DOI:
10.1039/C8RA07113F
(Review Article)
RSC Adv., 2018,
8, 40720-40730
Schiff base complex conjugates of bovine serum albumin as artificial metalloenzymes for eco-friendly enantioselective sulfoxidation†
Received
26th August 2018
, Accepted 23rd November 2018
First published on 5th December 2018
Abstract
Artificial metalloenzymes (BSA-ML) have been prepared by non-covalent insertion of transition metal Schiff-base complexes, ML (L = 2-hydroxynaphthalen-1-naphthaldehyde and 3,4-diaminobenzenesulfonic acid; M = Co, Mn, V, Fe, Cr), into bovine serum albumin (BSA) as the host protein and were characterized by UV-visible spectroscopy, ESI-TOF mass spectrometry and molecular docking studies. The catalytic activities of the BSA-ML in the selective oxidation of various prochiral sulfides in aqueous media, using H2O2 as oxidant, have been evaluated. During the optimization process, pH and the concentrations of catalyst and oxidant were found to have a remarkable influence on both yield and enantioselectivity. In certain cases, BSA-ML gave satisfactory results in the oxidation of organic sulfides to sulfoxides (up to 100% conversion, 100% chemoselectivity, 96% ee and 500 h−1 turnover frequency).
Introduction
Asymmetric selective oxidation of sulfides to sulfoxides is an important reaction that deserves much attention because enantiomerically pure sulfoxides are valuable compounds that have utility as powerful chiral auxiliaries,1–3 ligands,4,5 organocatalysts6,7 in asymmetric organic synthesis,8–10 and as active pharmaceutical ingredients.11–15 Traditionally, catalysis has been divided into three subdisciplines: heterogeneous, homogeneous, and enzymatic catalysis. Although promising metal catalysts16 have been developed and show encouraging practical potential, issues such as compound stability, efficacy, regio- and stereoselectivity, and environmental toxicity remain to be addressed.17 Enzymatic activity is reduced by the poor stability of enzymes outside their natural environment.18,19 In the past decade, artificial metalloenzymes have emerged as attractive alternatives that complement both homogeneous catalysts and enzymes.20–23 Artificial metalloenzymes are hybrid catalysts that incorporate non-natural metal cofactors into biological scaffolds.24–28 Compared to their natural counterparts, they are expected to have less complex structures but similar catalytic activity, better solubility in a wide range of solvents and higher stability, especially at high temperatures.29
Schiff-bases are a vital class of organic compounds.30 Schiff base compounds have been extensively exploited as ligands for mono- or multinuclear macrocyclic or macroacyclic metal complexes, which are useful as biomimetic catalysts31 in sulfoxidation.30,32–36 In particular, the use of transition metal complexes as catalysts,37 mainly based on titanium,38,39 vanadium,40,41 manganese,42,43 and iron,44,45 is an area of current interest.46,47
Serum albumins, the most abundant blood proteins in mammals, are globular, water-soluble, un-glycosylated transport proteins.48 They are not only able to recognize and accept a broad range of substrates,48 but can also discriminate between the enantiomers of a chiral molecule.49 This makes them promising candidates for the design of artificial metalloenzymes. Gross et al.50 embedded bis sulfonated metal corroles containing iron or manganese in to human, bovine, porcine, rabbit and sheep serum albumins. These new ‘‘hemozymes’’ were able to catalyze the sulfoxidation of thioanisole and some analogs in aqueous media. Using hydrogen peroxide as oxidant, sulfoxides were obtained in up to 74% ee and moderate yield. Bovine serum albumin (BSA) is the most widely used albumin because it is readily available and low cost.51,52 However, until very recently, little research has been conducted on the asymmetric oxidation of sulfides catalyzed by artificial metalloenzymes based on BSA.
Increasing global concerns in recent years have led to an exponential increase in the need for economically and environmentally compatible chemical processes.53 Consequently, the concept of green chemistry has emerged, which endorses the use of environmentally friendly reaction conditions.54 Sulfoxidation is generally achieved using oxidizing agents such as m-chloroperbenzoic acid,55,56 cumene hydroperoxide (CHP),57 NaClO,58 NaIO4,59 oxone,60 KMnO4 (ref. 61) and dimethyldioxirane.62 Many of these oxidants are known to have undesirable characteristics that include toxicity, formation of by-products, extended reaction times, and low yields.62 Reactions that use eco-friendly reagents and solvents such as hydrogen peroxide (H2O2) and water therefore offer great advantages.63–66
Inspired by the success of the BSA-cobalt(II) Schiff base complex hybrid that we previously designed as a biocatalyst for asymmetric sulfoxidation,36 we have attempted to expand the range of metal cofactors that can be used in the design of artificial metalloenzymes. Using BSA as the biomolecular scaffold, we have explored the application of these new catalysts in the enantioselective oxidation of different sulfides. Beside Co(III), Mn(III) and Fe(III) complexes we have reported,67 in the current, we report the synthesis and characterization of V(V) and Cr(III) complexes with a Schiff base ligand derived from 2-hydroxynaphthalen-1-naphthaldehyde and 3,4-diaminobenzenesulfonic acid. A series of BSA-ML Schiff base complex hybrids were then designed as biocatalysts. Continuing our efforts to develop simple and green routes, we have accomplished asymmetric sulfoxidation in the presence of water as solvent under mild conditions. Of the designed metalloenzymes, BSA-VL proved to be the most efficient, affording high yield and turnover frequency (TOF) for the conversion of all substrates tested into the corresponding sulfoxides. Up to 94% enantiomeric purity of 4-methoxyphenyl methyl sulfoxide was achieved.
Experimental section
Materials
BSA was purchased from Sigma-Aldrich (St. Louis, MO, USA). All of the sulfides were obtained from Energy Chemical (Shanghai, China). The preparation of 3,4-bis((2-hydroxynaphthalen-1-yl)methyleneamino)benzenesulfonic acid (NaH2L) has been described previously.67 Standard stock solutions (1 mM) of the ML complexes (M = Co, Mn, V, Fe, Cr) and BSA were prepared in dimethyl sulfoxide (DMSO) and 50 mM Tris (trihydroxymethyl aminomethane) buffer solution, pH 8.0, respectively.
Physical measurements methods
UV-visible spectra were recorded on a Cary 100 UV-visible spectrophotometer. The FT-IR spectra were recorded on a Perkin-Elmer Spectrum One FT-IR spectrophotometer with a germanium attenuated total reflection (ATR) accessory, a DTGS KBr detector and a KBr beam splitter ratio. Elemental analyses for C, H and N were carried out on a Model 2400 II, Perkin-Elmer elemental analyzer. ESI-MS (electrospray ionization mass spectrum) spectra were recorded on a Bruker HCT Electrospray Ionization Mass Spectrometer. Electrospray ionization-time of flight mass spectrometry (ESI-TOF MS) spectra was measured on an LCQ/AD Quadrupole Ion Trap ESI-MS. HPLC experiments were carried out using UV2302II/P2302II high performance liquid chromatograph. Optical rotations were measured with a WXG-4 polarimeter.
Synthesis of the complexes
All of the complexes were prepared by hydrothermal reaction. The preparations of ML (M = Co, Mn, Fe) have been described previously.67 A mixture of metal salt (0.3 mmol, V2O5 for VL and Cr(NO3)3·9H2O for CrL), NaH2L (0.3 mmol), and methanol (15 mL) was placed in a 25 mL Teflon-lined stainless steel vessel. The reaction mixtures were heated at high temperature (120 °C for VL and 80 °C for CrL) for 72 h and then cooled to room temperature at a controlled rate (10 °C h−1 for VL and 5 °C h−1 for CrL). The resulting crystalline products were washed with methanol and dried in air.
Complex VL. Color: atrovirens. Yield: 58% (based on V(V)). Anal. calc. for C28H17VN2O6S: C, 60.01; H, 3.06; N, 5.00. Found: C, 61.02; H, 3.65; N, 5.14%. EIS-MS (m/z): [C28H18VN2O6S]+, 561.03. FT-IR (KBr phase, cm−1): 3416s, 3135s, 1614m, 1597m, 1530s, 1357s, 1196s, 1039s, 829m, 751m, 701w, 563w.
Complex CrL. Color: reddish brown. Yield: 48% (based on Cr(III)). Anal. calc. for C29H23CrN2O7S: C,58.48; H, 3.89; N, 4.70. Found: C, 58.28; H, 3.73; N, 4.77%. EIS-MS (m/z): [C28H18CrN2O5S]+, 546.15. FT-IR (KBr phase, cm−1): 3121s, 1615w, 1533s, 1390s, 1357s, 1164s, 1036s, 824m, 752m, 697m.
