
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn iodine/DMSO-catalyzed sequential one-pot approach to 2,4,5-trisubstituted-1H-imidazoles from α-methylene ketones†
Janeeka Jayram and
Vineet Jeena*
and
Vineet Jeena*
School of Chemistry and Physics, University of KwaZulu-Natal, Scottsville, Pietermaritzburg, 3209, South Africa. E-mail: Jeenav1@ukzn.ac.za
First published on 7th November 2018
Abstract
A sequential one-pot approach to 2,4,5-trisubstituted imidazoles has been developed from α-methylene ketones and aldehydes. This methodology employs air-moisture stable reaction conditions and an inexpensive iodine/DMSO system affording a diverse range of known and novel (substrate scope) 2,4,5-trisubstituted imidazoles in moderate to excellent yields. The iodine/DMSO system was extended to the domino convergent synthesis of two functionalized intermediates, benzil and benzaldehyde, to produce the final product.
Introduction
2,4,5-Trisubstituted imidazoles occupy a special place in the realm of natural, pharmaceutical and synthetic organic chemistry, as this moiety exhibits numerous biological and pharmacological properties.1 This particular N-heterocyclic family has expanded its applications in various fields such as cosmetics,2 polymer chemistry,3 agro chemicals,4 materials chemistry (OLEDS, optical electronics, dye sensitized solar cells),5 photography as photosensitive compounds6 and industry as a corrosion inhibitor of certain transition metals.7 Accordingly, synthetic routes to access 2,4,5-trisubstituted imidazoles are of vital importance and thus, develop on a daily basis.The classical approach to assemble trisubstituted imidazoles involves the cyclocondensation of an α-diketone, aldehyde and ammonium acetate using transition metal catalysts or acidic media (Scheme 1a).8 However, several of these transformations involve harsh reaction conditions, multistep synthetic operations, tedious isolation procedures and low yields. In particular, while synthetically useful, such methodologies are limited to accessibility of starting materials which in turn restrict product diversity.9
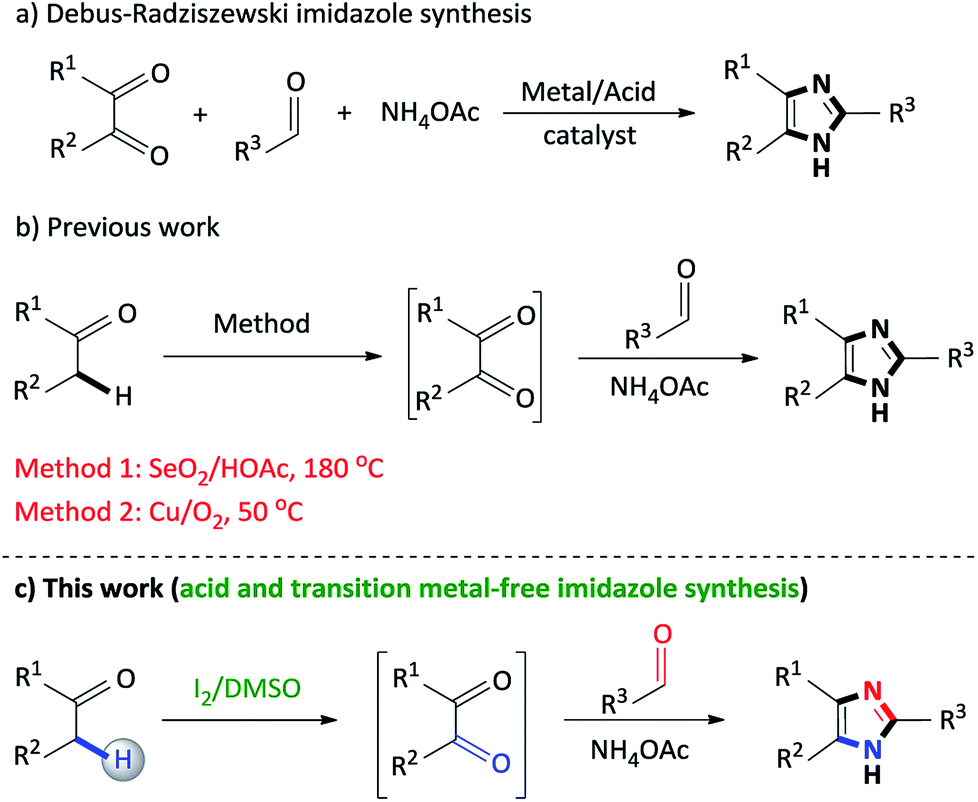 | ||
| Scheme 1 Synthetic routes toward the preparation of 2,4,5-trisubstituted imidazoles. | ||
Recently, the selective oxidation of non-activated carbon–hydrogen (C–H) bonds has become an area of profound interest in contemporary organic synthesis toward the formation of new carbon–carbon (C–C) and carbon–heteroatom (C–X, X = N, O, S, etc.) bonds.10 Specifically, the direct oxidation of unreactive benzylic Csp3–H bonds has received significant research interest to assemble C–N bonds, since it represents an atom-economical strategy.11 Within this context, numerous innovative systems for the oxidation of α-methylene ketones to diketones have been reported such as Cu(OAc)2/Ph3P,12 Cu(OAc)2·H2O/K2CO3,13 DMSO/KHCO3 (ref. 14) KOtBu/18-Crown-6/O2,15 Pd(OAc)2/triazole ligand16 and CuO/I2/DMSO.17
Our research group has recently developed a new type of one-pot domino process to synthesize 2,4,5-trisubstituted imidazoles through a SeO2/HOAc (Scheme 1b, Method 1) mediated oxidative cyclization from readily available α-methylene ketones and aldehydes.18 However, the notable disadvantages of this protocol is the stoichiometric use of toxic selenium dioxide, selenium waste, acidic media and high reaction temperatures. Thus, in an effort to address the factors above, a copper/O2 (Scheme 1b, Method 2)19 methodology was developed, however, while this approach proves its efficiency, the use of a transition metal catalyst detracts from this synthetic procedure. As a result, there is scope for further improvements toward the development of non-toxic, acid and metal-free syntheses using simple, inexpensive reagents and convenient procedures, in which the above mentioned drawbacks can be addressed.
In this context, the concept of transition metal-free C–H oxidation reactions is currently an active area of research and swiftly growing field in synthetic organic chemistry.20 Notably, in recent years, molecular iodine (I2) has proven to be an excellent catalyst for various organic transformations owing to its inexpensive, non-toxic properties and mild Lewis acidity.21 Additionally, dimethylsulfoxide (DMSO) is an inexpensive and environmentally friendly polar aprotic compound that performs a versatile role as a solvent, oxidant and oxygen source in various organic syntheses.22 In recent times, the iodine/DMSO combination has received considerable attention, in synthetic organic chemistry, as an effective and eco-friendly oxidative system as it has effected numerous organic transformations.23
In continuation of our studies on nitrogen heterocyclic synthesis,24 we herein report an improved, non-toxic, acid and transition metal free, I2/DMSO promoted coupling of benzylic Csp3–H α-methylene ketones with aldehydes to synthesize 2,4,5-trisubstituted imidazoles (Scheme 1c).
