
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Long noncoding RNA PCA3 regulates glycolysis, viability and apoptosis by mediating the miR-1/CDK4 axis in prostate cancer
Shuo Gu,
Xiaobing Niu,
Fei Mao and
Zongyuan Xu *
*
Department of Urology, The Affiliated Huaian No. 1 People's Hospital of Nanjing Medical University, No. 1, West Huanghe Road, Huaiyin District, Huaian City, Jiangsu Province 223300, P. R. China. E-mail: ldyyzhao@126.com; Tel: +86-139-5234-6996
First published on 8th November 2018
Abstract
Prostate cancer is one of the common tumor malignancies in men worldwide. Although long noncoding RNAs (lncRNAs) have been demonstrated to play essential roles in the progression of prostate cancer, the roles and potential mechanism of lncRNA prostate cancer antigen 3 (PCA3) remain poorly understood. In the present study, we investigated the role of PCA3 in aerobic glycolysis, viability and apoptosis in prostate cancer cells and probed the interaction between PCA3 and microRNA-1 (miR-1)/cyclin-dependent kinase 4 (CDK4). Here we found that PCA3 and CDK4 were up-regulated while miR-1 was down-regulated in prostate cancer tissues and cells. Moreover, knockdown of PCA3 inhibited aerobic glycolysis and viability and induced apoptosis in prostate cancer cells. Intriguingly, PCA3 was bound to miR-1 and inhibition of miR-1 reversed the regulatory effect of PCA3 knockdown on aerobic glycolysis, viability and apoptosis in prostate cancer cells. Besides, CDK4 was indicated as a target of miR-1 and it was regulated by PCA3 through functioning as a competing endogenous RNA (ceRNA) of miR-1 in prostate cancer cells. The results indicated that PCA3 might drive aerobic glycolysis, viability and apoptosis by regulating the miR-1/CDK4 axis in prostate cancer cells, providing a promising avenue for treatment of prostate cancer.
Introduction
Prostate cancer is one of the most common malignancies causing 1–2% of deaths in men.1 Much progress has been made in the diagnosis and treatment of prostate cancer in recent years.2 A recent study has demonstrated that the incidence of prostate cancer rises with age and many therapeutics have been used for prostate cancer treatment, such as radiotherapy, androgen-deprivation therapy, chemotherapy and immunotherapy.3 However, the effective treatment of patients with prostate cancer remains an open challenge. Aerobic glycolysis is reported as one of the major characteristics of cancer metabolism and contributes to cancer progression.4 A better understanding of the molecular mechanism that underlies aerobic glycolysis and tumor growth may be promising for treatment of prostate cancer.Long noncoding RNAs (lncRNAs) have been shown to play a pivotal role in tumorigenesis, metastasis, prognosis and diagnosis of prostate cancer.5 Moreover, lncRNAs have been reported to be associated with tumor metabolism in prostate cancer.6 For example, lncRNA-p21 displays an inhibitory effect on development of prostate cancer by affecting glycolysis.7 LncRNA prostate cancer-associated transcript 1 (PCAT1) has been reported to regulate development of cancers, such as prostate cancer and hepatocellular.8 Previous study has indicated many lncRNAs as oncogene in many cancers, including lncRNA prostate cancer antigen 3 (PCA3).9,10 PCA3 has been regarded as one of new biomarkers for prognosis and diagnosis of prostate cancer.11 Moreover, former work suggests that PCA3 is ectopic and has important clinical value for detection of prostate cancer.12 Besides, PCA3 has been suggested to positively correlate with risk and aggressiveness of prostate cancer.13 However, in-depth study of PCA3 participating in glycolysis and progression of prostate cancer is required.
Emerging evidence suggests that the lncRNAs/microRNAs (miRNAs)/mRNA interaction networks provide new insight for understanding the molecular mechanisms of prostate cancer.14 The available evidence indicates competing endogenous RNA (ceRNA) hypothesis, which suggests that lncRNA-related ceRNA networks play essential role in tumor progression through sponging miRNA to derepress mRNA expression.15 miR-1 has been demonstrated as a tumor suppressor and to be associated with prognosis of prostate cancer by regulating cell proliferation and motility.16 Moreover, miR-1 has been reported to be implicated in cell proliferation, migration and apoptosis in prostate cancer cells.17 Besides, bioinformatics analysis provides the putative link between PCA3 and miR-1. However, there is no direct evidence in support of miR-1 driving glycolysis in prostate cancer. Intriguingly, cyclin-dependent kinase 4 (CDK4) is shown to be one of target genes of miR-1 and associated with progression of esophageal squamous cell carcinoma cells.18 Moreover, CDK proteins have been reported to contribute to metabolic adaptation to tumor growth, including glycolytic metabolism.19 Notably, CDK4 has an impact on prostate cancer progression by regulating cell cycle, apoptosis, proliferation, migration and invasion.20 Hence, we hypothesized the PCA3/miR-1/CDK4 network might play a pivotal role in progression of prostate cancer. In the present study, we first measured the expressions of PCA3, miR-1 and CDK4 in prostate cancer tissues and cells. Moreover, we investigated the roles of PCA3 and miR-1 in glycolysis, viability and apoptosis of prostate cancer cells and probed the interaction between miR-1 and PCA3 or CDK4.
Materials and methods
Specimens
A total of 20 cancer samples and adjacent normal tissues were collected from 20 patients with prostate cancer at the Affiliated Huaian No. 1 People's Hospital of Nanjing Medical University. All samples were snap-frozen in liquid nitrogen immediately and stored at −80 °C until used. All patients had signed the informed consent and this study was conducted under the instructions approved by the Institutional Research Ethics Committee of the Affiliated Huaian No. 1 People's Hospital of Nanjing Medical University.Cell culture and transfection
The normal human prostate epithelial cells (RWPE-1), four prostate cancer cell lines (C4-2, 22Rv1, LNCaP and PC3) and 293T cells were obtained from American Tissue Culture Collection (ATCC, Manassas, VA, USA). All cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA) in a humidified incubator at 37 °C in 5% CO2 during the study.PCA3 overexpression plasmids were constructed by a ligation of pcDNA and amplified with the following primers (forward, 5′-GGATCCTAATACGACTCACTA TAGGGACAGAGGGGAGATTTG-3′; reverse, 5′-TCTAGATGAGGGTTAGAAAT ATGAAATGATT-3′). The si-RNA against PCA3 sequences were designed as forward: 5′-CAGAAUUCAUGCAGUGCAAdTdT-3′ and reverse: 5′-UUGCACUG CAUGAAUUCUG-3'. The negative control (NC) sequence (forward: 5′-UUCUCCG AACGUGUCACGUUUC-3′), and (reverse: 5′-GAAACGUGACACGUUCGGAG AA-3′). The PCA3, pcDNA, si-PCA3, si-NC, miR-1 mimics, anti-miR-1, miR-NC and anti-miR-NC were synthesized by Genepharma (Shanghai, China). A final concentration of 50 nM oligonucleotides or 2 μg of vectors were mixed with 10 μL Lipofectamine 2000 (Invitrogen) for 10 min and then the mixture was transfected into 22Rv1 and LNCaP cells via according to the manufacturer's protocol.