Crystal structure determination
X-Ray structure analysis was performed at 293 K using Agilent Supernova diffractometer (Mo, λ = 0.71073 Å). The structure was collected at 293(2) K and solved by direct methods using SHELxs-97 (ref. 68) and refined by full-matrix least-squares techniques against F2 with SHELxs-97.69 Anisotropic thermal parameters were assigned to all non-hydrogen atoms. The organic hydrogen atoms were generated geometrically, the hydrogen atoms of the water molecules were located from difference maps and refined with isotropic temperature factors. Analytical expressions of neutral-atom scattering factors were employed, and anomalous dispersion corrections were incorporated. The details of the crystal data were summarized in Table 1, and selected bond lengths and angles for complex CrL were listed in Table S1.†
Table 1 Crystal data and structure refinement for CrL
| Formula |
0.5(C58H40CrN4O14S2) |
γ (°) |
90.00° |
| Formula weight |
592.53 |
V (Å3) |
2522.2 (5) |
| Crystal system |
Orthorhombic |
Z |
4 |
| Space group |
Pnma |
Dc (g m−3) |
1.560 |
| a (Å) |
8.3302 (9) |
Goodness-of-fit on F2 |
1.061 |
| b (Å) |
23.634 (3) |
θ range for data collection (°) |
2.9 to 21.3° |
| c (Å) |
12.8113 (14) |
Reflections collected/unique |
25432/2266 [R(int) = 0.069] |
| α (°) |
90.00° |
Final R indices [I > 2σ(I)] |
R1 = 0.0476 ωR2 = 0.1251 |
| β (°) |
90.00° |
R indices (all data) |
R1 = 0.0758 ωR2 = 0.1370 |
Synthesis of hybrids
The standard stock solution of BSA (1 mL) was mixed with ML standard stock solution (1 mL) and incubated overnight at 0 °C. The mixture was dialyzed (3 cycles) in an ice bath, leading to the separation of the protein-complex hybrid from the complex solution. The resulting solution was then purified by size-exclusion chromatography (Sephadex G-75) and concentrated. The protein concentration was estimated by the Coomassie Brilliant Blue method.70
Molecular docking
Docking studies are regarded as a good approach to observe potential binding locations of a drug in a protein.71 The docking calculations were performed using AutoDock 4.2 (http://autodock.scripps.edu/, free software), which combines a rapid energy evaluation through pre-calculated grids of affinity potentials with a variety of search algorithms.72 The crystal structure of BSA (PDB ID: 3V03) was obtained from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). All water molecules were removed, and then polar hydrogen atoms were added to the BSA molecule.73 The partial atomic charges of BSA and ML were calculated using Gasteiger–Marsili74 and Kollman methods,75 respectively. The conformational search was selected by using the Lamarckian genetic algorithm (LGA) with the same parameters for each docking. Finally, according to the AutoDock scoring function, the dominating configuration of the BSA-ML with minimum binding energy was obtained.76,77 The minimum Gibbs free energy binding mode was visualized in PyMol (http://www.pymol.org/).
General procedure for enantioselective sulfoxidation
The catalytic selective oxidation of sulfide was carried out in a glass flask reactor (10 cm3) equipped with a magnetic stirrer and immersed in a thermostated bath. Sulfide (0.27 mmol) and the catalyst, BSA-ML (2.7 μmol), were added to 2 mL of phosphate buffer (PB) solution at pH 5.1. After stirring for 5 h, the oxidant (H2O2, 0.405 mmol, 30% w/w) was added to the solution with stirring. The reaction mixture was stirred for a further 20 h at ambient temperature (25 °C), and then quenched with sodium sulfite solution. The product and unreacted organic substrates were extracted with dichloromethane (5 × 2 mL), dried over anhydrous sodium sulfate and distilled under reduced pressure to remove excess solvent. The crude product was purified by column chromatography on silica gel. Control experiments were carried out under the same reaction conditions but without BSA in the reaction mixture. All of the products obtained in this study are known compounds and their spectroscopic data were identical to those reported in the literature.36 The enantiomeric excess (ee) values of the corresponding chiral sulfoxides were determined by HPLC analysis using a chiral column (Daicel, Chiralcel, OB-H) at room temperature. ee values and chemoselectivity were calculated using the formulas: ee% = [peak area (S − R)/(S + R)] × 100%; chemoselectivity% = [peak area SO/(SO + SO2)] × 100%, SO = sulfoxide, SO2 = sulfone. Conversions were based on sulfide substrate; yields were referred to isolated product after column chromatography and were based on substrate; turnover frequencies (TOF) were determined by dividing the concentration of product by the catalyst concentration per hour. The configuration of sulfoxides product from these reactions was proven by comparing the specific rotation with the literature values.36
Results and discussion
Description of the crystal structure
Single-crystal X-ray structural analysis shows that complex CrL crystallize in space groups of Pnma (Table 1) and the asymmetric unit consists of half a mirror symmetric [CrL(H2O)(CH3OH)] super-molecule. As illustrated in Fig. S1,† The central atom Cr1 lying on a mirror plane coordinates to two imino nitrogen atom (N1, N1A, symmetry codes: Ax, −y + 1/2, z) with the Cr–N distance is 1.991(3) Å, two phenolic oxygen atoms (O6, O6A) from one Schiff base ligand, an water atom (O4) and methanol atom (O5) with the Cr–O distances fall in the range 1.983(3)–1.994(4) Å. to form an distorted octahedral geometry. The adjacent [CrL(H2O)(CH3OH)] super-molecules further connect each other through H-bond interactions.
Spectroscopic characterization of the BSA-ML
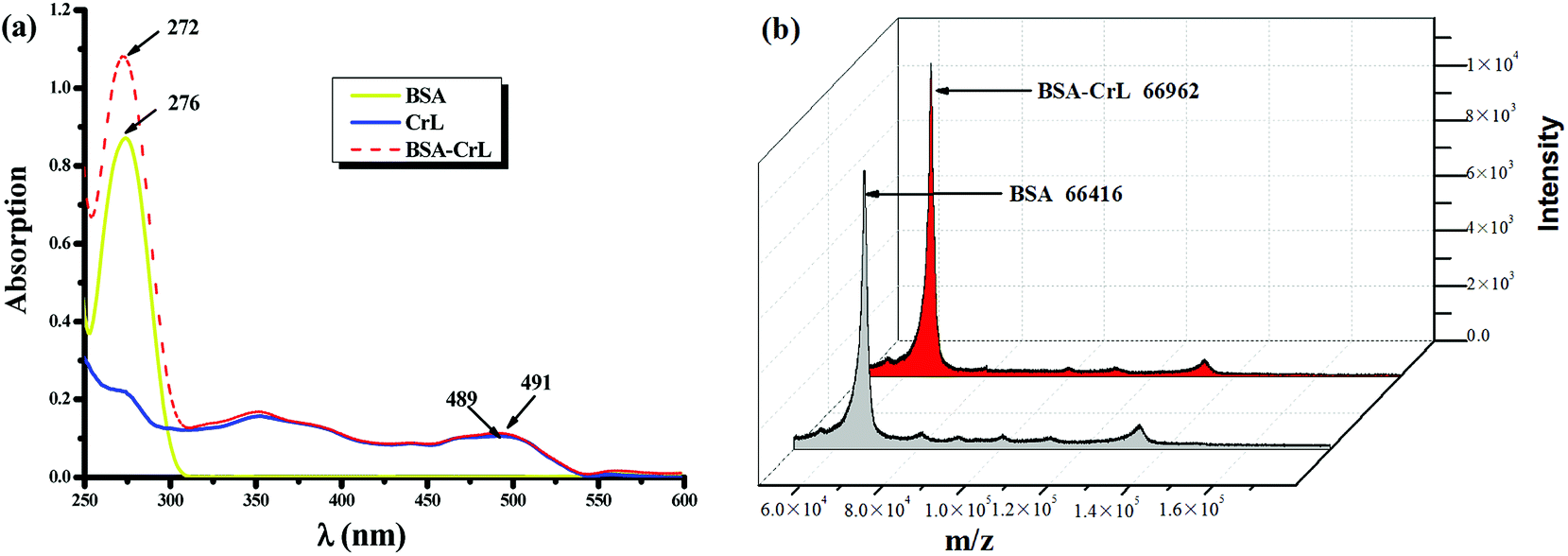
The binding interaction between BSA and ML complex was characterized using UV-visible spectroscopy and ESI-TOF mass spectrometry. BSA and BSA-ML hybrid were at equal concentrations and their UV-visible spectra were dominated by a single charge transfer (CT) band in the UV region. The UV-visible spectra of BSA-ML (M = Co, Mn, Fe) have been described previously,67 and the spectra of BSA-CrL and BSA-VL are shown in Fig. 1(a) and S2,† respectively. The spectra showed that attachment of CrL to BSA resulted in a shift of ëmax from 276 to 272 nm, accompanied by a substantial increase in its intensity, suggesting formation of the BSA-CrL hybrid (Fig. 1(a)). When comparing the CrL complex and BSA-CrL spectra, the presence of BSA induced only a slight shift of the band (2 nm) in the UV region, together with a slight increase in its intensity. For further analysis of the interaction between CrL and BSA, the ESI-TOF mass spectrum was recorded (Fig. 1(b)). The mass spectrum of a mixture of CrL complex and BSA gave two peaks, one of which corresponded to the calculated mass of the BSA-CrL hybrid (m/z 66961), in which 1 equiv. of the catalyst was attached to BSA (m/z 66416), indicating successful anchoring.78,79 The ESI-TOF spectra of the other four complexes are shown in the ESI (Fig. S3†).