Experimental
Typical procedure for the I2/DMSO-catalyzed reaction of α-methylene ketones, aldehydes and ammonium acetate
2-Phenylacetophenone (196 mg, 1.0 mmol) and iodine (126 mg, 0.5 mmol) were mixed in a round bottomed flask with 1 mL DMSO and heated to 100 °C for 2 hours. Thereafter, benzaldehyde (102 μL, 1.0 mmol), ammonium acetate (770 mg, 10 mmol) and ethanol (2 mL) were sequentially added and the mixture heated to 100 °C for a further 2 hours. After cooling, a Na2S2O3/ice water solution was added to the reaction mixture to yield the crude product which was filtered, air dried and recrystallized from ethanol to afford the desired product.The detailed characterization data for 3a–3w are provided in the ESI.†
Results and discussion
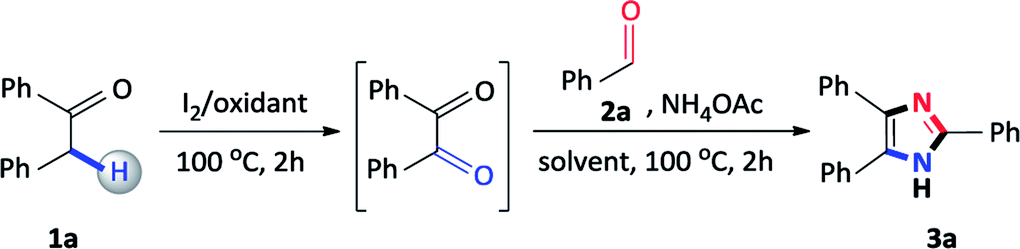
Initially, our studies commenced using benzyl phenyl ketone 1a and benzaldehyde 2a as a model substrate and the results are presented in Table 1. Firstly, 1a, 2a, ammonium acetate and I2 were added to DMSO and the mixture was heated at 100 °C for 4 hours, however, the reaction failed to proceed and no product was detected (Table 1, entry 1). Due to the failure of this reaction, we focused on sequential additions and accordingly, we attempted a reaction with 1a and I2 in DMSO, and heated at 100 °C for 2 hours, followed by the addition of 2a and ammonium acetate at 100 °C for a further 2 hours. Encouragingly, the expected 2,4,5-triphenylimidazole 3a was obtained in an isolated yield of 54% (Table 1, entry 2). Accordingly, we focused our studies on a sequential one-pot approach to prepare these scaffolds as this seemed to be the most viable synthetic route. In general, 2,4,5-trisubstituted imidazole syntheses proceed well in the presence of an alcohol solvent as it is known to favour the coupling of a diketone, aldehyde and ammonium acetate.25 Thus, in order to improve the yield, the reaction was attempted with methanol and iso-propanol affording 3a in 68% and 54% yield respectively (Table 1, entries 3 and 4). Finally, ethanol was shown to be the best performing additive solvent as the yield significantly increased to 82% (Table 1, entry 5). Attempts were made to decrease the amount of iodine, however, 0.25 and 0.10 equivalents lowered the yield of the desired product to 74% and 32% respectively (Table 1, entries 6 and 7). Furthermore, performing the reaction in the absence of iodine resulted in no product formation thus, confirming its importance in this multicomponent reaction (Table 1, entry 8). The reaction also failed to produce the expected 2,4,5-triphenylimidazole 3a when DMSO was replaced with alternative oxidants such as dimethylformamide (DMF), acetonitrile (CH3CN) and toluene (PhMe) confirming its vital importance in the reaction (Table 1, entries 9–11). Many synthetic procedures use varying amounts of ammonium acetate depending on the reaction conditions employed to produce the desired product in maximum yield.26 Thus, to complete the study, the amount of ammonium acetate was reduced to 5.0 and 2.5 equivalents, however, the yield of 3a considerably decreased to 62% and 26% (Table 1, entries 12 and 13). To conclude, the conditions described in entry 5 were found to be optimal, allowing for maximum conversion to the desired product 3a.
|
||||
|---|---|---|---|---|
| Entry | I2 (equiv.) | Oxidant (1 mL) | Solvent (2 mL) | Yieldb (%) |
| a Reaction conditions: Step I: 1a (1 mmol), I2 (0.5 mmol), 100 °C for 2 h; Step II: 2a (1 mmol), NH4OAc (10 mmol), 100 °C for 2 h.b Isolated yield.c One-pot reaction.d NH4OAc (5.0 equiv.).e NH4OAc (2.5 equiv.). | ||||
| 1c | 0.5 | DMSO | — | — |
| 2 | 0.5 | DMSO | — | 54 |
| 3 | 0.5 | DMSO | MeOH | 68 |
| 4 | 0.5 | DMSO | i-PrOH | 54 |
| 5 | 0.5 | DMSO | EtOH | 82 |
| 6 | 0.25 | DMSO | EtOH | 74 |
| 7 | 0.10 | DMSO | EtOH | 32 |
| 8 | — | DMSO | EtOH | — |
| 9 | 0.5 | DMF | EtOH | — |
| 10 | 0.5 | CH3CN | EtOH | — |
| 11 | 0.5 | PhMe | EtOH | — |
| 12d | 0.5 | DMSO | EtOH | 62 |
| 13e | 0.5 | DMSO | EtOH | 26 |
Having identified the optimized reaction conditions, the applicability of the devised system was evaluated by varying the α-methylene ketone and aldehyde (Scheme 2). The various electron-withdrawing and electron-donating para-substituted benzaldehydes all underwent a smooth transformation with 1a to provide the corresponding 2,4,5-trisubstituted imidazoles 3b–3d in good to excellent yields (80–86%). Similar results were obtained upon substitution at the meta and ortho position of benzaldehyde producing good yields of 63–75% (Schemes 2 and 3e–3g). In order to expand the scope of our methodology, a bulkier substrate, 2-naphthaldehyde was evaluated and also coupled smoothly with 1a to afford 2-(naphthalene-2-yl)-4,5-diphenyl-1H-imidazole 3h in a 74% yield. We further extended our methodology to heterocyclic and aliphatic aldehydes such as furfural, cyclohexanecarboxaldehyde and 2-ethylbutyraldehyde to produce the desired imidazoles 3i–3k albeit, in moderate yields with prolonged reaction time. Next, reactions of benzaldehyde with substituted α-methylene ketones were investigated. Unfortunately, propiophenone and 2-butanone were not compatible with this system, and only starting material was recovered. Benzyl phenyl ketone 1a substituted with para-Cl or Br-groups reacted smoothly to furnish 2,4,5-trisubstituted imidazoles 3n–3o in good to excellent yields (78–84%). Similarly, coupling 4-chloro-2-phenylacetophenone with para substituted benzaldehydes, under the optimized reaction conditions, successfully afforded the corresponding heterocyclic imidazoles 3p–3r in good yields (66–76%). Compounds 3n–3r were isolated as a mixture of tautomers due to the presence of the fluid hydrogen on the nitrogen atom.