RNA extraction and quantitative reverse transcription PCR (qRT-PCR)
Total RNA was extracted from specimens or cells using Trizol reagent (Thermo Fisher, Wilmington, DE, USA) and analyzed with a NanoDrop Spectrophotometer (NanoDrop, Wilmington, DE, USA) following the manufacturer's instructions. For PCA3 and CDK4 detection, first strand cDNA was synthesized by TaqMan Reverse Transcription Kit and then used for qRT-PCR using SYBR green (Toyobo, Tokyo, Japan) with β-actin as internal control. For miR-1 detection, TaqMan microRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) was used for synthesization of cDNA, followed by amplification instructions with SYBR green and U6 was regarded as a housekeeping gene. All primers were listed as: PCA3 (forward, 5′-ACACAGGAAGCACAAAAGG-3′; reverse, 5′-GATGACCCAAGATGGCGG C-3′), CDK4 (forward, 5′-GTGGAAACTCTGAAGCCGAC-3'; reverse, 5′-CAGA GATTCGCTTGTGTGGG-3′), β-actin (forward, 5′-AGCAGCATCGCCCCAAA GTT-3′; reverse, 5′-GGGCACGAAGGCTCATCATT-3′); miR-1, (forward, 5′-GGGGTGGAATGTAAAGAA-3′; reverse, 5′-TGCGTGTCGTGGAGTC-3′); U6 (forward, 5′-GCTTCGGCAGCACATATACTAAAAT-3′; reverse, 5′-CGCTTCAC GAATTTGCGTGTCAT-3′). Results were evaluated with 2−ΔΔCt method.Measurement of glucose consumption and lactate production
Transfected 22Rv1 and LNCaP cells were starved in 96-well plate with serum-free medium overnight. After three washes in PBS, cells were collected and used for analyses of glucose consumption and lactate production by using Glucose Uptake Colorimetric Assay Kit or Lactate Assay Kit (Sigma, St. Louis, MO, USA) according to the manufacturer's instructions, respectively. The concentrations of glucose and lactate were measured by using microplate reader (Molecular Devices, Palo Alto, CA, USA) and normalized by the total protein concentration determined by bicinchoninic acid (BCA) assay kit (Thermo Fisher).Cell viability assay
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay was performed to measure cell viability. Transfected 22Rv1 and LNCaP cells were seeded into 96-well plates at the density of 0.5 × 104 cells per well for 24, 48 or 72 h. Each sample was prepared in triplicate and the reaction was repeated three times. Then cells were treated with 10 μL MTT (5 mg mL−1; Sigma) and maintained for 4 h at 37 °C. Then remove the supernatant and dimethyl sulfoxide (DMSO, 100 μL per well; Sigma) was added to cells to dissolve the formazan. The absorbance was measured at 490 nm with a microplate reader (Bio-Rad, Hercules, CA, USA).Cell apoptosis assay
For cell apoptosis assay, flow cytometry was conducted into prostate cancer cells by using Annexin V-FITC/propidium iodide (PI) detection kit (Sigma). Transfected 22Rv1 and LNCaP cells were resuspended in binding buffer and treated with Annexin V-FITC and PI for 20 min without light. Every sample was prepared in triplicate. The positive cells were analyzed through a flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).Luciferase assays
Putative binding sites of miR-1 and 3′-untranslated regions (3′-UTR) sequences of PCA3 or CDK4 were predicted through the miRcode 11 (http://www.mircode.org/) and TargetScan Release 7.2 (http://www.targetscan.org/vert_72/) online. The full-length PCA3 or CDK4 were amplified and cloned into pGL3 luciferase reporter vector (Promega, Madison, WI, USA) to generate the wide-type plasmid (PCA3-WT or CDK4-WT). Mutations of 3′-UTR sequences of PCA3 or CDK4 were afforded through Quik Change Multi Site-Directed Mutagenesis kit (Agilent Technologies, Little Falls, CA, USA), respectively. Before transfection, 293T cells were seeded into 24-well plates at a density of 1 × 105 cells per well for 24 h. Then 50 ng of WT or MUT luciferase reporter plasmids, 10 ng of Renilla luciferase vector with 50 nM miR-1 mimics, miR-NC, or PCA3 were co-transfected in 293T cells using Lipofectamine 2000 following the manufacturer's protocols. At 48 h after transfection, cells were collected for luciferase activities analysis using Luciferase Assay Kit (Promega) and Renilla luciferase activities were used for normalization.RNA immunoprecipitation (RIP)
The RIP was conducted in 22Rv1 cells by using Magna RNA-binding protein immunoprecipitation kit (Millipore, Billerica, MA, USA). In brief, 22Rv1 cells were lysed in cell lysis buffer and then incubated in RIP buffer containing magnetic beads conjugated with antibody against Argonaute 2 (anti-Ago2) or anti-IgG for 2 h at 4 °C. After washed with PBS, the RNA-protein-beads complexes were treated with proteinase K (Sigma) and isolated by Trizol reagent. qRT-PCR was performed in purified products to evaluate the abundance of PCA3 or miR-1.Biotin pull down assay
Biotin pull down analysis was conducted in 22Rv1 cells to probe the interaction between PCA3 and miR-1 using RNA-Protein Pull Down Kit (Thermo Fisher). PCA3-WT, PCA3-MUT, miR-1-WT, miR-1-MUT or corresponding NC was labelled with biotin and transfected into 22Rv1 cells. After the transfection, cells were lysed and then incubated with magnetic beads for 4 h at 4 °C. Then the beads were eluted with biotin elution buffer and miR-1 or PCA3 abundance was measured by qRT-qPCR.Western blotting
Transfected 22Rv1 and LNCaP cells were lysed in RIPA lysis buffer (Thermo Fisher) containing 1% protease inhibitor and then total proteins were quantified by BCA protein assay kit (Sigma) according to the manufacturer's instructions. Samples were boiled at 98 °C for 5 min and then loaded onto SDS-PAGE gel, followed by transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). The membranes were blocked with 5% non-fat milk in Tris–buffer saline containing 0.1% Tween 20 (TBST) for 2 h at room temperature and then incubated with primary monoclonal antibodies against CDK4 (ab137675, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2000 dilution, Abcam, Cambridge, UK) or β-actin (ab227387, 1:10000 dilution, Abcam) overnight at 4 °C. After three washes in TBST, membranes were hatched with secondary antibodies (ab205718, 1:20000 dilution, Abcam) conjugated by horseradish peroxidase (HRP) for 2 h at room temperature. Signals were visualized by using enhanced chemiluminescence (ECL) chromogenic substrate (GE Healthcare, Amersham, UK) and analyzed by using Image Lab software (Bio-Rad, Hercules, CA, USA).