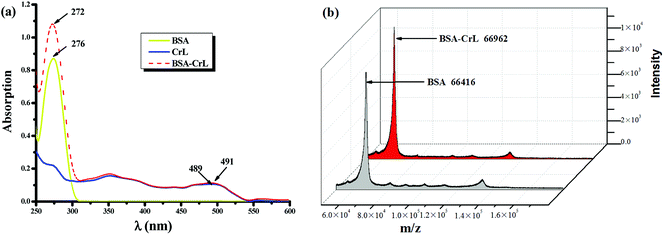 |
| | Fig. 1 (a) UV-visible spectra of 20 ìM BSA (yellow solid line), 20 ìM CrL complex (blue solid line) and 20 ìM BSA-CrL hybrid (red dashed line) in 0.05 M PBS buffer pH 7.45; (b) ESI-TOF MS spectrum of BSA and BSA-CrL. | |
Molecular docking studies
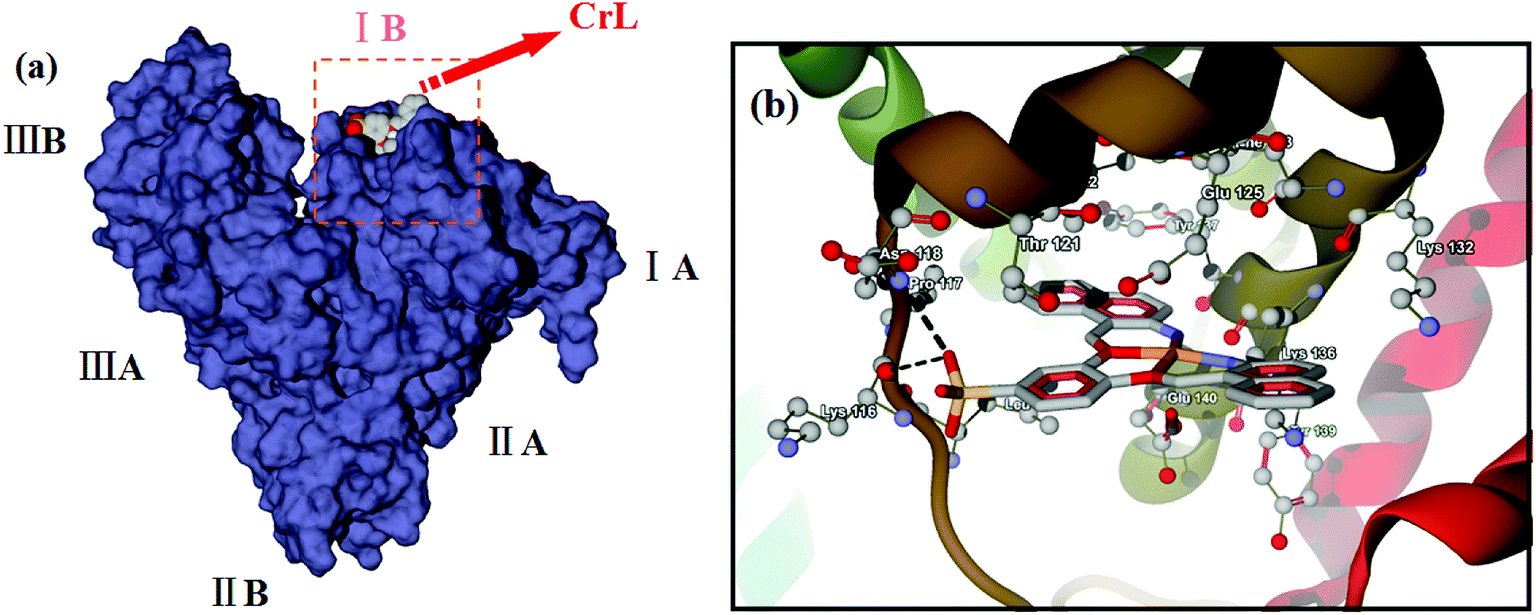
Molecular docking has become a useful tool in recent years for understanding the binding interactions between synthesized compounds and biological macromolecules. To explore the potential binding site of CrL complex on BSA, a molecular docking study was performed. BSA is a globular protein composed of three homologous domains (I, II, III), each of which is divided into subdomains A and B.80,81 Fig. 2(a) shows the lowest energy binding mode found for CrL binding to BSA. Similarly to artificial metalloenzymes that we have reported previously,36 the binding site of CrL was located in subdomain IB (site I), which contains a hydrophobic, polar environment. A close-up view of this docked form is shown in Fig. 2(b). The amino acid residues closest to the ligand, including Glu-125, Lys-136 and Thr-121 etc., suggest hydrophobic and van der Waals interactions to the compound,82,83 consistent with the spectroscopic data.
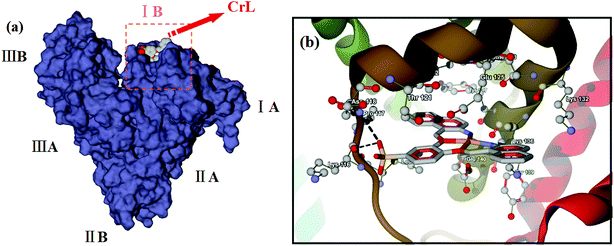 |
| | Fig. 2 (a) Is the lowest energy binding mode of CrL to BSA; (b) is the close-up view of binding site of CrL on BSA corresponding to (a), and the selected amino acid residues are shown by stick model. | |
Catalytic activity in asymmetric sulfoxidation
Initially, the oxidation reactions were carried out with thioanisole as a model substrate and H2O2 as oxidant in water at room temperature (25 °C). The value of the hybrid was demonstrated by comparing the catalytic properties of ML with the BSA-ML hybrid. The results are presented in Table 2. When experiments were run in the absence of BSA, the reactions proceeded with low chemoselectivity and almost zero enantioselectivity (entries 1–5). In contrast, when the reaction was run in the presence of BSA-ML as catalyst, the ee values increased significantly (entries 6–10) and thioanisole sulfoxide was obtained as the major product with high chemoselectivity (95–100%). These results strongly suggest that the BSA scaffold enhances both the reactivity and enantioselectivity of metal catalysts. The protein scaffold provides a hydrophobic pocket that may favor collision with substrate.84 Similarly, many reports36,85–88 have found that the host protein has an influence on reactivity and enantioselectivity.