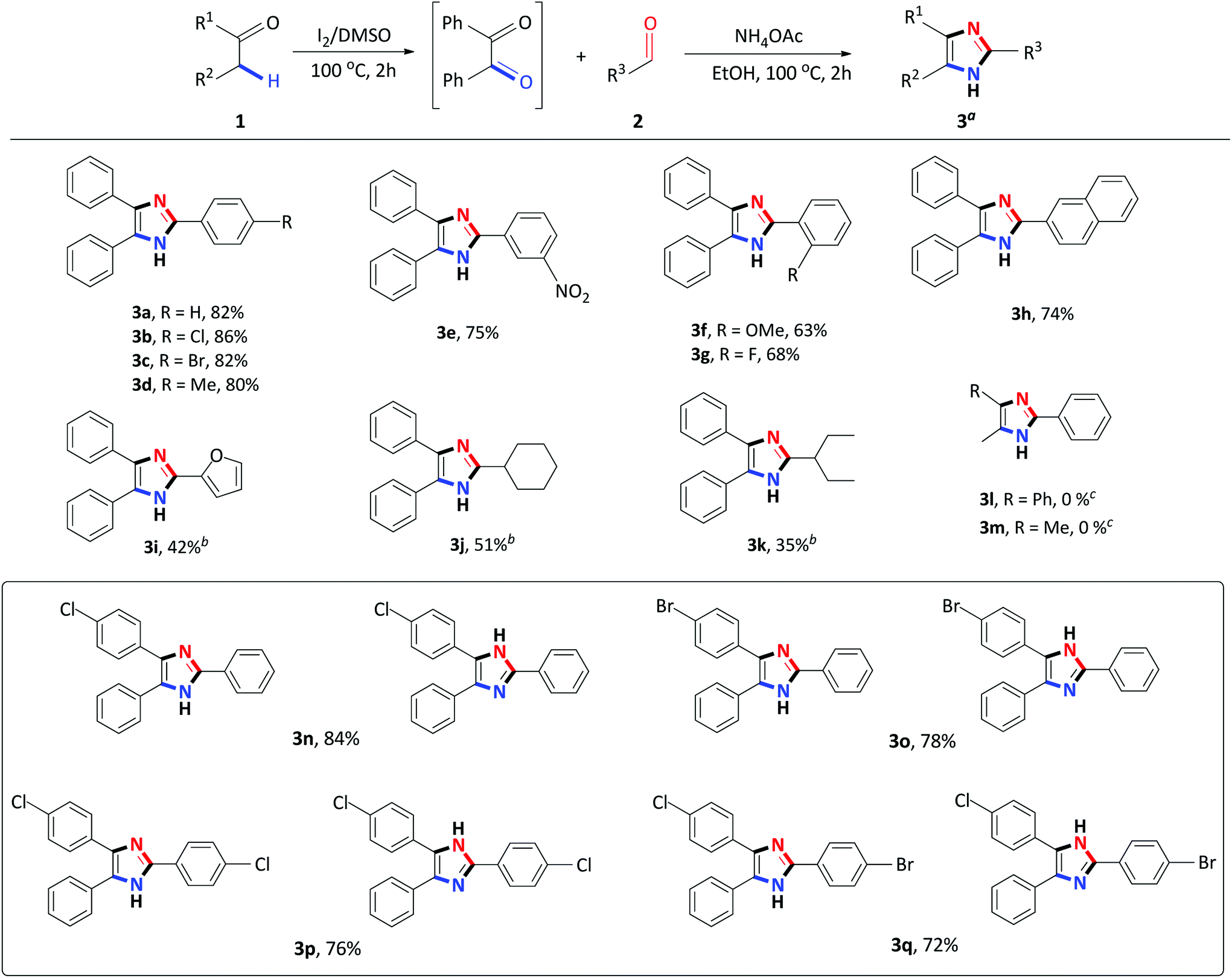 | ||
| Scheme 2 Step I: 1 (1 mmol), I2 (0.5 mmol), DMSO (1 mL), 100 °C for 2 h; Step II: 2 (1 mmol), NH4OAc (10 mmol), EtOH (2 mL), 100 °C for 2 h. aIsolated yield. bStep II: 24 h. cStep I: 24 h. | ||
Recently, several 2,4,5-trisubstituted imidazoles were shown to have activity against malarial strains owing to the novelty of the chemotype.27 As a result, in order to enhance the diversity and thus the possible biological relevance of the imidazole scaffold, we decided to establish substrate scope with this strategy which is not known in literature. As a result, a series of novel 2,4,5-trisubstituted imidazoles 3r–3w were synthesized as a mixture of tautomers in good to excellent yields (53–85%) (Scheme 3). Consequently, this atom-economical synthetic procedure overcomes the limitations of previous methodologies which are restricted to the accessibility of starting materials, thus creating a new avenue to synthesize numerous novel imidazole derivatives yielding drug like properties. Next, we turned our attention to the domino convergent synthesis of two suitably functionalized intermediates, benzil and benzaldehyde, to converge onto the final product 3a. As previously mentioned, the I2/DMSO system is able to effect numerous organic transformations, hence, we rationalized that this system could simultaneously oxidize the benzyl phenyl ketone 1a and benzyl alcohol 4a, in the same reaction vessel, to produce the diketone and aldehyde which will subsequently form the desired imidazole using our devised system. To test this hypothesis we mixed benzyl phenyl ketone 1a and benzyl alcohol 4a in the presence of I2/DMSO and heated the mixture at 100 °C for 24 hours to form the corresponding diketone and aldehyde. Thereafter, ammonium acetate and ethanol were added to the reaction and heated at 100 °C for a further 2 hours to afford 3a in an isolated yield of 48% (Scheme 4). This result illustrates that the I2/DMSO system can successfully oxidize two different substrates simultaneously, in the same reaction vessel, for convergent synthesis into the desired product. We further anticipate that this result would inspire the design for more efficient and eco-friendly domino convergent syntheses for the preparation of complex products from simple materials via this innovative synthetic strategy.