:2000 dilution, Abcam, Cambridge, UK) or β-actin (ab227387, 1:10000 dilution, Abcam) overnight at 4 °C. After three washes in TBST, membranes were hatched with secondary antibodies (ab205718, 1:20000 dilution, Abcam) conjugated by horseradish peroxidase (HRP) for 2 h at room temperature. Signals were visualized by using enhanced chemiluminescence (ECL) chromogenic substrate (GE Healthcare, Amersham, UK) and analyzed by using Image Lab software (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data were expressed as the mean ± standard error of mean from three independent experiments. Student's t test was conducted to analyze the significant differences between groups by using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA, USA). The statistically significant was considered as p < 0.05 in all graphs.Results
PCA3 was significantly enhanced in prostate cancer tissues and cells
To investigate the role of PCA3 in prostate cancer, the abundance of PCA3 was first measured in prostate cancer tissues and cells. As a result, a strong increase of PCA3 abundance was shown in prostate cancer tissues compared with that in normal control (Fig. 1A). Moreover, the patients were classified by Gleason score and higher level of PCA3 was exhibited in patients with Gleason score ≥ 7 than that in Gleason score < 7, suggesting that upregulated PCA3 was positively correlated with Gleason score of the patients (Fig. 1B). In addition, PCA3 expression was also markedly increased in C4-2, 22Rv11, LNCaP and PC3 cells, especially in 22Rv1 and LNCaP cells (Fig. 1C). Hence, 22Rv1 and LNCaP cells were used for further experiments in vitro.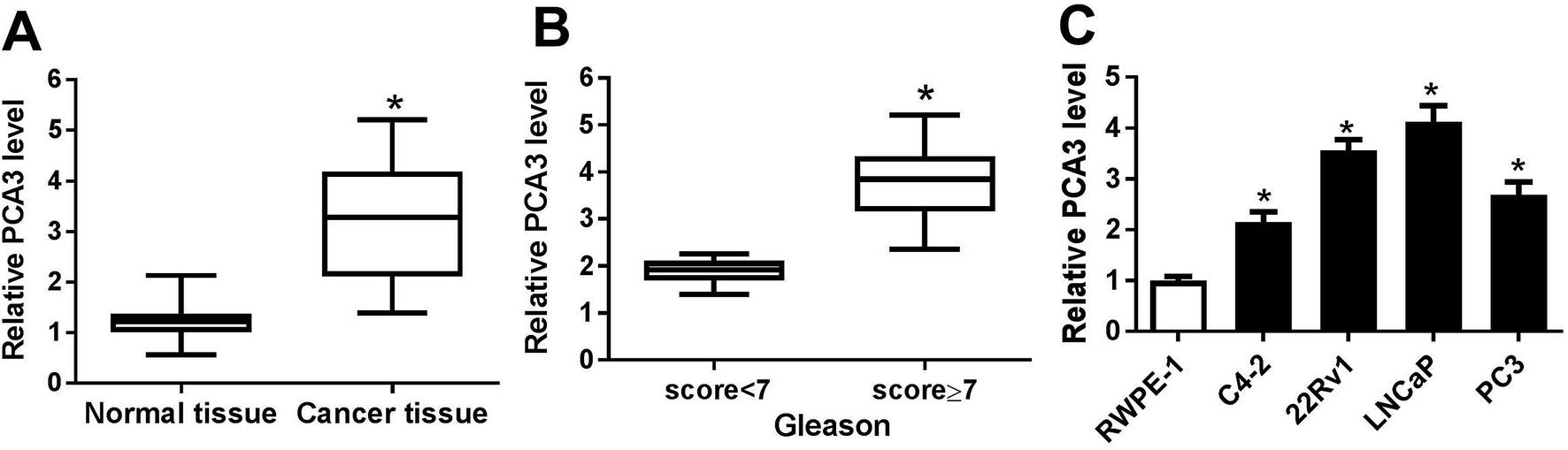 | ||
| Fig. 1 (A) The expression of PCA3 was measured in prostate cancer tissues and normal. (B) The level of PCA3 was detected in tissues from patients with different Gleason score of prostate cancer. (C) The abundance of PCA3 was examined in prostate cancer cells (C4-2, 22Rv1, LNCaP, PC3) and RWPE-1. *p < 0.05. | ||
Abrogation of PCA3 inhibited aerobic glycolysis, viability and promoted apoptosis in prostate cancer cells
To explore the potential roles of PCA3 in progression of prostate cancer, the effect of PCA3 inhibition on aerobic glycolysis, viability and apoptosis was investigated in 22Rv1 and LNCaP cells. As expected, the PCA3 abundance was effectively suppressed in si-PCA3-transfected 22Rv1 and LNCaP cells compared with that in si-NC-treated cells (Fig. 2A). Deficiency of PCA3 led to great loss of glucose consumption and lactate production in 22Rv1 and LNCaP cells, indicating that inhibition of PCA3 blocked the aerobic glycolysis (Fig. 2B and C). Moreover, knockdown of PCA3 dramatically impaired viability of 22Rv1 and LNCaP cells at 24, 48 and 72 h after transfection (Fig. 2D). Besides, abrogation of PCA3 induced great apoptosis in 22Rv1 as well as LNCaP cells (Fig. 2E).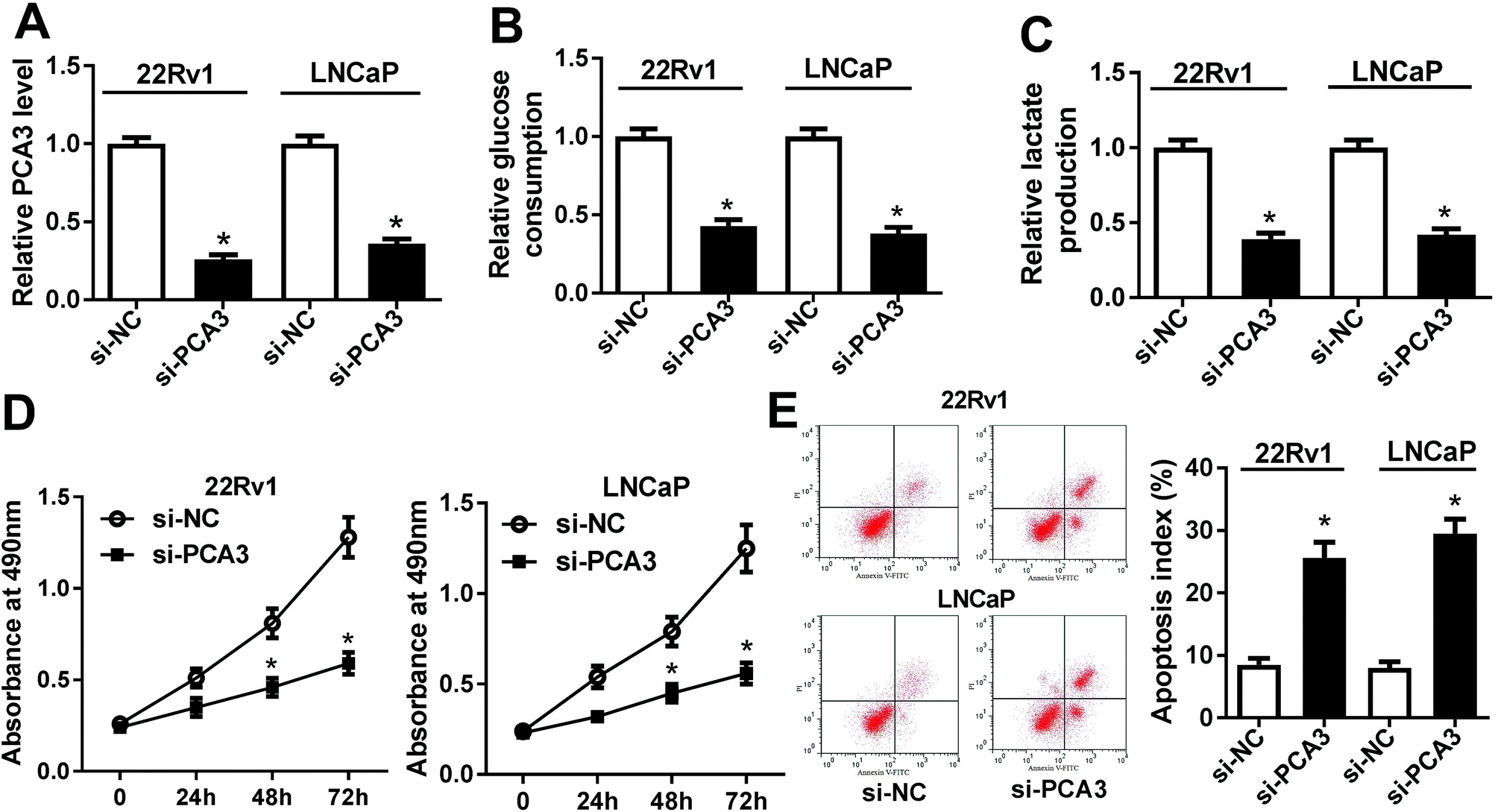 | ||
| Fig. 2 (A) The expression of PCA3 was measured in 22Rv1 and LNCaP cells with si-PCA3 or si-NC transfection. (B and C) The glucose consumption and lactate production were investigated in si-PCA3 or si-NC-treated 22Rv1 and LNCaP cells. (D) The cell viability was detected in 22Rv1 and LNCaP cells transfected with si-PCA3 or si-NC. (E) The effect of PCA3 inhibition on cell apoptosis was evaluated in 22Rv1 and LNCaP cells transfected with si-PCA3 or si-NC. *p < 0.05. | ||
