997, 38, 3699–3702 CrossRef CAS.
- M. Zaidlewicz, J. R. Binkul and W. Sokół, J. Organomet. Chem., 1999, 580, 354–362 CrossRef CAS.
- S. C. Pellegrinet, M. A. Silva and J. M. Goodman, J. Comput.-Aided Mol. Des., 2004, 18, 209–214 CrossRef CAS PubMed.
- H. C. Brown and P. K. Jadhav, J. Org. Chem., 1984, 49, 4089–4091 CrossRef CAS.
- U. S. Racherla, Y. Liao and H. C. Brown, J. Org. Chem., 1992, 57, 6614–6617 CrossRef CAS.
- S. C. Pellegrinet, unpublished results.
- U. P. Dhokte, R. Soundararajan, P. V. Ramachandran and H. C. Brown, Tetrahedron Lett., 1996, 37, 8345–8348 CrossRef CAS.
- To the best of our knowledge, the only example of alkylhalovinylboranes that has been described to date is chloromethylvinylborane, but its reactivity as Diels–Alder dienophile has not been investigated. C. D. Good and D. M. Ritter, J. Am. Chem. Soc., 1962, 84, 1162–1166 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09W, Gaussian, Inc., 2009 Search PubMed.
- S. C. Pellegrinet, M. A. Silva and J. M. Goodman, J. Am. Chem. Soc., 2001, 123, 8832–8837 CrossRef CAS PubMed.
- A. Rastelli, R. Gandolfi, M. Sarzi-Amadé and B. Carboni, J. Org. Chem., 2001, 66, 2449–2458 CrossRef CAS PubMed.
- M. A. Silva, S. C. Pellegrinet and J. M. Goodman, J. Org. Chem., 2002, 67, 8203–8209 CrossRef CAS PubMed.
- M. A. Silva, S. C. Pellegrinet and J. M. Goodman, Arkivoc, 2003, 2003, 556–565 Search PubMed.
- M. A. Silva, S. C. Pellegrinet and J. M. Goodman, J. Org. Chem., 2003, 68, 4059–4066 CrossRef CAS PubMed.
- D. Hallooman, D. Cudian, M. Ríos-Gutiérrez, L. Rhyman, I. A. Alswaidan, M. I. Elzagheid, L. R. Domingo and P. Ramasami, ChemistrySelect, 2017, 2, 9736–9743 CrossRef CAS.
- A. Atalay and R. Abbasoglu, J. Phys. Org. Chem., 2018, e3893, DOI:10.1002/poc.3893.
- B. Mennucci and J. Tomasi, J. Chem. Phys., 1996, 106, 5151–5158 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211–7218 CrossRef CAS.
- A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066–4073 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: a Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, Cambridge, UK, New York, 2005 Search PubMed.
- Analysis of the computed global and local reactivity indices of the reactants is in line with FMO results (see the ESI†).
- The Diels–Alder reactions can take place by different mechanisms: synchronical mechanism, biradicaloidal one step-mechanism, stepwise biradical mechanism (non-polar mechanisms) or one step-two stage mechanism, stepwise zwitterionic mechanism (polar mechanisms). The Diels–Alder reactions under study can be classified as one step-two stage reactions. For example see: (a) R. Jasiński, React. Kinet., Mech. Catal., 2016, 119, 49–57 CrossRef; (b) D. A. Singleton, B. E. Schulmeier, C. Hang, A. A. Thomas, S.-W. Leung and S. R. Merrigan, Tetrahedron, 2001, 57, 5149–5160 CrossRef CAS; (c) R. Jasiński, M. Kubik, A. Łapczuk-Krygier, A. Kącka, E. Dresler and A. Boguszewska-Czubara, React. Kinet., Mech. Catal., 2014, 113, 333–345 CrossRef; (d) R. Jasiński, Comput. Theor. Chem., 2014, 1046, 93–98 CrossRef; (e) R. Jasiński, J. Fluorine Chem., 2018, 206, 1–7 CrossRef.
- The computed values of electron density transfer (GEDT) for the TSs (see the ESI†) also suggested that the Diels–Alder reactions under study can be classified as polar Diels–Alder reactions (P-DA).
- P. L. Pisano, A. M. Sarotti and S. C. Pellegrinet, Tetrahedron Lett., 2009, 50, 6121–6125 CrossRef CAS.
- A. M. Sarotti, P. L. Pisano and S. C. Pellegrinet, Arkivoc, 2011, 2011, 343–357 Search PubMed.
- J. D. Bonk and M. A. Avery, Tetrahedron: Asymmetry, 1997, 8, 1149–1152 CrossRef CAS.
- N. Grimblat, A. M. Sarotti, P. L. Pisano and S. C. Pellegrinet, New J. Chem., 2016, 40, 1966–1969 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR spectra, computational methods, FMOs and global and local reactivity indices of reactants, optimized geometries of the transition structures (TSs) not included in the paper and charge transfer (GEDT) of TSs, cartesian coordinates, absolute energies including zero-point energy corrections, free energies and number of imaginary frequencies of all the stationary points reported in the paper and values of imaginary frequencies of all transition structures. See DOI: 10.1039/c8ra07089j |
| This journal is © The Royal Society of Chemistry 2018 |