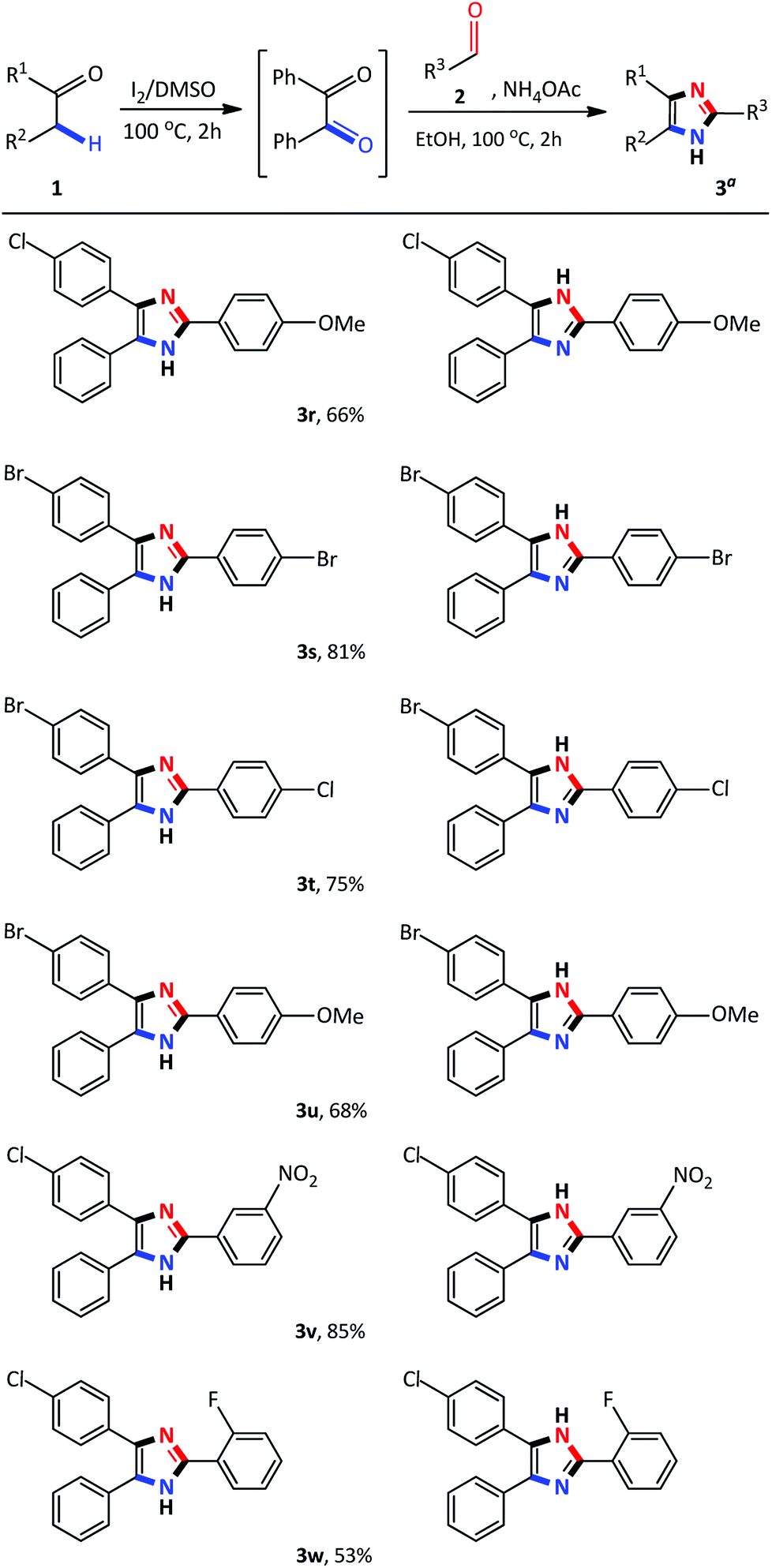 | ||
| Scheme 3 Synthesis of novel 2,4,5-trisubstituted imidazoles. Step I: 1 (1 mmol), I2 (0.5 mmol), DMSO (1 mL), 100 °C for 2 h; Step II: 2 (1 mmol), NH4OAc (10 mmol), EtOH (2 mL), 100 °C for 2 h. aIsolated yield. | ||
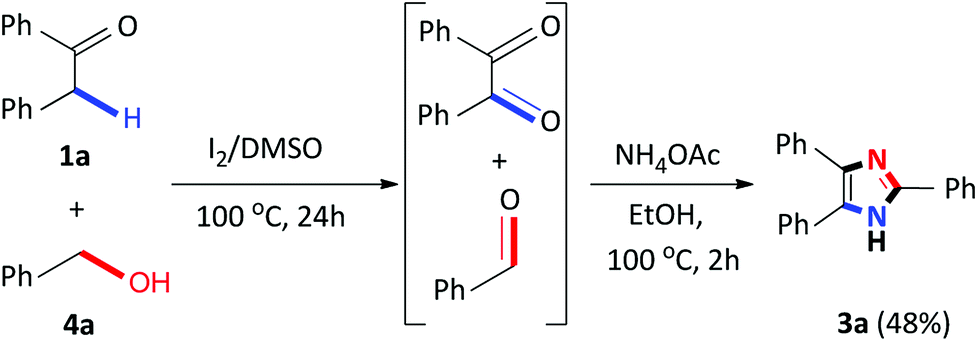 | ||
| Scheme 4 Domino convergent synthesis of 2,4,5-triphenyl imidazole 3a. | ||
To investigate a plausible reaction mechanism, a series of control experiments were performed (Scheme 5). In experiment 1, benzyl phenyl ketone 1a was oxidized, in the presence of I2/DMSO to benzil 5a (96%) which indicates that the diketone is an intermediate in the reaction. Under the iodine-catalysis conditions, the addition of the radical inhibitor (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) afforded the capture adduct 6a in 46% (experiment 2). The TEMPO-trapped reaction strongly supports the formation of 6a, in which the radical is located at the α-position of benzyl phenyl ketone. In order to rule out the role of hydroxyl and peroxide radicals as an intermediate, a reaction of benzyl phenyl ketone 1a with butylated hydroxytoluene (BHT)28 in the presence of I2 in DMSO was performed (experiment 3). In this reaction, the diketone was obtained in 90% yield, thereby proving non participation of peroxide radicals in our reaction. Mechanistically, we proposed that an α-iodinated species is the reactive intermediate in the oxidation of 1a to afford the benzil intermediate 5a. Accordingly, 2-iodo-1,2-diphenylethanone 7a was synthesized29 and subject to oxidation in DMSO affording benzil in 97% yield (experiment 4). Having completed the iodination reaction, we sought to determine the source of oxygen in the oxidation reaction. Accordingly, there are three potential oxygen sources in the reaction system: molecular oxygen in air, a trace amount of water in the solvent DMSO and, DMSO itself. The reaction proceeded well under a nitrogen atmosphere affording 5a in 95% yield indicating that oxygen from the air does not participate in the reaction (experiment 5). When the reaction was performed in a toluene/water biphasic mixture, the hydroxylation product 8a and benzil intermediate 5a was obtained in 22% and 16% yield respectively, indicating that water plays a minor role in the oxidation reaction to afford 8a which is further oxidized to provide 5a (experiment 6). Furthermore, the reaction of 7a in anhydrous DMSO under a nitrogen atmosphere afforded the diketone intermediate in 98% yield (experiment 