Table 2 Enantioselective oxidation of thioanisolea
| Entry |
Catalyst |
Conversion (%) |
Yield (%) |
Chemoselectivity (%) |
ee (%) |
Reactions were performed in PB (2 mL, pH 5.1) at room temperature for 20 h. The ratios of H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thioanisole:catalyst (2.7 μmol) were 150:100:1. The sulfoxide of thioanisole was in the R configuration. :thioanisole:catalyst (2.7 μmol) were 150:100:1. The sulfoxide of thioanisole was in the R configuration. |
| 1 |
CoL |
65 |
41 |
64 |
<5 |
| 2 |
MnL |
54 |
39 |
72 |
<5 |
| 3 |
VL |
62 |
40 |
65 |
<5 |
| 4 |
FeL |
66 |
50 |
77 |
<5 |
| 5 |
CrL |
59 |
44 |
75 |
<5 |
| 6 |
BSA-CoL |
88 |
88 |
100 |
31 |
| 7 |
BSA-MnL |
70 |
70 |
100 |
32 |
| 8 |
BSA-VL |
86 |
81 |
95 |
27 |
| 9 |
BSA-FeL |
90 |
90 |
100 |
16 |
| 10 |
BSA-CrL |
79 |
77 |
98 |
15 |
Influence of pH
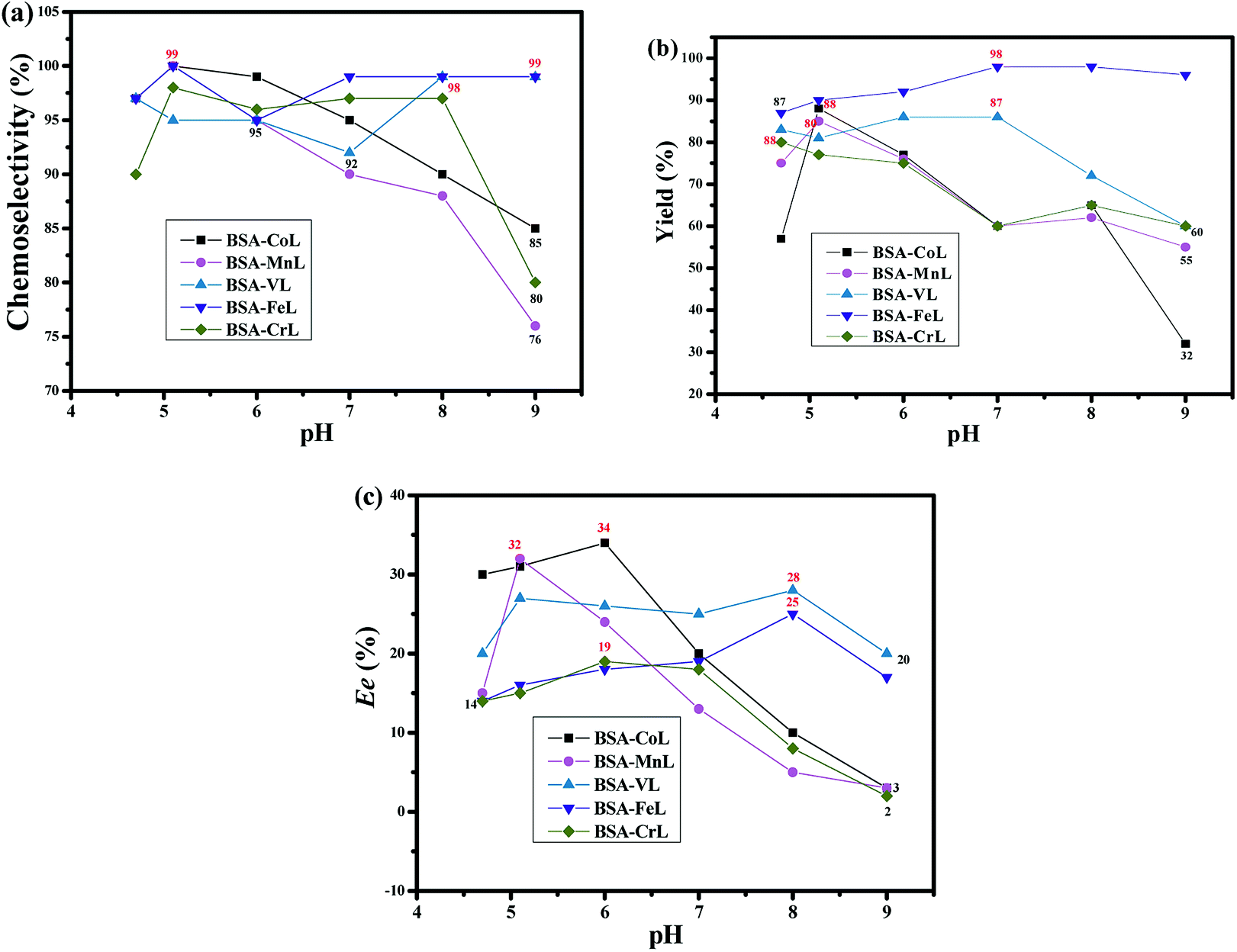
The optimal reaction conditions were determined by conducting reactions at different pH values, as well as various substrate, oxidant and catalyst concentrations. Firstly, the effect of pH values was determined in PB (pH 4.7–9) at room temperature and the results are shown in Fig. 3. The pH of the reaction medium had a highly significant effect on the activity of BSA-ML (M = Co, Mn, Cr) and ee in the oxidation of thioanisole. In the case of BSA-FeL, the pH value did not have a significant effect. In particular, the chemoselectivity was high (95–100%, Fig. 3(a)). Optimal chemoselectivities, yields and ee values were obtained at more acidic pH values with BSA-ML (M = Co, Mn, Cr), while the highest values with BSA-VL and BSA-FeL were obtained at non-acidic pH values under the same reaction conditions (Fig. 3(a)–(c)). The BSA-ML (M = Co, Mn, Cr) complexes would be easily decomposed under extreme pH conditions, which limits the available range under basic conditions. Different pH values were selected depending on the optimum enantioselectivity (pH 6 for BSA-CoL and BSA-CrL, pH 5.1 for BSA-MnL, and pH 8 for BSA-VL and BSA-FeL) (Fig. 3(c)).
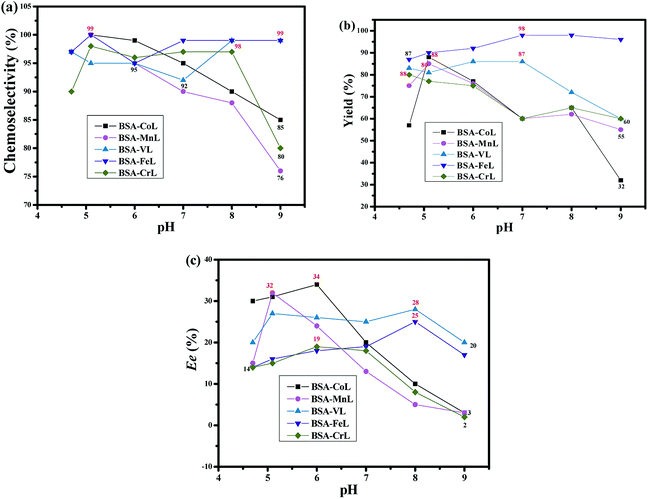 |
| | Fig. 3 (a) Chemoselectivity, (b) yield and (c) ee values under different pH values on the oxidation of thioanisole in PB at room temperature for 20 h. The mol ratios of H2O2:thioanisole:BSA-ML (2.7 μmol) were 150:100:1. The sulfoxide of thioanisole was in the R configuration. | |
Influence of substrate concentration
Full details are presented in Table 3. Overall, changes to the substrate concentration did not increase catalytic activity or the ee value. Good chemoselectivities (≥90%) were obtained when the thioanisole concentration was increased over the range of 0.034–0.338 mM. However, the clearest difference was observed with BSA-MnL (Table 3, entries 7–12). Higher concentrations of thioanisole reduced the yield and ee values, with almost no changes in the sulfoxide selectivity (>95%). The ee values increased significantly from 8% to 32% when the substrate concentration was increased from 0.034 to 0.135 mM. Accordingly, 0.068 mM (for BSA-CoL), 0.135 mM (for BSA-MnL and BSA-CrL) and 0.203 mM (for BSA-VL and BSA-FeL) were chosen as the optimum concentrations for further experiments (Table 3, note numbers in bold).
Table 3 Effect of substrate concentration on the oxidation of thioanisolea
| Entry |
Catalyst |
Concentration (mM) |
Conversion (%) |
Yield (%) |
Chemoselectivity (%) |
ee (%) |
| Reactions were performed in PB (2 mL) at room temperature for 20 h. The ratios of H2O2:thioanisole: BSA-ML were 150:100:1. pH = 6. pH = 5.1. pH = 8; the sulfoxide of thioanisole was in the R configuration. |
| 1 |
BSA-CoLb |
0.034 |
80 |
76 |
95 |
35 |
| 2 |
BSA-CoLb |
0.068 |
95 |
93 |
98 |
40 |
| 3 |
BSA-CoLb |
0.135 |
78 |
77 |
99 |
34 |
| 4 |
BSA-CoLb |
0.203 |
77 |
77 |
100 |
34 |
| 5 |
BSA-CoLb |
0.270 |
75 |
75 |
100 |
32 |
| 6 |
BSA-CoLb |
0.338 |
74 |
74 |
100 |
32 |
| 7 |
BSA-MnLc |
0.034 |
88 |
79 |
90 |
8 |
| 8 |
BSA-MnLc |
0.068 |
90 |
86 |
95 |
10 |
| 9 |
BSA-MnLc |
0.135 |
80 |
80 |
100 |
32 |
| 10 |
BSA-MnLc |
0.203 |
84 |
80 |
95 |
16 |
| 11 |
BSA-MnLc |
0.270 |
80 |
75 |
94 |
18 |
| 12 |
BSA-MnLc |
0.338 |
75 |
71 |
94 |
16 |
| 13 |
BSA-VLd |
0.034 |
50 |
49 |
98 |
25 |
| 14 |
BSA-VLd |
0.068 |
78 |
78 |
100 |
30 |
| 15 |
BSA-VLd |
0.135 |
75 |
72 |
99 |
28 |
| 16 |
BSA-VLd |
0.203 |
75 |
75 |
100 |
34 |
| 17 |
BSA-VLd |
0.270 |
81 |
81 |
100 |
33 |
| 18 |
BSA-VLd |
0.338 |
76 |
75 |
99 |
33 |
| 19 |
BSA-FeLd |
0.034 |
95 |
95 |
100 |
17 |
| 20 |
BSA-FeLd |
0.068 |
99 |
94 |
95 |
18 |
| 21 |
BSA-FeLd |
0.135 |
99 |
98 |
99 |
25 |
| 22 |
BSA-FeLd |
0.203 |
97 |
95 |
98 |
27 |
| 23 |
BSA-FeLd |
0.270 |
90 |
85 |
94 |
27 |
| 24 |
BSA-FeLd |
0.338 |
86 |
77 |
90 |
25 |
| 25 |
BSA-CrLb |
0.034 |
75 |
70 |
94 |
14 |
| 26 |
BSA-CrLb |
0.068 |
75 |
71 |
95 |
15 |
| 27 |
BSA-CrLb |
0.135 |
78 |
75 |
96 |
19 |
| 28 |
BSA-CrLb |
0.203 |
82 |
74 |
90 |
18 |
| 29 |
BSA-CrLb |
0.270 |
76 |
70 |
92 |
17 |
| 30 |
BSA-CrLb |
0.338 |
52 |
47 |
90 |
12 |
Influence of oxidant and catalyst concentrations
Having optimized the pH and substrate concentration for the oxidation of thioanisole, we next examined the effect of H2O2 and catalyst stoichiometry (with respect to substrate) on the conversion, selectivity and ee of the product (Tables 4 and 5). It was evident for all complexes except BSA-CrL that a greater amount of H2O2 increased conversion during 20 h, but decreased product selectivity by giving the over-oxidized sulfone product in excess. There was little change in the ee of the desired product. In the case BSA-CrL, increasing the oxidant concentration from 1 to 2.5 equivalents reduced conversion and selectivity, but increased the enantiomeric excess (Table 4, entries 17–20). Similar to the artificial enzyme that we reported previously, this effect may be due to oxidative destruction of the catalyst, BSA-CrL, combined with a kinetic resolution process due to excess oxidant.36 Systematic variation of the oxidant: substrate molar ratio revealed the best compromise between catalytic activity and sulfoxide ee value (Table 4, note numbers in bold).