PCA3 sponged miR-1 in prostate cancer cells
To address the underlying mechanism allows PCA3 involved in progression of prostate cancer, bioinformatics analysis was conducted by miRcode 11 online. The software described the putative binding sites of PCA3 and miR-1, suggesting that PCA3 might bound to miR-1 (Fig. 3A). Hence, luciferase activity, RIP and biotin pull down assays were performed to validate the prediction. As a result, a great loss of luciferase activity was observed in 293T cells co-transfected with PCA3-WT and miR-1 compared with that in cells treated with PCA3-MUT and miR-1, whereas the luciferase activity of cells showed little change in response to miR-NC treatment (Fig. 3B). Moreover, Ago2 treatment showed notably elevated levels of PCA3 and miR-1 compared with IgG group in 22Rv1 cells (Fig. 3C). In addition, high expression of miR-1 was displayed in 22Rv1 cells transfected with bio-probe-PCA3-WT compared with that in NC treatment, while bio-probe-PCA3-WT transfection failed to show efficacy (Fig. 3D). Meanwhile, 22Rv1 cells with bio-miR-1-WT transfection showed higher PCA3 enrichment, whereas the efficacy was lost with respect to bio-miR-1-MUT (Fig. 3D). Besides, the abundance of miR-1 was measured in prostate cancer tissues and cells, respectively. Results indicated that miR-1 was significantly impaired in cancer tissues and cells, and it was negatively correlated with Gleason score (Fig. 3E–G).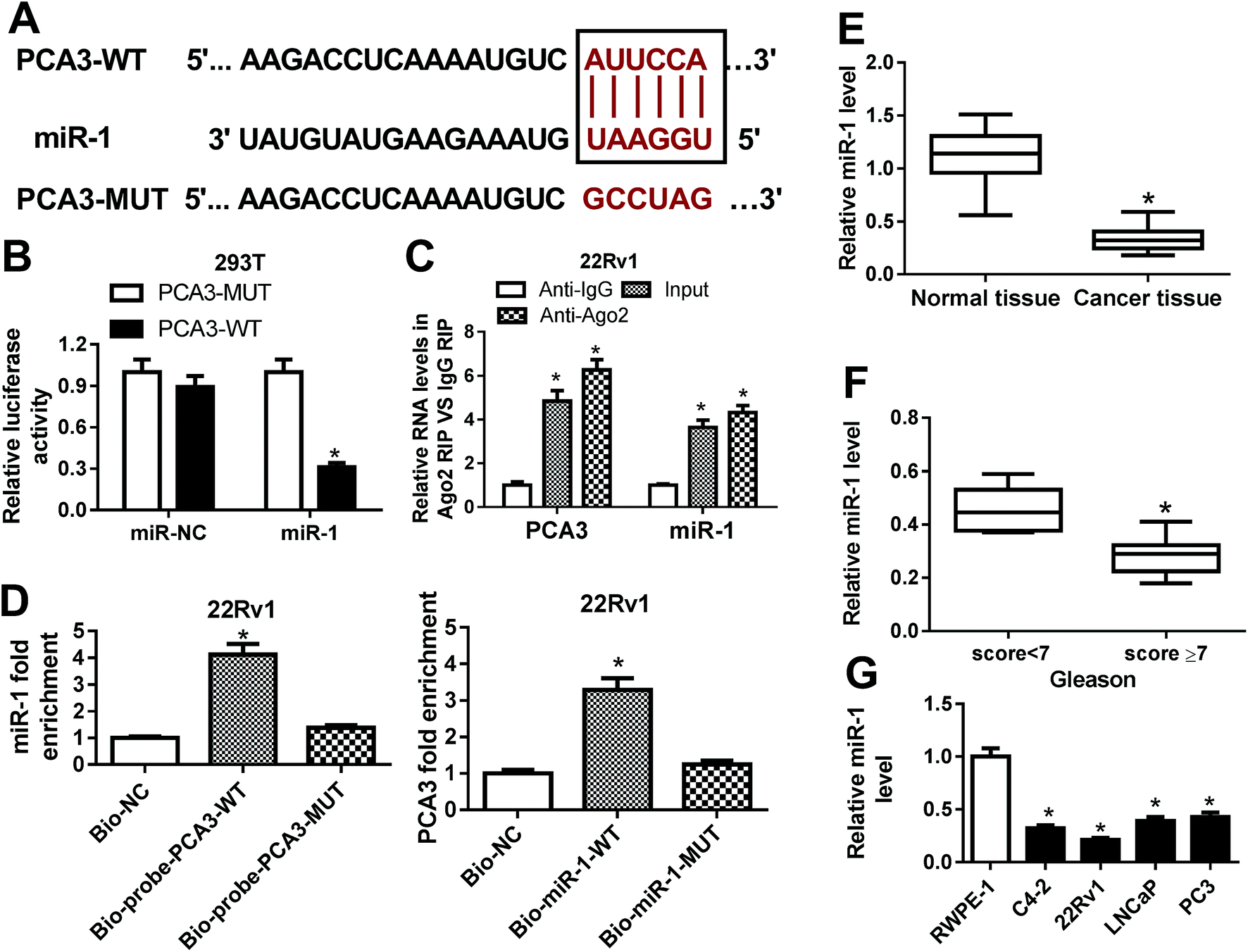 | ||
| Fig. 3 (A) The potential binding sites of PCA3 and miR-1 were predicted by miRcode 11 software online. (B) The luciferase activity was measured in 293T cells co-transfected with PCA3-MUT or PCA3-WT and miR-NC or miR-1. (C) The interaction between PCA3 and miR-1 was investigated by RIP in 22Rv1 cells. (D) The biotin pull down assay was conducted in 22Rv1 cells. (E) The expression of mir-1 was measured in prostate cancer and normal samples. (F) The level of miR-1 was detected in prostate cancer patients with Gleason score < 7 or ≥7. (G) The abundance of miR-1 was examined in prostate cancer cells. *p < 0.05. | ||
Knockdown of miR-1 reversed the effect of PCA3 interference on aerobic glycolysis, viability and apoptosis in prostate cancer cells
Seeing that PCA3 interacted with miR-1, we next investigated whether miR-1 was implicated in PCA3-mediated progression of prostate cancer in vitro. 22Rv1 and LNCaP cells were transfected with si-PCA3, anti-miR-1 or their corresponding NC. As a result, interference of PCA3 induced miR-1 expression, whereas deficiency of miR-1 attenuated the abundance in 22Rv1 and LNCaP cells (Fig. 4A). Moreover, knockdown of miR-1 reversed the inhibitory effect of PCA3 depletion on glucose consumption in 22Rv1 and LNCaP cells compared with anti-miR-NC treatment (Fig. 4B). Similarly, lactate production displayed same trends in response to transfection of si-PCA3 or (and) anti-miR-1 in 22Rv1 and LNCaP cells (Fig. 4C). Additionally, knockdown of miR-1 overturned the PCA3 abrogation-mediated inhibition of viability in 22Rv1 and LNCaP cells (Fig. 4D). Besides, absence of miR-1 ablated si-PCA3-induced apoptosis in 22Rv1 and LNCaP cells transfected with si-PCA3 and anti-miR-1 compared with that transfected with si-PCA3 and anti-miR-NC (Fig. 4E).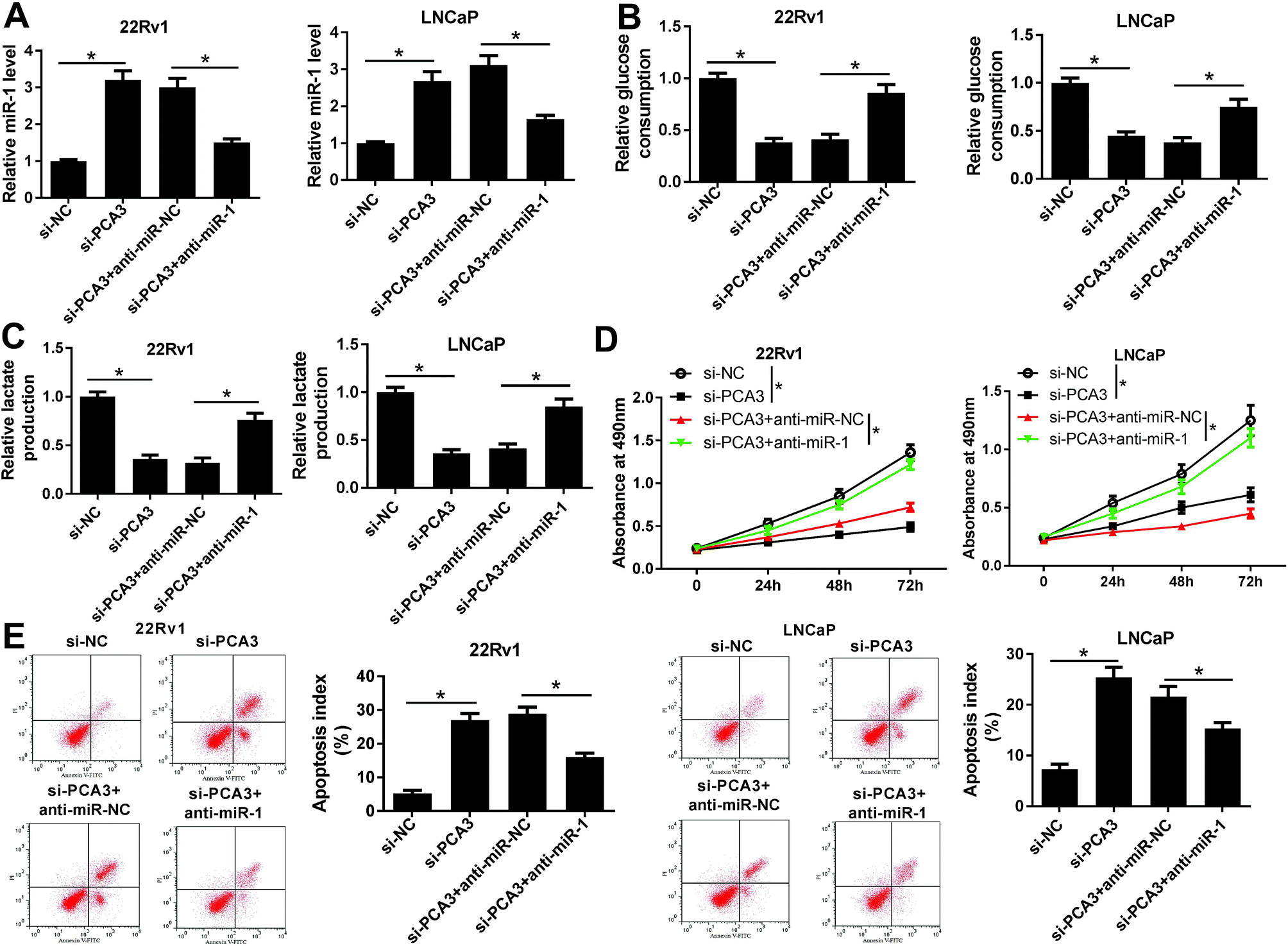 | ||