6b). This preliminary result confirms that majority of the incorporated oxygen in the benzil intermediate is indeed coming from DMSO. Finally, in order to understand the effect of iodine in the coupling reaction, 1a, 2a and ammonium acetate was reacted in ethanol to afford the corresponding 2,4,5-triphenylimidazole 3a, however, no product was obtained (experiment 7a). Moreover, the addition of iodine resulted in 92% formation of 3a indicating that iodine plays a role in the coupling reaction (experiment 7b). On the basis of the above observations, we propose a plausible mechanism of consecutive iodination/oxidation/cyclization for the synthesis of 2,4,5-trisubstituted imidazoles (Scheme 6). The reaction proceeds with an iodine assisted proton abstraction from the methylene position of 1 to generate the benzyl radical A.30 Subsequently, iodination affords the corresponding α-iodinated intermediate, 2-iodo-1,2-diphenylethanone B, which reacts with DMSO to generate the active intermediate C. Two possible pathways could be proposed for the reaction of C to form the benzil intermediate 5.31 Based on control experiment 6, the minor pathway (green) involves a water attack on the sulfur cation of C to form the hydroxylation intermediate D, regenerating DMSO and HI for further cycle, which subsequently forms 5 through intermediate E.32 On the other hand, the major pathway (purple) involves proton abstraction from the α-carbon of C, followed by the removal of HI and DMS, affording the desired benzil intermediate 5. Accordingly, the oxidation of B is not solely a DMSO catalyzed reaction, since the catalytic water still exists, but majorly relies on the DMSO to react with the α-iodinated intermediate to afford the diketone intermediate 5. In terms of the coupling reaction,33 iodine is capable of binding to the carbonyl oxygen of the diketone intermediate 5 and aldehyde 2, owing to its mild Lewis acidity, thus increasing the reactivity of the substrates,34 which is supported by control experiment 7b. Moreover, iodine facilitates the formation of the imine intermediates H and I which condense to form the desired imidazole 3.
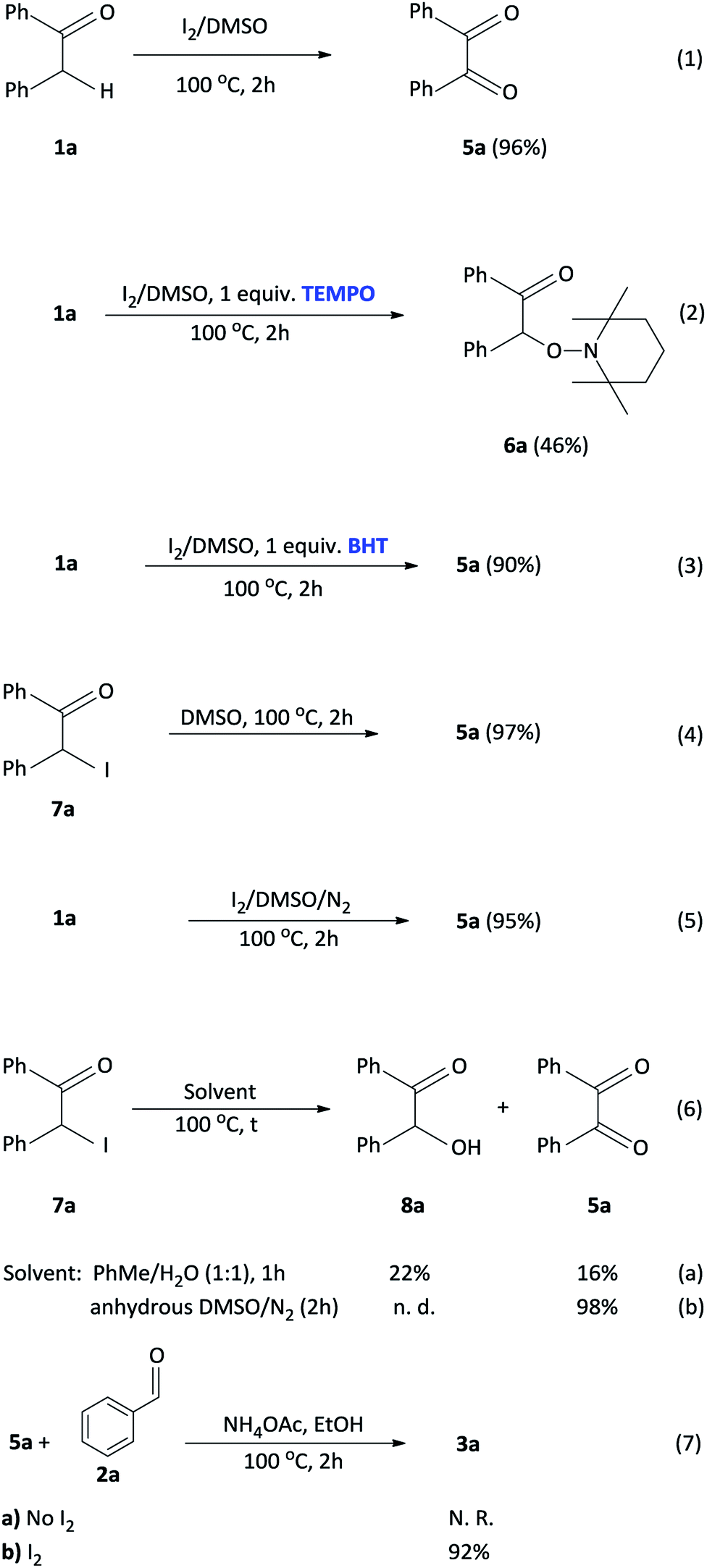 | ||
| Scheme 5 Control experiments. | ||
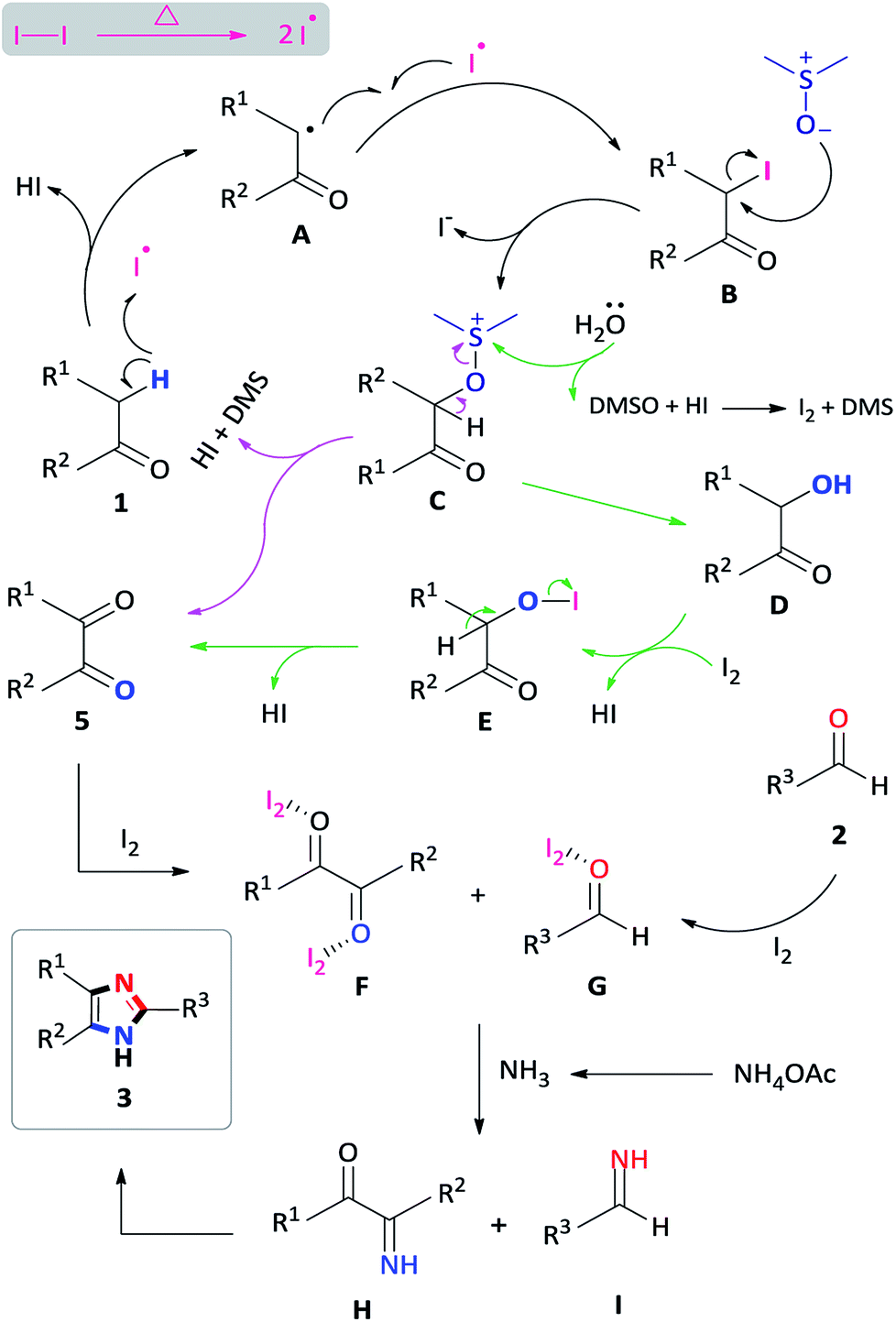 | ||
| Scheme 6 Plausible mechanism for imidazole synthesis. | ||
Conclusion