Table 4 Effect of oxidant concentration on the oxidation of thioanisolea
| Entry |
Catalyst |
H2O2 (equiv.) |
Conversion (%) |
Yield (%) |
Chemoselectivity (%) |
ee (%) |
| Reactions were performed in PB (2 mL) at room temperature for 20 h; The ratio of thioanisole:BSA-ML was 100:1. pH = 6; the concentration of thioanisole was 0.068 mM. pH = 5.1; the concentration of thioanisole was 0.135 mM. pH = 8; the concentration of thioanisole was 0.203 mM. pH = 6; the concentration of thioanisole was 0.135 mM. The sulfoxide of thioanisole was in the R configuration. |
| 1 |
BSA-CoLb |
1 |
74 |
73 |
99 |
36 |
| 2 |
BSA-CoLb |
1.5 |
95 |
93 |
98 |
40 |
| 3 |
BSA-CoLb |
2 |
96 |
83 |
86 |
38 |
| 4 |
BSA-CoLb |
2.5 |
98 |
89 |
75 |
40 |
| 5 |
BSA-MnLc |
1 |
78 |
73 |
94 |
27 |
| 6 |
BSA-MnLc |
1.5 |
80 |
80 |
100 |
32 |
| 7 |
BSA-MnLc |
2 |
85 |
77 |
90 |
28 |
| 8 |
BSA-MnLc |
2.5 |
86 |
71 |
82 |
28 |
| 9 |
BSA-VLd |
1 |
75 |
75 |
100 |
34 |
| 10 |
BSA-VLd |
1.5 |
77 |
73 |
95 |
33 |
| 11 |
BSA-VLd |
2 |
84 |
80 |
94 |
34 |
| 12 |
BSA-VLd |
2.5 |
92 |
74 |
80 |
35 |
| 13 |
BSA-FeLd |
1 |
77 |
75 |
98 |
27 |
| 14 |
BSA-FeLd |
1.5 |
86 |
85 |
99 |
23 |
| 15 |
BSA-FeLd |
2 |
90 |
77 |
85 |
24 |
| 16 |
BSA-FeLd |
2.5 |
96 |
73 |
76 |
25 |
| 17 |
BSA-CrLe |
1 |
84 |
82 |
98 |
15 |
| 18 |
BSA-CrLe |
1.5 |
78 |
75 |
96 |
19 |
| 19 |
BSA-CrLe |
2 |
76 |
68 |
89 |
24 |
| 20 |
BSA-CrLe |
2.5 |
64 |
48 |
75 |
36 |
Table 5 Effect of catalyst concentration on the oxidation of thioanisolea
| Entry |
Catalyst |
Concentration (μM) |
Conversion (%) |
Yield (%) |
Chemoselectivity (%) |
ee (%) |
| Reactions were performed in PB (2 mL) at room temperature for 20 h. pH = 6; the ratio of H2O2:thioanisole (0.068 mM) was 150:100. pH = 5.1; the ratio of H2O2:thioanisole (0.135 mM) was 150:100. pH = 8; the ratio of H2O2:thioanisole (0.203 mM) was 100:100. pH = 6; the ratio of H2O2:thioanisole (0.135 mM) was 200:100. The sulfoxide of thioanisole was in the R configuration. |
| 1 |
BSA-CoLb |
0.34 |
84 |
83 |
99 |
32 |
| 2 |
BSA-CoLb |
0.68 |
95 |
93 |
98 |
40 |
| 3 |
BSA-CoLb |
1.02 |
92 |
87 |
95 |
35 |
| 4 |
BSA-CoLb |
1.36 |
88 |
84 |
96 |
36 |
| 5 |
BSA-MnLc |
0.68 |
68 |
65 |
96 |
30 |
| 6 |
BSA-MnLc |
1.35 |
80 |
80 |
100 |
32 |
| 7 |
BSA-MnLc |
2.03 |
75 |
71 |
95 |
30 |
| 8 |
BSA-MnLc |
2.70 |
66 |
61 |
92 |
26 |
| 9 |
BSA-VLd |
1.02 |
70 |
70 |
100 |
30 |
| 10 |
BSA-VLd |
2.03 |
75 |
75 |
100 |
34 |
| 11 |
BSA-VLd |
3.05 |
74 |
70 |
94 |
32 |
| 12 |
BSA-VLd |
4.06 |
55 |
52 |
95 |
28 |
| 13 |
BSA-FeLd |
1.02 |
65 |
64 |
98 |
24 |
| 14 |
BSA-FeLd |
2.03 |
77 |
75 |
98 |
27 |
| 15 |
BSA-FeLd |
3.05 |
70 |
67 |
95 |
23 |
| 16 |
BSA-FeLd |
4.06 |
59 |
57 |
96 |
23 |
| 17 |
BSA-CrLe |
0.68 |
74 |
68 |
92 |
15 |
| 18 |
BSA-CrLe |
1.35 |
76 |
68 |
89 |
24 |
| 19 |
BSA-CrLe |
2.03 |
66 |
59 |
90 |
24 |
| 20 |
BSA-CrLe |
2.70 |
64 |
58 |
91 |
20 |
The concentration of catalyst is an important factor, as previously reported by H. R. Khavasi et al.89 For all catalysts, the conversion of thioanisole increased when the proportion of catalyst was increased from 0.5 mol% to 1 mol% of substrate. A higher concentration of catalyst reduced conversion and ee value with almost no changes in sulfoxide selectivity (>89%) (Table 5). The optimal amount of catalyst can be explained considering the reports of Marchena et al.,90 who indicated that the H2O2 decomposition rate increased at higher catalyst loading. The lower availability of oxidant explains the decreased conversion when the catalyst loading is increased. A catalyst loading of 1 mol% of substrate was therefore selected.