| Fig. 4 (A) The effect of inhibition of PCA3 and miR-1 on miR-1 expression was measured in transfected 22Rv1 and LNCaP cells. (B and C) The effect of miR-1 depletion on si-PCA3-mediated glucose consumption and lactate production in 22Rv1 and LNCaP cells. (D) Cell viability was investigated in 22Rv1 and LNCaP cells transfected with si-NC, si-PCA3, si-PCA3+anti-miR-NC or si-PCA3+anti-miR-1. (E) Cell apoptosis was analyzed in 22Rv1 and LNCaP cells transfected with si-NC, si-PCA3, si-PCA3+anti-miR-NC or si-PCA3+anti-miR-1. *p < 0.05. | ||
CDK4 was a target of miR-1 in prostate cancer cells
Given that functional miRNAs were afforded by regulating target genes, a promising target of miR-1 was expected for better understanding of the mechanism in prostate cancer. Bioinformatics analysis revealed the potential binding sites of miR-1 and CDK4 by TargetScan Release 7.2 online (Fig. 5A). Hence, luciferase activity was measured in 293T cells and results showed that miR-1 overexpression led to a strong reduction of luciferase activity compared with miR-NC treatment in CDK4-WT-transfected cells, whereas the efficacy was lost in response to CDK4-MUT transfection (Fig. 5B). Moreover, CDK4 mRNA expression was strikingly enhanced in prostate cancer tissues and positively associated with the Gleason score (Fig. 5C and D). Similarly, the prostate cancer cells also showed highly expressed CDK4 compared with RWPE-1 cells at mRNA level (Fig. 5E). Notably, addition of miR-1 effectively inhibited CDK4 protein level, whereas absence of miR-1 specially induced the abundance of CDK4 protein in 22Rv1 cells (Fig. 5F).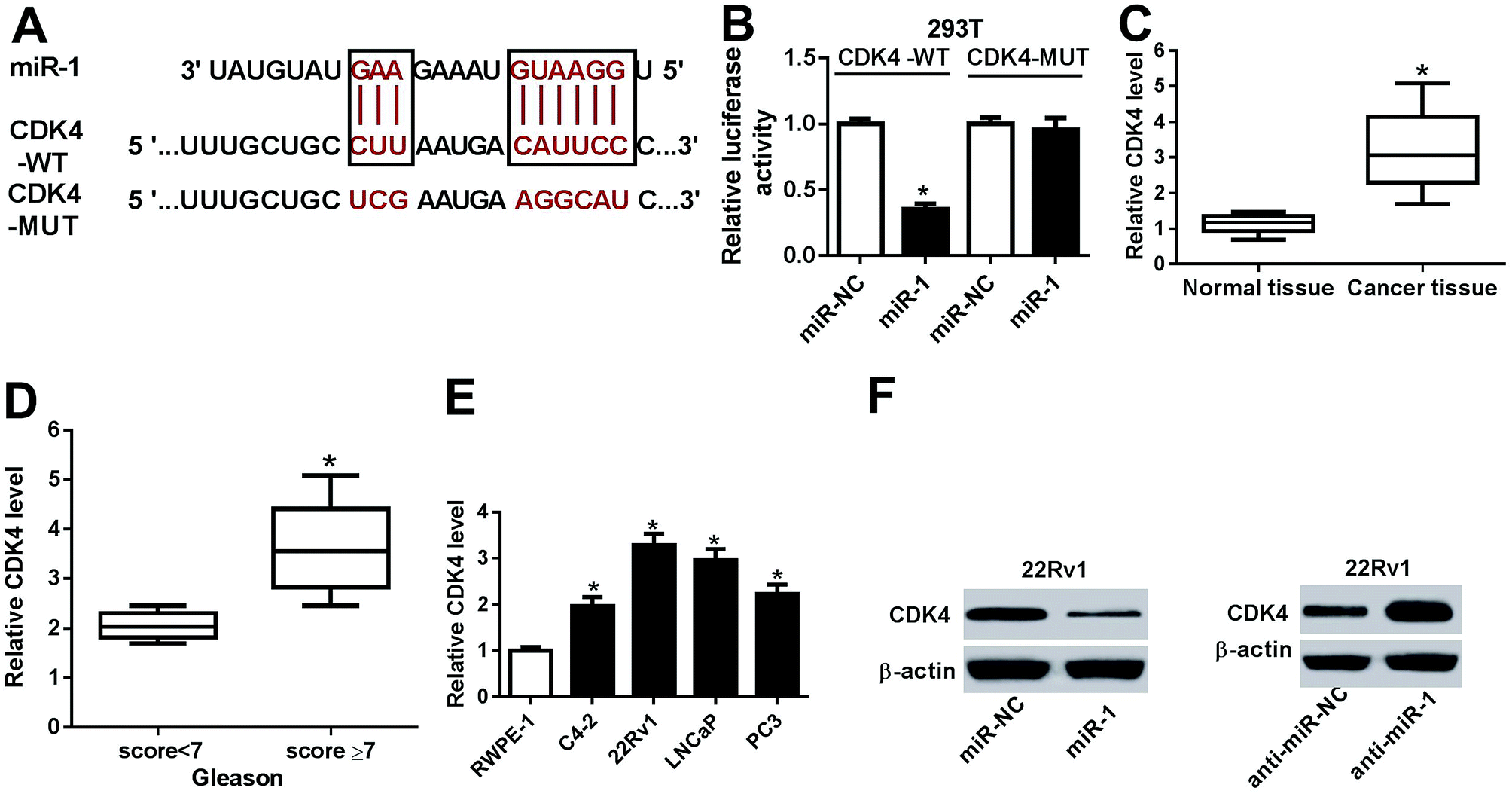 | ||
| Fig. 5 (A) The putative binding sites of miR-1 and CDK4 were provided by TargetScan Release 7.2 online. (B) The luciferase activity was measured in 293T cells co-transfected with CDK4-WT or CDK4-MUT and miR-NC or miR-1. (C) The mRNA expression of CDK4 was measured in prostate cancer tissues or normal. (D) The level of CDK4 mRNA was detected in prostate cancer patients with Gleason score < 7 or ≥7. (E) The abundance of CDK4 was examined at mRNA level in prostate cancer cells. (F) The abundance of CDK4 was measured at protein level in 22RV1 cells transfected with miR-1 mimics, inhibitors or corresponding NC. *p < 0.05. | ||
PCA3 functioned as a ceRNA of miR-1 to induce CDK4 protein expression in prostate cancer cells
Having established the interaction between miR-1 and PCA3 or CDK4, we further investigated whether PCA3 served as a ceRNA of miR-1 to regulate CDK4 expression in prostate cancer cells. To evaluate the luciferase activity, CDK4-WT, miR-1 or (and) PCA3 were transfected into 293T cells. In the CDK4-WT-transfected 293T cells, miR-1 overexpression remarkably suppressed the luciferase activity, whereas introduction of PCA3 reversed the luciferase activity (Fig. 6A). Moreover, the effect of PCA3 and miR-1 on CDK4 protein expression was investigated in 22Rv1 and LNCaP cells. Results showed that PCA3 overexpression promoted CDK4 protein expression in 22Rv1 cells compared with pcDNA treatment, which was weakened by presence of miR-1 (Fig. 6B). However, inhibition of PCA3 and miR-1 showed opposite effect in LNCaP cells, revealed by impeded CDK4 by si-PCA3 and rescued CDK4 by anti-miR-1 at protein level (Fig. 6C).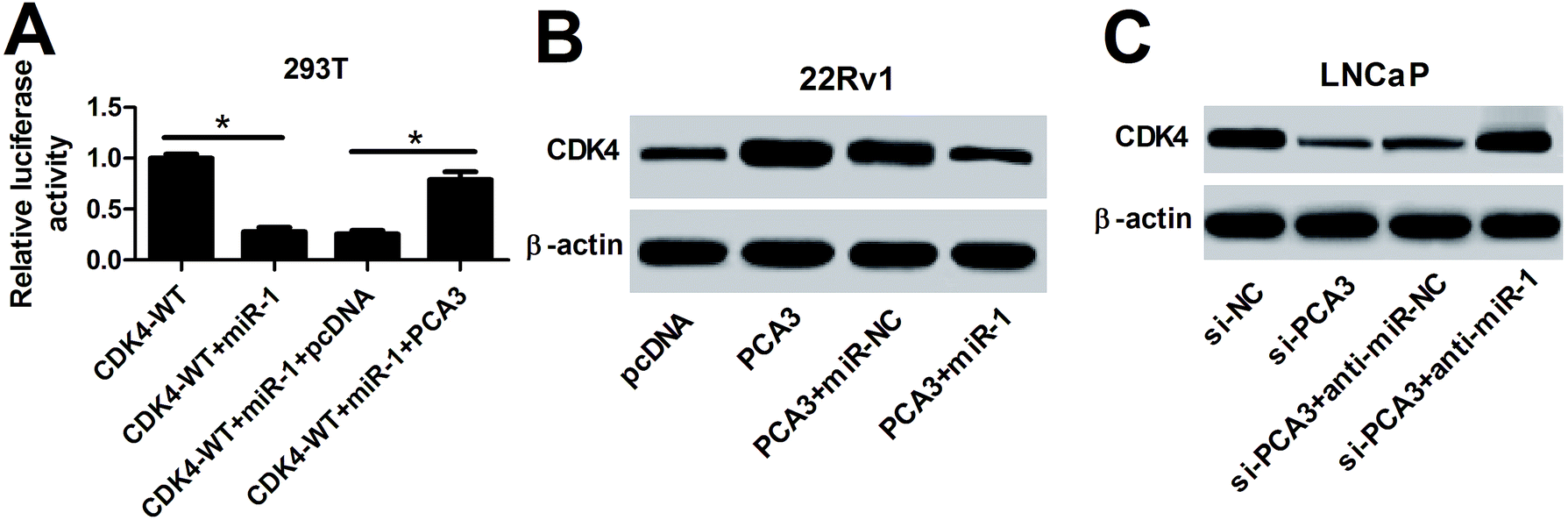 | ||