In summary, an improved, non-toxic, acid and transition metal free, sequential one-pot approach to 2,4,5-trisubstituted imidazoles was developed using α-methylene ketones instead of the traditional diketone. This environmentally friendly I2/DMSO system provides access to various substituted 2,4,5-trisubstituted imidazoles in moderate to excellent yields, under mild conditions and short reaction times. Moreover, a substrate scope was established by synthesizing novel 2,4,5-trisubstituted imidazoles, in good to excellent yields, which could potentially have biological properties. In addition, this approach could effect a domino convergent synthesis of the desired imidazole albeit in a moderate yield, through the simultaneous oxidation of benzyl phenyl ketone and benzyl alcohol. Supplementary studies expanding the scope of this methodology as well as in-depth mechanistic experiments are currently underway in our laboratories and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Research Foundation (NRF) of South Africa for a postgraduate bursary (JJ) and a Thuthuka research grant (VJ).Notes and references
- (a) M. Roue, I. Domart-Coulon, A. Ereskovsky, C. Djediat, T. Perez and M. L. BourguetKondracki, J. Nat. Prod., 2010, 73, 1277–1282 CrossRef CAS PubMed; (b) L. Zhang, X. M. Peng, G. L. Damu, R. X. Geng and C. H. Zhou, Med. Res. Rev., 2014, 34, 340–437 CrossRef CAS PubMed; (c) B. Cui, B. L. Zheng, K. He and Q. Y. Zheng, J. Nat. Prod., 2003, 66, 1101–1103 CrossRef CAS PubMed; (d) S. Tsukamoto, T. Kawabata, H. Kato, T. Ohta, H. Rotinsulu, R. E. P. Mangindaan, R. W. M. Van Soest, K. Ukai, H. Kobayashi and M. Namikoshi, J. Nat. Prod., 2007, 70, 1658–1660 CrossRef CAS PubMed; (e) Z. Jin, Nat. Prod. Rep., 2009, 26, 382–445 RSC; (f) S. Baroniya, Z. Anwer, P. K. Sharma, R. Dudhe and N. Kumar, Der Pharmacia Sinica, 2010, 1, 172–182 CAS; (g) I. Ali, M. N. Lone and H. Y. Aboul-Enein, Med. Chem. Commun., 2017, 8, 1742–1773 RSC.
- (a) M. A. Babizhayev, Life Sci., 2006, 78, 2343–2357 CrossRef CAS PubMed; (b) M. A. Babizhayev, G. M. Nikolayev, J. G. Nikolayeva and Y. E. Yegorov, Am. J. Ther., 2012, 19, 69–89 CrossRef PubMed.
- (a) W. Wang, X. Ji, A. Kapur, C. Zhang and H. Mattoussi, J. Am. Chem. Soc., 2015, 137, 14158–14172 CrossRef CAS PubMed; (b) J. V. Olsson, D. Hult, Y. Cai, S. Garcia-Gallego and M. Malkoch, Polym. Chem., 2014, 5, 6651–6655 RSC.
- (a) L. Chen, B. Zhao, Z. Fan, X. Liu, Q. Wu, H. Li and H. Wang, J. Agric. Food Chem., 2018, 66, 7319–7327 CrossRef CAS PubMed; (b) J.-H. Choi, N. Abe, A. Tanaka, K. Fushimi, Y. Nishina, A. Morita, Y. Kiriiwa, R. Motohashi, D. Hashizume, H. Koshino and H. Kawagishi, J. Agric. Food Chem., 2010, 158, 9956–9959 CrossRef PubMed.