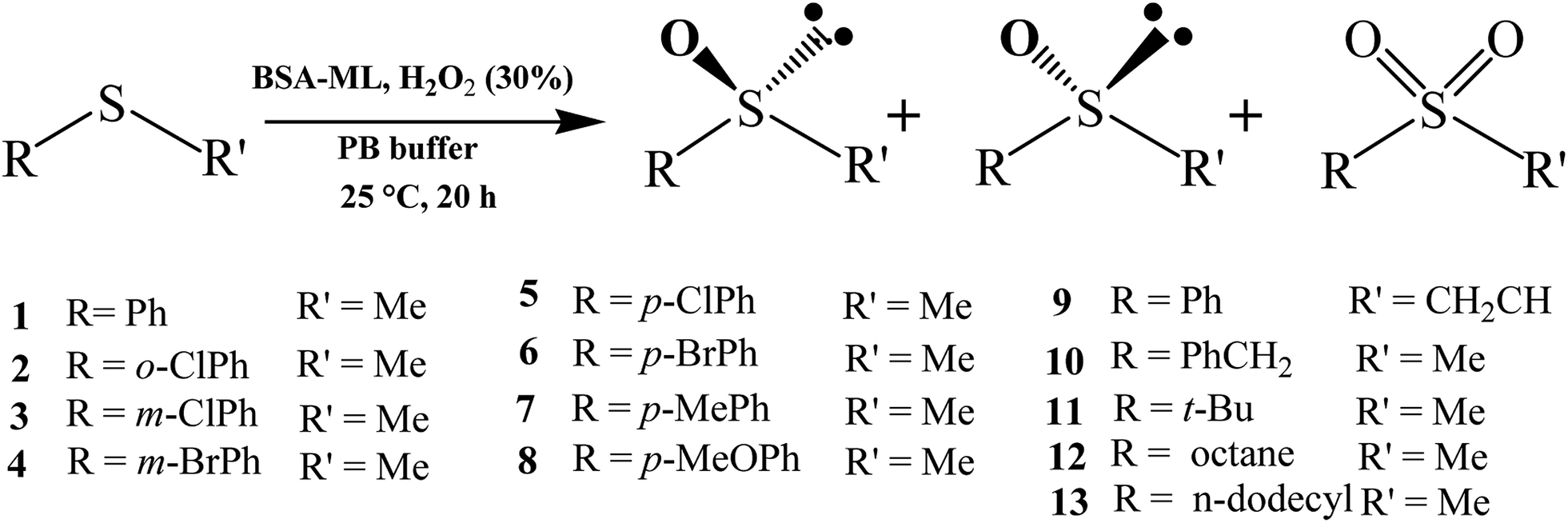
Scope of catalysis
After optimization of the reaction conditions, we successfully oxidized a variety of sulfides using hybrid BSA-ML as the catalyst (Scheme 1).
 |
| | Scheme 1 Enantioselectivity of various prochiral sulfides oxidation using BSA-ML as the catalyst. | |
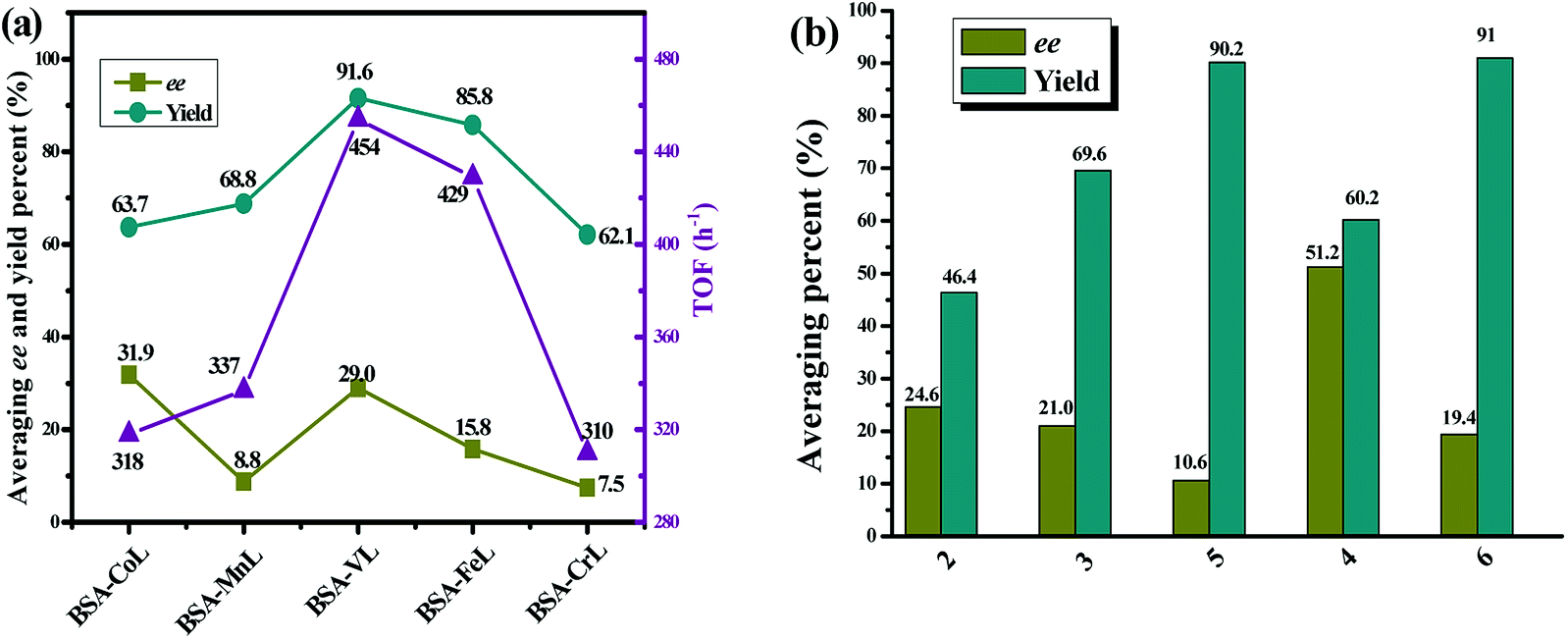
As shown in Table S2,† the ML complex did not greatly affect the chemoselectivity for all substrates. In 92% of cases, chemoselectivities of ≥90% were obtained with BSA-ML hybrids. As in natural metalloenzymes, the role of the BSA scaffold is to drive the selectivity and protect the inorganic catalyst. The cavity may release the reaction product, sulfoxide, before it is over-oxidized into sulfone, leading to the observed high selectivity.91 In particular, good chemoselectivities were achieved even for sulfides with branched or longer alkyl groups instead of aryl substituents (substrates 11–13). Throughout the series of substrates, the results indicated a considerable effect on three important aspects: enantioselectivity, chemical yield and TOF. The ee values for all substrates averaged 31.9%, 8.8%, 29.0%, 15.8%, and 7.5% for conjugates of BSA with CoL, MnL, VL, FeL and CrL, respectively. A similar effect was observed for the yield of sulfoxide, but the average values were significantly higher: 63.7%, 68.8%, 91.6%, 85.8% and 62.1%. Compared to BSA-CoL/MnL/CrL (average TOFs < 350 h−1), BSA-VL and BSA-FeL afforded good TOFs (average values of 454 h−1 and 429 h−1, respectively, Fig. 4(a)). Pleasingly, under certain reaction conditions BSA-VL/FeL/CrL completely and selectively provided the appropriate sulfoxide within 20 h with high TOFs (BSA-VL for sulfides 3 and 5; BSA-FeL for sulfide 7; BSA-CrL for sulfide 5) (Table S2,† note the marks in bold). Another aspect of the metal effect was that the identity of the major enantiomer obtained with BSA conjugated to different ML was frequently opposite (e.g., BSA-CoL/VL (R) vs. BSA-FeL (S) for substrate 7; BSA-FeL (S) vs. BSA-VL/MnL/CrL (R) for substrate 8; BSA-CoL/FeL (R) vs. BSA-MnL/CrL (S) for substrate 9; BSA-CoL (R) vs. BSA-VL/FeL (S) for substrate 12) (Table S2,† note the red marks).
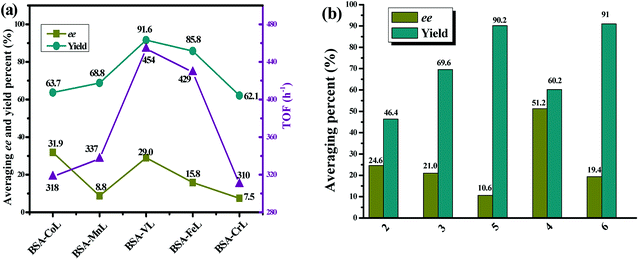 |
| | Fig. 4 (a) Comparison of averaging ee values (dark yellow), averaging yields (dark cyan) and turnover frequencies (purple) in the sulfoxidation reactions catalyzed by BSA-ML; (b) comparison of averaging ee values (dark yellow), averaging yields (dark cyan) in the sulfoxidation for different substrates. Reactions were performed in the optimal reaction conditions for different BSA-ML system. | |
The data in Table S2† also suggest that both steric and electronic factors have a pronounced effect on the enantioselectivity of the reaction. The average ee obtained for all BSA-ML with substrates 2, 3 and 5 were 24.6%, 21.0% and 10.6%, respectively. Similarly, the products from the oxidation of substrates 4 and 6 were obtained with ee values of 51.2% and 19.4%, respectively (Fig. 4(b)). The results indicated that increasing the steric bulk of the aromatic moiety of the sulfide led to an increase in enantioselectivity. This is consistent with the results obtained with the artificial metalloenzyme reported previously.36 In contrast, a lower yield was observed (Fig. 4(b)). In addition, it is note worthy that disappointing enantiomeric excesses (<25%, except for BSA-CoL with sulfide 10) were obtained with sterically bulky sulfides (sulfides 9–10) and those with branched or longer alkyl groups instead of aryl substituents (sulfides 11–13) (Table S2†), while the yields were moderate to high (average 45.8–92.8% for sulfides 9–13). Sulfides containing electron-donating Me (sulfide 7) and OMe (sulfide 8) at the para position on the phenyl gave sulfoxides with much lower enantioselectivity (average 6.7%, except for BSA-VL with sulfide 8) and higher yield (average 93.2%) than those observed for thioanisole (average 31.4% ee and 78.2% yield) with all BSA-ML. Interestingly, the introduction of electron-withdrawing Cl (sulfide 5) and Br (sulfide 6) groups at the para position resulted in a similar effect, both of which gave relatively lower average ee (15.0%) and higher average yield (95.2%). Similarly, higher TOFs (>400 h−1, except for BSA-CoL with sulfide 5) were obtained for the sulfoxidation of substrates with electron-donating or electron-with drawing groups on the phenyl and consequently more easily oxidized. This suggests that the effect of substrate is predominantly steric rather than electronic.