| Fig. 6 (A) The luciferase activity was investigated in CDK4-WT-transfected 293T cells treated with miR-1 or (and) PCA3. (B) The protein level of CDK4 was measured in 22Rv1 cells transfected with PCA3 or (and) miR-1. (C) The effect of inhibition of PCA3 and miR-1 on CDK4 protein abundance was investigated in LNCaP cells. *p < 0.05. | ||
Discussion
LncRNAs have been suggested to play essential roles in progression of prostate cancer through interacting with miRNAs and decoying proteins.21 Recently, lncRNAs-mediated regulatory networks show great promise for better understanding the mechanism of prostate cancer progression.22 In this study, we focused on the role and mechanism of PCA3 in prostate cancer progression and first provided the view that PCA3/miR-1/CDK4 axis was involved in glycolysis, viability and apoptosis in prostate cancer cells.PCA3, as a promising biomarker, has been reported to be highly expressed in prostate cancer.12,23 Similarly, we also found the elevated expression of PCA3 in prostate cancer tissues and cells. Moreover, high expression of PCA3 was associated with severity of prostate cancer. These results indicated that PCA3 might serve as an oncogene in prostate cancer progression. Nevertheless, the interaction between PCA3 and glycolysis in prostate cancer was not clear. Hence, we explored and found that inhibition of PCA3 suppressed glycolysis in prostate cancer cells. Besides, we also revealed that knockdown of PCA3 impeded cell viability but inflicted the apoptosis of prostate cancer cells. This is also in agreement with sever such reports which uncovered that interference of PCA3 limited the cell growth and viability but induced cell apoptosis in prostate cancer.24,25 However, the mechanism by which PCA3 presents the oncogenic function needs to be further probed. Former efforts indicated the importance of ceRNA networks which showed that PCA3 affected the pathogenesis and progression by sponging miR-96 or miR-1261 in prostate cancer.26,27 However, the evidence is absence of the interaction between PCA3 and miR-1 in prostate cancer. Therefore, we next probed the potential function of miR-1 on PCA3-mediated progression of prostate cancer.
miRNA-mediated interaction has been reported to have essential roles in cell glycolytic metabolism and provided new avenues for therapeutics of prostate cancer.28 miR-1, a tumor suppressive miRNA, has been shown to be down-regulated in prostate cancer.29 Seeing that the miRNA-targeting mechanisms is highly conservative among species and phylogeny, 293T, one of immortal cell lines, has been widely used transfection for luciferase activity. Hence, we also validated the interaction in 293T cells by luciferase activity. In our study, we indicated reduction of miR-1 in prostate cancer tissues and cells. Notably, we found that miR-1 was negatively regulated by PCA3 in prostate cancer cells, suggesting that miR-1 might be required for PCA3-mediated progression of prostate cancer. Therefore, the restoration experiments were conducted in turn and results showed that depletion of miR-1 reversed the inhibitory effect of PCA3 abrogation on glycolysis, which was consistent with a such effort supporting miR-1 blocked tumor glycolysis and proliferation.30 Moreover, we found that knockdown of miR-1 attenuated the regulatory effect of PCA3 inhibition on cell viability and apoptosis, which is similar to the former study indicating the pro-survival and anti-apoptotic effects of miR-1 silencing in prostate cancer.17 Besides, accruing literature revealed miR-1 played essential roles in lncRNA-mediated regulatory network by regulating target genes abundances, such as Hes 1 and HuR.31,32 To better understand the mechanism, a promising target of miR-1 is needed to be explored in this study.