- (a) P. Abhishek, C. J. Kulkarni, A. B. Tonzola and A. J. Samson, Chem. Mater., 2004, 16, 4556–4573 CrossRef; (b) Z. Wang, P. Lu, S. Chen, Z. Gao, F. Shen, W. Zhang, Y. Xu, H. S Kwok and Y. Ma, J. Mater. Chem., 2011, 21, 5451–5456 RSC; (c) K. Takagi, K. Kusafuka, Y. Ito, K. Yamauchi, K. Ito, R. Fukuda and M. Ehara, J. Org. Chem., 2015, 80, 7172–7183 CrossRef CAS PubMed; (d) D. Kumar, K. R. J. Thomas, C.-P. Lee and K.-C. Ho, J. Org. Chem., 2014, 79, 3159–3172 CrossRef CAS PubMed; (e) X. Chen, C. Jia, Z. Wan and X. Yao, Dyes Pigm., 2014, 104, 48–56 CrossRef CAS.
- (a) H. Yamashita and J. Abe, J. Phys. Chem. A, 2011, 115, 13332–13337 CrossRef CAS PubMed; (b) K. Motoh and J. Abe, J. Phys. Chem. A, 2011, 115, 4650–4656 CrossRef PubMed.
- (a) L. Zhang, Y. He, Y. Zhou, R. Yang, Q. yang, D. Qing and Q. Niu, Petroleum, 2015, 1, 237–243 CrossRef; (b) M. M. Antonijevic and M. B. Petrovic, Int. J. Electrochem. Sci., 2008, 1, 1–28 Search PubMed.
- Selected recent examples include: (a) A. Bamoniri, B. F. Mirjalili, S. Nazemian and N. Y. Mahabadi, Bulg. Chem. Commun., 2014, 46, 79–84 CAS; (b) A. Maleki, H. Movahed and R. Paydar, RSC Adv., 2016, 6, 13657–13665 RSC; (c) A. Teimouri and A. N. Chermahini, J. Mol. Catal. A: Chem., 2011, 346, 39–45 CrossRef CAS; (d) B. Sadeghi, B. B. F. Mirjalili and M. M. Hashemi, Tetrahedron Lett., 2008, 49, 2575–2577 CrossRef CAS; (e) A. R. Karimi, Z. Alimohammadi and M. M. Amini, Mol. Diversity, 2010, 14, 635–641 CrossRef CAS PubMed; (f) R. H. Shoar, G. Rahimzadeh, F. Derikvand and M. Farzaneh, Synth. Commun., 2010, 40, 1270–1275 CrossRef CAS; (g) S. A. Dake, M. B. Khedkar, G. S. Irmale, S. J. Ukalgaonkar, V. V. Thorat, S. A. Shintre and R. P. Pawar, Synth. Commun., 2012, 42, 1509–1520 CrossRef CAS; (h) H. R. Shaterian and M. Ranjbar, J. Mol. Liq., 2011, 160, 40–49 CrossRef CAS.
- (a) D. Yang, D. Fokas, J. Li, L. Yu and C. M. Baldino, Synthesis, 2005, 47–56 Search PubMed; (b) K. Bahrami, M. M. Khodaei and F. Naali, J. Org. Chem., 2008, 73, 6835–6837 CrossRef CAS PubMed; (c) L. J. Goossen and T. Knauber, J. Org. Chem., 2008, 73, 8631–8634 CrossRef CAS PubMed.
- (a) Y. Ashikari, T. Nokami and J.-I. Yoshida, J. Am. Chem. Soc., 2011, 133, 11840–11843 CrossRef CAS PubMed; (b) Y. Ashikari, T. Nokami and J.-I. Yoshida, Org. Lett., 2012, 14, 938–941 CrossRef CAS PubMed; (c) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed; For selected recent reviews see: (d) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2364 CrossRef CAS PubMed; (e) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381–3430 RSC; (f) J. Mlochowski and H. Wójtowicz-Mlochowski, Molecules, 2015, 20, 10205–10243 CrossRef CAS PubMed.
- (a) J. Xie, C. Pan, A. Abdukader and C. Zhu, Chem. Soc. Rev., 2014, 43, 5245–5256 RSC; (b) M. L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901–910 RSC; (c) K. Wu, Z. Huang, X. Qi, Y. Li, G. Zhang, C. Liu, H. Yi, L. Meng, E. E. Bunel, J. T. Miller, C.-W. Pao, J.-F. Lee, Y. Lan and A. Lei, Sci. Adv., 2015, 1, e1500656 CrossRef PubMed; (d) S. Guo, Q. Zhang, H. Li, H. Guo and W. He, Nano Res., 2017, 10, 3261–3267 CrossRef CAS; (e) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC.
- S. Cacchi, G. Fabrizi, A. Goggiamani, A. Lazzetti and R. Verdiglione, Synthesis, 2013, 45, 1701–1707 CrossRef CAS.
- J.-W. Yu, S. Mao and Y.-Q. Wang, Tetrahedron Lett., 2015, 56, 1575–1580 CrossRef CAS.
- R. Chebolu, A. Bahuguna, R. Sharma, V. K. Mishra and P. C. Ravikumar, Chem. Commun., 2015, 51, 15438–15441 RSC.
- H. Wang, Z. Wang, H. Huang, J. Tan and K. Xu, Org. Lett., 2016, 18, 5680–5683 CrossRef CAS PubMed.
- G. Urgoitia, A. Maiztegi, R. SanMartin, M. T. Herrrero and E. Dominguez, RSC Adv., 2015, 5, 103210–103217 RSC.
- M. Lian, Q. Li, Y. Zhu, G. Yin and A. Wu, Tetrahedron, 2012, 68, 9598–9605 CrossRef CAS.
- V. Jeena and M. Mazibuko, Heterocycles, 2017, 94, 1909–1922 CrossRef CAS.
- J. Jayram and V. Jeena, Green Chem., 2017, 19, 5841–5845 RSC.
- (a) R. Zhou, H. Liu, H. Tao, X. Yu and J. Wu, Chem. Sci., 2017, 8, 4654–4659 RSC; (b) U. K. Das, L. J. W. Shimon and D. Milstein, Chem. Commun., 2017, 53, 13133–13136 RSC; (c) Y. Z. Yan, Y. H. Zhang, C. T. Feng, Z. G. Zha and Z. Y. Wang, Angew. Chem., Int. Ed., 2012, 51, 8077–8081 CrossRef CAS PubMed; (d) Y. P. Zhu, Z. Fei, M. C. Liu, F. C. Jia and A. X. Wu, Org. Lett., 2013, 15, 378–381 CrossRef CAS PubMed; (e) Y. Z. Yan, Y. Xu, B. Niu, H. F. Xie and Y. Q. Liu, J. Org. Chem., 2015, 80, 5581–5587 CrossRef CAS PubMed; (f) C. Mukhopadhyay and P. K. Tapaswi, Green Chem. Lett. Rev., 2015, 5, 109–120 CrossRef.