Conclusions
In summary, we have successfully synthesized and characterized five artificial metalloenzymes consisting of a ML cofactor bound to BSA. All of the prepared BSA-ML tested in this study showed catalytic activity for sulfide oxidation under environmentally friendly and mild conditions (using H2O 2 as the oxidant) in aqueous media. This study may drive further research into the use of BSA, a cheap an abundant protein, in oxidation reactions. More significantly, the mild reaction conditions make the present protocol very useful to address environmental concerns and the industrial need for green, sustainable chemistry. The pH and concentrations of catalyst and oxidant were varied to improve conversion, selectivity and enantioselectivity for thioanisole. In the oxidation of different sulfides, the observed activity depended mainly on the nature of the ML and substrates. The highest catalytic activity was obtained with BSA-VL, which gave satisfactory catalytic activity in certain cases (yield, ee and TOF up to 100%, 96% and 500 h−1, respectively). The next steps will be to develop immobilization strategies that may contribute to the stability of artificial enzymes, while allowing then to be recycled.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (21361003, 21861005), the Natural science Foundation of Guangxi (2016GXNSFDA380005, 2016GXNSFFA380010, 2016GXNSFBA380237), Middle-aged and young teachers in colleges and universities in Guangxi basic ability promotion project (KY2016LX480), New Century Ten, Hundred, Thousand Talents Project in Guangxi, The Innovative Team & Outstanding Talent Program of Colleges and Universities in Guangxi (2017-38) and the Foundation of Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, IRT 1225.
References
- Z.-H. Zhang, X.-S. Yang, Q.-Q. Zhang, L. Wang, M.-Y. He, Q. Chen and X.-F. Huang, RSC Adv., 2016, 6, 104036–104040 RSC.
- S. Otocka, M. Kwiatkowska, L. Madalińska and P. Kiełbasiński, Chem. Rev., 2017, 117, 4147–4181 CrossRef CAS PubMed.
- H. Gholami, J. Zhang, M. Anyika and B. Borhan, Org. Lett., 2017, 19, 1722–1725 CrossRef CAS.
- M. Mellah, A. Voituriez and E. Schulz, Chem. Rev., 2007, 107, 5133–5209 CrossRef CAS.
- S. M. G. Pires, M. M. Q. Simões, I. C. M. S. Santos, S. L. H. Rebelo, M. M. Pereira, M. G. P. M. S. Neves and J. A. S. Cavaleiro, Appl. Catal., A, 2012, 439–440, 51–56 CrossRef CAS.
- A. Massa, A. V. Malkov, P. Kocovsky and A. Scettri, Tetrahedron Lett., 2003, 44, 7179–7181 CrossRef CAS.
- S. Kobayashi, C. Ogawa, H. Konishi and M. Sugiura, J. Am. Chem. Soc., 2003, 125, 6610–6611 CrossRef CAS.
- J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19–31 CrossRef CAS.
- S. Doherty, J. G. Knight, M. A. Carroll, A. R. Clemmet, J. R. Ellison, T. Backhouse, N. Holmes, L. A. Thompson and R. A. Bourne, RSC Adv., 2016, 6, 73118–73131 RSC.
- Y.-Y. Zhang, R. Tan, G.-W. Zhao, X.-F. Luo, C. Xing and D.-H. Yin, J. Catal., 2016, 335, 62–71 CrossRef CAS.
- S. Rodríguez, N. Haddad, R. P. Frutos, N. Grinberg, D. Krishnamurthy and C. H. Senanayake, Org. Process Res. Dev., 2017, 21, 444–447 CrossRef.
- P. K. Bera, P. Kumari, S. H. R. Abdi, N. H. Khan, R. I. Kureshy, P. S. Subramanianab and H. C. Bajaj, RSC Adv., 2014, 4, 61550–61556 RSC.
- D. Linde, M. Cañellas, C. Coscolín, I. Davó-Siguero, A. Romero, F. Lucas, F. J. Ruiz-Dueñas, V. Guallar and A. T. Martínez, Catal. Sci. Technol., 2016, 6, 6277–6285 RSC.
- C. J. Carrasco, F. Montilla and A. Galindo, Catal. Commun., 2016, 84, 134–136 CrossRef CAS.
- C. Shen, J. Qiao, L.-W. Zhao, K. Zheng, J.-Z. Jin and P.-F. Zhang, Catal. Commun., 2017, 92, 114–118 CrossRef CAS.
- J. Fujisaki, K. Matsumoto, K. Matsumoto and T. Katsuki, J. Am. Chem. Soc., 2011, 133, 56–61 CrossRef CAS PubMed.
- C. Herrero, N. Nguyen-Thi, F. Hammerer, F. Banse, D. Gagné, N. Doucet, J.-P. Mahy and R. Ricoux, Catalysts, 2016, 6, 202–214 CrossRef.
- L. Rondot, E. Girgenti, F. Oddon, C. Marchi-Delapierre, A. Jorge-Robin and S. Ménage, J. Mol. Catal. A: Chem., 2016, 416, 20–28 CrossRef CAS.
- V. Mojr, M. Buděšínský, R. Cibulka and T. Kraus, Org. Biomol. Chem., 2011, 9, 7318–7326 RSC.
- T. Heinisch and T. R. Ward, Acc. Chem. Res., 2016, 49, 1711–1721 CrossRef CAS PubMed.
- Y. Okamoto, V. Köhler, C. E. Paul, F. Hollmann and T. R. Ward, ACS Catal., 2016, 6, 3553–3557 CrossRef CAS.
- M. Bersellini and G. Roelfes, Dalton Trans., 2017, 46, 4325–4330 RSC.
- M.-P. Cheng, Y.-H. Li, J. Zhou, G.-Q. Jia, S.-M. Lu, Y. Yang and C. Li, Chem. Commun., 2016, 52, 9644–9647 RSC.
- D. F. Sauer, S. Gotzen and J. Okuda, Org. Biomol. Chem., 2016, 14, 9174–9183 RSC.
- M. Hoarau, C. Hureau, E. Gras and P. Faller, Coord. Chem. Rev., 2016, 308, 445–459 CrossRef CAS.
- C. Marchi-Delapierre, L. Rondot, C. Cavazza and S. Ménage, Isr. J. Chem., 2015, 55, 61–75 CrossRef CAS.
- T. Heinisch and T. R. Ward, ChemInform, 2015, 46, 3406–3418 CrossRef.
- M. Bersellini and G. Roelfes, Org. Biomol. Chem., 2017, 15, 3069–3073 RSC.
- Q. Zhang, X.-X. He, A.-L. Han, Q.-X. Tu, G.-Z. Fang, J.-F. Liu, S. Wang and H.-B. Li, Nanoscale, 2016, 8, 1685–16856 Search PubMed.
- A. Pordea, D. Mathis and T. R. Ward, J. Organomet. Chem., 2009, 694, 930–936 CrossRef CAS.
- R. Golbedaghi, M. Rezaeivala, M. Khalili, B. Notash and J. Karimi, J. Mol. Struct., 2016, 1125, 144–148 CrossRef CAS.
- J.-L. Zhang, D. K. Garner, L. Liang, Q. Chen and Y. Lu, Chem. Commun., 2008, 14, 1665–1667 RSC.
- R. Ricoux, M. Allard, R. Dubuc, C. Dupont, J.-D. Maréchalc and J.-P. Mahy, Org. Biomol. Chem., 2009, 7, 3208–3211 RSC.
- Q. Raffy, R. Ricoux and J.-P. Mahy, Tetrahedron Lett., 2008, 49, 1865–1869 CrossRef CAS.
- E. Sansiaume-Dagousset, A. Urvoas, K. Chelly, W. Ghattas, J.-D. Maréchal, J.-P. Mahy and R. Ricoux, Dalton Trans., 2014, 43, 8344–8354 RSC.
- J. Tang, F.-P. Huang, Y. Wei, H.-D. Bian, W. Zhang and H. Liang, Dalton Trans., 2016, 45, 8061–8072 RSC.
- P. Subramaniam, S. Anbarasan, S. S. Devi and A. Ramdass, Polyhedron, 2016, 119, 14–22 CrossRef CAS.