Intriguingly, CDK4, a cyclin-dependent kinase, was implicated as a direct target of miR-1.18 CDK4 was reported to be associated with cell apoptosis and glycolytic pathway in prostate cancer.33 Furthermore, CDK4 has been shown to positively correlated with glycolysis and tumor growth in cancer cells.34 More particularly, CDK4 was involved in the regulatory pathway which mediated cell cycle, apoptosis, proliferation, migration, invasion and tumor growth in prostate cancer.20 In our study, we found that CDK4 was increased and regulated by PCA3 and miR-1 in prostate cancer, revealing the promising regulatory network of PCA3/miR-1/CDK4 in prostate cancer progression. However, there was absence of the direct role of CDK4 in glycolysis, viability and apoptosis of prostate cancer cells. Hence, the functional CDK4 is needed to be elucidated in further study and continuing preclinical research via animal model are urgently expected to improve in future.
Conclusions
In conclusion, the expressions of PCA3 and CDK4 were enhanced while miR-1 abundance was decreased in prostate cancer tissues and cells. Moreover, abrogation of PCA3 blocked aerobic glycolysis, viability and promoted apoptosis in prostate cancer cells. In addition, PCA3 was interacted with miR-1 and depletion of miR-1 attenuated knockdown of PCA3-mediated inhibition of aerobic glycolysis as well as viability and promotion of apoptosis in prostate cancer cells. Besides, CDK4 was directly targeted by miR-1 and it was regulated by PCA3 through functioning as a ceRNA of miR-1 in prostate cancer cells. Collectively, we elucidated that PCA3 knockdown inhibited aerobic glycolysis, viability while induced apoptosis through functioning as a ceRNA of miR-1 to regulate CDK4 protein level in prostate cancer cells, indicating PCA3 as a promising biomarker for treatment of prostate cancer.Ethical statement
All experiments were performed in accordance with the NIH (National Institutes of Health) guidelines, and experiments were approved by the Institutional Research Ethics Committee of the Affiliated Huaian No. 1 People's Hospital of Nanjing Medical University. Informed consents were obtained from human participants of this study.Conflicts of interest
The authors have no conflict of interest to declare.Abbreviations
| LncRNA | Long noncoding RNA |
| PCA3 | Prostate cancer antigen 3 |
| miRNA | MicroRNA |
| CDK4 | Cyclin-dependent kinase 4 |
| PCAT1 | Prostate cancer-associated transcript 1 |
| qRT-PCR | Quantitative reverse transcription PCR |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide |
| RIP | RNA immunoprecipitation |
| ceRNA | Competing endogenous RNA |
| DMEM | Dulbecco's modified Eagle medium |
| NC | Negative control |
| DMSO | Dimethyl sulfoxide |
| PI | Propidium iodide |
| 3′-UTR | 3′-Untranslated regions |
| Ago2 | Argonaute 2 |
| PVDF | Polyvinylidene difluoride |
| TBST | Tris–buffer saline containing 0.1% Tween 20 |
| HRP | Horseradish peroxidase |
| ECL | Enhanced chemiluminescence |
Acknowledgements
Not applicable.References
- G. Attard, C. Parker, R. Eeles, F. Schröder, S. Tomlins, I. Tannock, C. Drake and J. de Bono, Lancet, 2016, 387, 70–82 CrossRef.