- (a) Y.-D. Wu, X. Geng, Q. Gao, J. Zhang, X. Wu and A.-X. Wu, Org. Chem. Front., 2016, 3, 1430–1434 RSC; (b) K. K. D. R. Viswanadham, M. P. Reddy, P. Sathyanarayana, O. Ravi, R. Kant and S. R. Bathula, Chem. Commun., 2014, 50, 13517–13520 RSC; (c) C. Chen, C. Chen, B. Li, J. Tao and J. Peng, Molecules, 2012, 17, 12506–12520 CrossRef CAS PubMed; (d) B. P. Bandgar, S. V. Bettigeri and N. S. Joshi, Synth. Commun., 2004, 34, 1447–1453 CrossRef CAS; (e) X. Yi, L. Jiao and C. Xi, Org. Biomol. Chem., 2016, 14, 9912–9918 RSC.
- (a) S. Song, X. Huang, Y. –F. Liang, C. Tang, X. Li and N. Jiao, Green Chem., 2015, 17, 2727–2731 RSC; (b) J.-C. Xiang, Q.-H. Gao and A.-X. Wu, in Solvents as reagents in organic synthesis: reactions and applications, ed. X.-F. Wu, Wiley-VCH Verlag GmbH & Co., Germany, 2017, ch. 7, pp. 315–349 Search PubMed.
- For selected recent review see: A. Monga, S. Bagchi and A. Sharma, New J. Chem., 2018, 52, 1551–1576 RSC.
- (a) J. Jayram and V. Jeena, Heterocycles, 2016, 92, 2213–2224 CrossRef CAS; (b) S. Naidoo and V. Jeena, Heterocycles, 2016, 92, 1655–1664 CrossRef CAS.
- (a) G. A. Price, A. K. Brisdon, K. R. Flower, R. G. Pritchard and P. Quayle, Tetrahedron Lett., 2014, 55, 151–154 CrossRef CAS; (b) D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 596–600 RSC; (c) M. Kidwai, P. Mothsra, V. Bansal and R. Goyal, Monatshefte für Chemie, 2006, 137, 1189–1194 CrossRef CAS.
- (a) S. Balalaie and A. Arabanian, Green Chem., 2000, 2, 274–276 RSC; (b) Y. Xu, L. F. Wan, H. Salehi, W. Deng and X. Qing, Heterocycles, 2004, 63, 1613–1618 CrossRef CAS; (c) A. Parveen, A. Ahmed and S. K. Ahmed, Res. J. Pharm., Biol. Chem. Sci., 2010, 1, 943–951 CAS; (d) E. Chauveau, C. Marestin, F. Schiets and R. Mercier, Green Chem., 2010, 12, 1018–1022 RSC; (e) M. Esmaeilpour, J. Javidi and M. Zandi, New J. Chem., 2015, 39, 3388–3398 RSC; (f) Z. Zarnegar and J. Safari, New J. Chem., 2014, 38, 4555–4565 RSC; (g) J. Safari and Z. Zarnegar, Ultrason. Sonochem., 2013, 20, 740–746 CrossRef CAS PubMed.
- (a) K. J. Wicht, J. M. Combrinck, P. J. Smith, R. Hunter and T. J. Egan, ACS Med. Chem. Lett., 2017, 8, 201–205 CrossRef CAS PubMed; (b) F.-j. Gamo, L. M. Sanz, J. Vidal, C. de Cozar, E. Alvarez, J. L. Lavandera, D. E. Vanderwall, D. V. S. Green, V. Kumar, S. Hasan, J. R. Brown, C. E. Peishoff, L. R. Cardon and J. F. Garcia-Bustos, Nature, 2010, 465, 305–312 CrossRef CAS PubMed; (c) K. Y. Fong, R. D. Sandlin and D. W. Wright, Int. J. Parasitol.: Drugs Drug Resist., 2015, 5, 84–91 Search PubMed; (d) D. Plouffe, A. Brinker, C. McNamara, K. Henson, N. Kato, K. Kuhen, A. Nagle, F. Adrian, J. T. Matzen, P. Anderson, T.-g. Nam, N. S. Gray, A. Chatterjee, J. Janes, S. F. Yan, R. Trager, J. S. Caldwell, P. G. Schultz, Y. Zhou and E. A. Winzeler, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9059–9064 CrossRef CAS PubMed.
- (a) C. R. Lambert, H. S. Black and T. G. Truscott, Free Radical Biol. Med., 1996, 21, 395–400 CrossRef CAS; (b) H. S. Black, Front. Biosci., 2002, 7, 1044–1055 CrossRef.
- V. Estevez, M. Villacampa and J. C. Menendez, Chem. Commun., 2013, 49, 591–593 RSC.
- (a) J. Gromada and K. Matyjaszewski, Macromolecules, 2001, 34, 7664–7671 CrossRef CAS; (b) H.-Y. Huang, H.-R. Wu, F. Wei, D. Wang and L. Liu, Org. Lett., 2015, 17, 3702–3705 CrossRef CAS PubMed.
- (a) H.-L. Li, X.-L. An, L.-S. Ge, X. Luo and W.-P. Deng, Tetrahedron, 2015, 71, 3247–3252 CrossRef CAS; (b) Y. Liang, K. Wu, S. Song, X. Li, X. Huang and N. Jiao, Org. Lett., 2015, 17, 876–879 CrossRef CAS PubMed.
- (a) D. von de Heiden, S. Bozkus, M. Klussmann and M. Breugst, J. Org. Chem., 2017, 82, 4037–4043 CrossRef PubMed; (b) C. Xie, Z. Zhang, B. Yang, G. Song, H. Gao, L. Wen and C. Ma, Tetrahedron, 2015, 71, 1831–1837 CrossRef CAS.
- S. S. K. Boominathan, C.-Y. Chen, P.-J. Huang, R.-J. Hou and J.-J. Wang, New J. Chem., 2015, 39, 6914–6918 RSC.
- (a) M. Kidwai, P. Mothsra, V. Bansal, R. K. Somvanshi, A. S. Ethayathulla, S. Dey and T. P. Singh, J. Mol. Catal. A: Chem., 2007, 265, 177–182 CrossRef CAS; (b) S. S. Ali, Arch. Appl. Sci. Res., 2010, 2, 392–397 CAS; (c) L. Wu, B. Niu, W. Li and F. Yan, Bull. Korean Chem. Soc., 2009, 30, 2777–2778 CrossRef CAS; (d) S. Patil, J. Patil and S. Dharap, WJPR, 2015, 4, 2476–2483 CAS; (e) H. P. Kalmode, K. S. Vadagaonkar and A. C. Chaskar, RSC Adv., 2014, 4, 60316–60326 RSC; (f) H. P. Kalmode, K. S. Vadagaonkar and A. C. Chaskar, Synthesis, 2015, 47, 429–438 CrossRef CAS; (g) R. Deshidi, M. Kumar, S. Devari and B. A. Shah, Chem. Commun., 2014, 50, 9533–9535 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and copies of spectroscopic data. See DOI: 10.1039/c8ra07238h |
| This journal is © The Royal Society of Chemistry 2018 |