- K. P. Bryliakov and E. P. Talsi, Eur. J. Org. Chem., 2008, 19, 3369–3376 CrossRef.
- L. Postigo, M. Ventura, T. Cuenca, G. Jimenez and B. Royo, Catal. Sci. Technol., 2015, 5, 320–324 RSC.
- C. Drago, L. Caggiano and R. F. W. Jackson, Angew. Chem., Int. Ed., 2005, 44, 7221–7223 CrossRef CAS PubMed.
- A. Lazar, P. Sharma and A. P. Singh, Microporous Mesoporous Mater., 2013, 170, 331–339 CrossRef CAS.
- W. Dai, J. Li, B. Chen, G. Li, Y. Lv, L. Wang and S. Gao, Org. Lett., 2013, 15, 5658–5661 CrossRef CAS PubMed.
- H. Srour, J. Jalkh, P. L. Maux, S. Chevance, M. Kobeissi and G. Simonneaux, J. Mol. Catal. A: Chem., 2013, 370, 75–79 CrossRef CAS.
- J. Legros and C. Bolm, Chem.–Eur. J., 2005, 11, 1086–1092 CrossRef CAS PubMed.
- H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940–8941 CrossRef CAS PubMed.
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and S. Joost, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- H. Srour, P. L. Maux, S. Chevance and G. Simonneaux, Coord. Chem. Rev., 2013, 257, 3030–3050 CrossRef CAS.
- J.-P. Mahy, J.-D. Maréchal and R. Ricoux, Chem. Commun., 2015, 51, 2476–2494 RSC.
- D. C. M. Albanese and N. Gaggero, RSC Adv., 2015, 5, 10588–10598 RSC.
- A. Mahammed and Z. Gross, J. Am. Chem. Soc., 2005, 127, 2883–2887 CrossRef CAS PubMed.
- Y.-J. Hu, Y. Ou-Yang, Y. Zhang and Y. Liu, Protein J., 2010, 29, 234–241 CrossRef CAS PubMed.
- M. Ohashi, T. Koshiyama, T. Ueno, M. Yanase, H. Fujii and Y. Watanabe, Angew. Chem., Int. Ed., 2003, 42, 1005–1008 CrossRef CAS PubMed.
- R. K. Sharma, A. Pandey and S. Gulati, Polyhedron, 2012, 45, 86–93 CrossRef CAS.
- C. Herrero, N. Nguyen-Thi, F. Hammerer, F. Banse, D. Gagné, N. Doucet, J. Mahy and R. Ricoux, Catalysts, 2016, 6, 202 CrossRef.
- N. K. Jana and J. G. Verkade, Org. Lett., 2003, 5, 3787–3790 CrossRef CAS PubMed.
- R. J. Griffin, A. Henderson, N. J. Curtin, A. Echalier, J. A. Endicott, I. R. ardcastle, D. R. Newell, N. E. M. Noble, L. Z. Wang and B. T. Golding, J. Am. Chem. Soc., 2006, 128, 6012–6013 CrossRef CAS PubMed.
- R. S. Varma and K. P. Naicker, Org. Lett., 1999, 1, 189–192 CrossRef CAS.
- N. Fukuda and T. Ikemoto, J. Org. Chem., 2010, 75, 4629–4631 CrossRef CAS PubMed.
- S. Lovat, M. Mba, H. C. L. Abbenhuis, D. Vogt, C. Zonta and G. Licini, Inorg. Chem., 2009, 48, 4724–4728 CrossRef CAS PubMed.
- B. Yu, A.-H. Liu, L.-N. He, B. Li, Z.-F. Diao and Y.-N. Li, Green Chem., 2012, 34, 957–962 RSC.
- P. Hanson, R. A. A. J. Hendrickx and J. R. L. Smith, New J. Chem., 2010, 34, 65–84 RSC.
- J. G. W. Gokel, H. M. Gerdes and D. M. Dishong, J. Org. Chem., 1980, 45, 3634–3639 CrossRef.
- P. G. Reddy, N. Mamidi and C. P. Pradeep, CrystEngComm, 2016, 18, 4272–4276 RSC.
- J. Fujisaki, K. Matsumoto, K. Matsumoto and T. Katsuki, J. Am. Chem. Soc., 2011, 133, 56–61 CrossRef CAS PubMed.
- K. Kamata, T. Hirano, R. Ishimoto and N. Mizuno, Dalton Trans., 2010, 39, 5509–5518 RSC.
- G. La Sorella, L. Sperni, G. Strukul and A. Scarso, Adv. Synth. Catal., 2016, 358, 3443–3449 CrossRef CAS.
- X.-M. Zhang, J. Tang, L.-N. Wang, D. Yao, Q. Yu, F.-P. Huang and H.-D. Bian, Polyhedron, 2017, 133, 433–440 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXS-97, Program for X-ray Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- H. Tang, Z.-H. Shi, N.-G. Li, Y.-P. Tang, Q.-P. Shi, Z.-X. Dong, P.-X. Zhang and J.-A. Duan, Arch. Pharmacal Res., 2015, 38, 1789–1801 CrossRef CAS PubMed.
- Z. Li, H. Yan, G.-L. Chang, M. Hong, J.-M. Dou and M.-J. Niu, J. Photochem. Photobiol., B, 2016, 163, 403–412 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- R. Tiwari, K. Mahasenan, R. Pavlovicz, C. L. Li and W. Tjarks, J. Chem. Inf. Comput. Sci., 2009, 49, 1581–1589 CrossRef CAS PubMed.
- J.-H. Shi, Y.-Y. Zhu, J. Wang and J. Chen, Luminescence, 2015, 30, 44–52 CrossRef CAS PubMed.
- J.-H. Shi, J. Chen, J. Wang, Y.-Y. Zhu and Q. Wang, Spectrochim. Acta, Part A, 2015, 149, 630–637 CrossRef CAS PubMed.
- J. R. Carey, S. K. Ma, T. D. Pfister, D. K. Garner, H. K. Kim, J. A. Abramite, Z.-L. Wang, Z.-J. Guo and Y. Lu, J. Am. Chem. Soc., 2004, 126, 10812–10813 CrossRef CAS PubMed.
- T. Ueno, T. Koshiyama, S. Abe, N. Yokoi, M. Ohashi, H. Nakajima and Y. Watanabe, J. Organomet. Chem., 2007, 692, 142–147 CrossRef CAS.
- J.-J. Ao, L. Gao, T. Yuan and G.-F. Jiang, Chemosphere, 2015, 119, 590–600 CrossRef CAS PubMed.
- Y. Teng, L.-Y. Zou, M. Huang and W.-S. Zong, J. Mol. Recognit., 2015, 28, 232–238 CrossRef CAS PubMed.
- Z. Li, H. Yan, G.-L. Chang, M. Hong, J.-M. Dou and M.-J. Niu, J. Photochem. Photobiol., B, 2016, 163, 403–412 CrossRef CAS PubMed.
- J.-H. Shi, Y.-Y. Zhu, J. Wang and J. Chen, Luminescence, 2015, 30, 44–52 CrossRef CAS PubMed.
- L. Rondot, E. Girgenti, F. Oddon, C. Marchi-Delapierre, A. Jorge-Robin and S. Ménage, J. Mol. Catal. A: Chem., 2016, 416, 20–28 CrossRef CAS.
- J.-L. Zhang, D. K. Garner, L. Liang, Q. Chen and Y. Lu, Chem. Commun., 2008, 14, 1665–1667 RSC.
- P. Rousselot-Pailley, C. Bochot, C. Marchi-Delapierre, A. Jorge-Robin, L. Martin, J. C. Fontecilla-Camps, C. Cavazza and S. Ménage, ChemBioChem, 2009, 10, 545–552 CrossRef CAS PubMed.
- M.-P. Cheng, Y.-H. Li, J. Zhou, G.-Q. Jia, S.-M. Lu, Y. Yang and C. Li, Chem. Commun., 2016, 52, 9644–9647 RSC.
- A. Mahammed and Z. Gross, J. Am. Chem. Soc., 2005, 127, 2883–2887 CrossRef CAS PubMed.
- R. Rahimi, A. A. Tehrani, M. A. Fard, B. M. M. Sadegh and H. R. Khavasi, Catal. Commun., 2009, 11, 232–235 CrossRef CAS.
- C. L. Marchena, C. Saux, R. Dinamarca, G. Pecchi and L. Pierella, RSC Adv., 2016, 6, 102015–102022 RSC.
- L. Rondot, E. Girgenti, F. Oddon, C. Marchi-Delapierre, A. Jorge-Robin and S. Ménage, J. Mol. Catal. A: Chem., 2016, 416, 20–28 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1854581. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra07113f |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Meiyi Luoc and
Fuping Huang*b
*ab,
Meiyi Luoc and
Fuping Huang*b