- M. Litwin and H. Tan, JAMA, 2017, 317, 2532–2542 CrossRef PubMed.
- G. Jha, V. Anand, A. Soubra and B. Konety, Nat. Rev. Clin. Oncol., 2014, 11, 354–364 CrossRef CAS PubMed.
- M. Vander Heiden, L. Cantley and C. Thompson, Science, 2009, 324, 1029–1033 CrossRef CAS PubMed.
- T. Xu, C. Lin, S. Cheng, J. Min, L. Li, X. Meng, C. Huang, L. Zhang, Z. Deng and J. Li, Mol. Cancer, 2018, 17, 103 CrossRef PubMed.
- C. Hung, L. Wang, Y. Yu, H. Chen, S. Srivastava, G. Petrovics and H. Kung, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18697–18702 CrossRef CAS PubMed.
- X. Wang, Y. Xu, X. Wang, C. Jiang, S. Han, K. Dong, M. Shen and D. Xu, Cell Proliferation, 2017, 50, e12395 CrossRef PubMed.
- Y. Ren, J. Shang, J. Li, W. Liu, Z. Zhang, J. Yuan and M. Yang, J. Biol. Chem., 2017, 292, 17939–17949 CrossRef CAS PubMed.
- E. Bozgeyik, E. Bayraktar, A. Chavez-Reyes and C. Rodriguez-Aguayo, Mol. Biol., 2016, 5, 3 Search PubMed.
- J. Shih, L. Wang, C. Huang, H. Kung and C. Hsieh, Int. J. Mol. Sci., 2015, 16, 28943–28978 CrossRef CAS PubMed.
- D. Chistiakov, V. Myasoedova, A. Grechko, A. Melnichenko and A. Orekhov, Semin. Cancer Biol., 2018, 52, 9–16 CrossRef CAS PubMed.
- Y. Cui, W. Cao, Q. Li, H. Shen, C. Liu, J. Deng, J. Xu and Q. Shao, Sci. Rep., 2016, 6, 25776 CrossRef CAS PubMed.
- J. Lai, L. Moya, J. An, A. Hoffman, S. Srinivasan, J. Panchadsaram, C. Walpole, J. Perry-Keene, S. Chambers, M. Lehman, C. Nelson, J. Clements and J. Batra, Sci. Rep., 2017, 7, 16862 CrossRef PubMed.
- Y. Ye, S. Li and S. Wang, PLoS One, 2018, 13, e01V98055 CrossRef PubMed.
- L. Salmena, L. Poliseno, Y. Tay, L. Kats and P. Pandolfi, Cell, 2011, 146, 353–358 CrossRef CAS PubMed.
- R. Hudson, M. Yi, D. Esposito, S. Watkins, A. Hurwitz, H. Yfantis, D. Lee, J. Borin, M. Naslund, R. Alexander, T. Dorsey, R. Stephens, C. Croce and S. Ambs, Nucleic Acids Res., 2012, 40, 3689–3703 CrossRef CAS PubMed.
- J. Chang, W. Xu, X. Du and J. Hou, OncoTargets Ther., 2018, 11, 3461–3473 CrossRef PubMed.
- S. Jiang, C. Zhao, X. Yang, X. Li, Q. Pan, H. Huang, X. Wen, H. Shan, Q. Li, Y. Du and Y. Zhao, Int. J. Mol. Med., 2016, 38, 113–122 CrossRef CAS PubMed.
- X. Escoté and L. Fajas, Cancer Lett., 2015, 356, 171–175 CrossRef PubMed.
- D. Roberto, L. Klotz and V. Venkateswaran, Prostate, 2018 DOI:10.1002/pros.23728.
- C. Klinge, Endocr.-Relat. Cancer, 2018, 25, 259–282 Search PubMed.
- Z. Du, T. Sun, E. Hacisuleyman, T. Fei, X. Wang, M. Brown, J. Rinn, M. Lee, Y. Chen, P. Kantoff and X. Liu, Nat. Commun., 2016, 7, 10982 CrossRef CAS PubMed.
- Z. Yang, L. Yu and Z. Wang, Chin. J. Cancer Res., 2016, 28, 65–71 CAS.
- L. Ferreira, A. Palumbo, K. de Mello, C. Sternberg, M. Caetano, F. de Oliveira, A. Neves, L. Nasciutti, L. Goulart and E. Gimba, BMC Cancer, 2012, 12, 507 CrossRef CAS PubMed.
- C. Lin, J. Wang, Y. Wang, P. Zhu, X. Liu, N. Li, J. Liu, L. Yu and W. Wang, Anticancer Res., 2017, 37, 4303–4310 Search PubMed.
- Y. Li and Z. Yang, Pathol., Res. Pract., 2018, 18, 30854–30859 Search PubMed.
- J. He, B. Li, Z. Han, M. Zou, L. Wang, Y. Lv, J. Zhou, M. Cao, Y. Li and J. Zhang, Tumor Biol., 2016, 37, 16163–16176 CrossRef CAS PubMed.
- K. Kasomva, A. Sen, M. Paulraj, S. Sailo, V. Raphael, K. Puro, S. Assumi and S. Ignacimuthu, Int. J. Biochem. Cell Biol., 2018, 102, 109–116 CrossRef CAS PubMed.
- B. Kumar, A. Rosenberg, S. Choi, K. Fox-Talbot, A. De Marzo, L. Nonn, W. Brennen, L. Marchionni, M. Halushka and S. Lupold, Sci. Rep., 2018, 8, 7189 CrossRef PubMed.
- W. Xu, Z. Zhang, K. Zou, Y. Cheng, M. Yang, H. Chen, H. Wang, J. Zhao, P. Chen, L. He, X. Chen, L. Geng and S. Gong, Cell Death Discovery, 2017, 8, e2761 CrossRef CAS PubMed.
- J. Zheng, D. Yi, Y. Liu, M. Wang, Y. Zhu and H. Shi, Am. J. Transl. Res., 2017, 9, 3696–3704 Search PubMed.
- D. Yu, C. Zhang and J. Gui, OncoTargets Ther., 2017, 10, 2609–2619 CrossRef PubMed.
- D. Milanesa, M. Choudhury, C. Mallouh, H. Tazaki and S. Konno, Eur. Urol., 2000, 37, 728–734 CrossRef CAS PubMed.
- H. Wang, B. Nicolay, J. Chick, X. Gao, Y. Geng, H. Ren, H. Gao, G. Yang, J. Williams, J. Suski, M. Keibler, E. Sicinska, U. Gerdemann, W. Haining and T. Roberts, et al., Nature, 2017, 546, 426–430 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |