
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAggregation prevention: reduction of graphene oxide in mixed medium of alkylphenol polyoxyethylene (7) ether and 2-methoxyethanol†
Heng Sua,
Chaocan Zhang *a,
Xi Lib,
Lili Wua and
Yanjun Chena
*a,
Xi Lib,
Lili Wua and
Yanjun Chena
aDepartment of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, China. E-mail: polymers@whut.edu.cn; suhengwhut@163.com; poly_wl@whut.edu.cn; yanjunchen@whut.edu.cn; Fax: +86 878 63157; Tel: +86 878 63157
bDepartment of Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan 430070, China. E-mail: chemlixi@whut.edu.cn
First published on 22nd November 2018
Abstract
Graphene has attracted great interest due to its extensive applications in optoelectronic and electronic circuits and devices. However, reduction of graphene oxide (GO) to graphene is a process in which hydrophilic GO converts to hydrophobic graphene. Very little is known about the aggregation of graphene and the cause of performance degradation by general chemical reduction methods as the single reaction medium presents difficulty in satisfying the good dispersion of hydrophilic GO and hydrophobic graphene simultaneously. In this paper, we report a mixed medium of alkylphenol polyoxyethylene (7) ether (OP-7) and 2-methoxyethanol (EGM) for the preparation of graphene. The strong polar nature of EGM provides a good dispersion environment for GO, while the π–π interaction between the π-electrons in nonionic surfactant OP-7 aromatic ring structure and the π-electrons in graphene make the hydrophobic graphene well dispersed and prevent aggregation. Moreover, the reduction temperature is not high and the reduction time is short. The electrical conductivity of graphene without high-temperature treatment reached 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 S m−1. We have found the potential reduction mechanism of graphene and fundamentally solved the problem of aggregation. Our findings make it possible to process graphene materials using low-cost mixed medium processing techniques, providing a valuable reference for the large-scale preparation of graphene.
000 S m−1. We have found the potential reduction mechanism of graphene and fundamentally solved the problem of aggregation. Our findings make it possible to process graphene materials using low-cost mixed medium processing techniques, providing a valuable reference for the large-scale preparation of graphene.
Introduction
Graphene is a one-atom-thick planar sheet of sp2-hybridized carbon atoms arranged hexagonally. Graphene has extraordinary electrical, thermal, mechanical and other properties due to its unique two-dimensional crystal structure,1,2 and it has emerged as an attractive material in electronics, optoelectronics and capacitors.3,4 Among its various properties, the distinctive electronic properties of graphene has great potential applications in high-electron-mobility transistors,5 supercapacitors6–8 and solar cells.9,10 Up to now, several fabrication routes for the production of graphene have been established, such as mechanical exfoliation,11–13 chemical reduction of graphene oxide solution,14–17 epitaxial growth18,19 and chemical vapor deposition.20,21 The chemical reduction method plays an important role in the prospect of potential industrialization.The chemical reduction method involves oxidizing graphite to form GO, and then reducing GO. Preparation of GO is conducted by methods such as Hummers method,22 Brodie method23 and Staudenmaier method24 and then, the reduction of GO (RGO) is realized by using hydrazine,25 hydrohalic acid26 and sodium borohydride27 as reducing agents. Until now, hydrazine hydrate, the most widely used reducing agent, was used in aqueous medium as a reductant to reduce GO (RGOH2O). Previous studies have reported that RGO shows a distinct sharp peak between the 23.0° and 24.9° region of the XRD spectrum,27–32 indicating that they have an ordered layer structure, and the electrical conductivity at room temperature is generally between 200 and 7200 S m−1.25,29,33–36 The value of conductivity fluctuates greatly, but the reason for this has not been studied in literature. In our opinion, ideal single-layer graphene should not have a typical layered structure, so there should be no sharp peak in the XRD spectrum. The edge of the graphite sheet is preferentially oxidized during the graphite oxidation stage, resulting in the oxidation degree of GO being higher than that in the middle region.37,38 Therefore, the edge portion of GO will be preferentially reduced to hydrophobic graphene structure, while the middle region will be reduced slowly. Finally, the structure of RGO is hydrophobic in the marginal area, while the intermediate area is hydrophilic and due to the interaction forces between hydrophobic regions, RGO tends to overlap and aggregate into a layered structure. This shows that the reduction of GO to RGO is a process where hydrophilic GO gets converted to hydrophobic RGO. Not only polar but also non-polar single reaction medium presents difficulties in satisfying the requirements of good dispersion for RGO. Furthermore, preventing aggregation during the reduction phase is the key to preparing RGO with excellent performance.
In this study, we designed a mixed medium that can simultaneously satisfy the well-dispersed hydrophilic GO and also prevent superimposed aggregation during the reduction process: OP-7 and EGM were mixed and used as the reaction medium and hydrazine hydrate was used as the reducing agent to reduce GO (RGOOP-7/EGM). The strong polar nature of EGM provided a good dispersion environment for GO, while the π–π interaction between the π-electrons in the OP-7 aromatic ring structure and the π-electrons in the RGO enabled the effective dispersion of hydrophobic graphene in the reaction medium and prevented superimposed aggregation. The reduction routes and aggregation prevention mechanism are shown in Fig. 1. As a result, the XRD spectrum of the as-prepared RGOOP-7/EGM shows a broad diffraction peak between the 22.0° and 26.9°, indicating that there is no typical layered structure. Furthermore, the reduction temperature is not high and the reduction time is short. The electrical conductivity of graphene prepared without high-temperature treatment was 14000 S m−1.
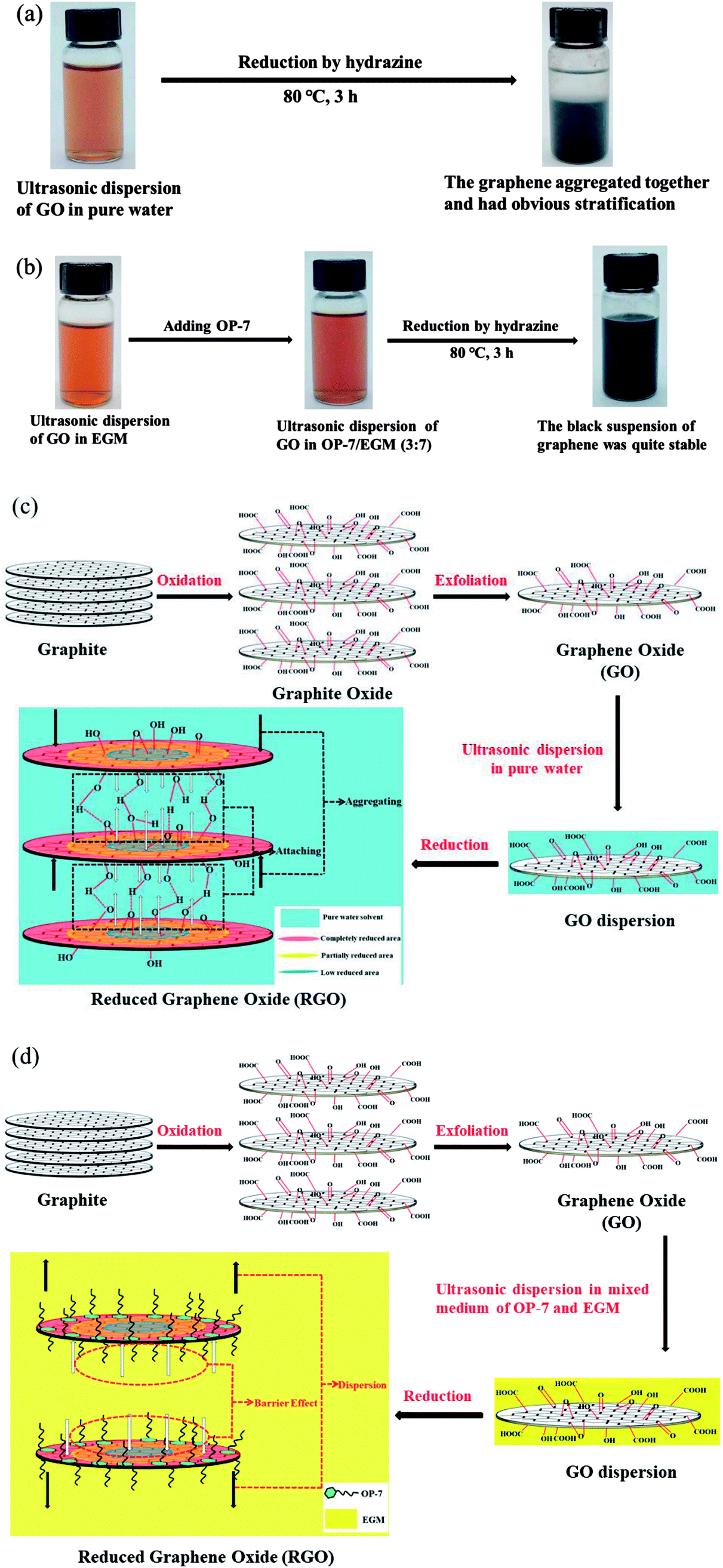 | ||
| Fig. 1 Reduction routes of GO in pure water (a) and in the mixed reaction medium (OP-7 and EGM) (b); the cause of aggregation in pure water (c) and the aggregation prevention mechanism in the mixed reaction medium (OP-7 and EGM) (d). | ||
Experimental section
Chemicals and materials
Natural crystalline flake graphite (NG) (99.85% purity, 325 mesh), sulfuric acid (H2SO4, 98%), phosphoric acid (H3PO4, ≥ 85%), hydrochloric acid (HCl, 36%), anhydrous ethanol (≥99.7%), 2-methoxyethanol (EGM ≥99%), hydrazine hydrate (N2H4·H2O, 85%), hydrogen peroxide (H2O2, 30%), potassium permanganate (KMnO4) and barium chloride (BaCl2) were purchased from Sinopharm Chemical Reagent Co., Ltd. Alkylphenol polyoxyethylene (7) ether (OP-7) was purchased from Wen Hua Chemical Reagent Factory. Pure water with a frequency of 50 Hz (KQ-50, Kunshan Ultrasonic Instruments Co.) was used in all experiments.Synthesis and dispersion of GO
GO was synthesized from natural crystalline flake graphite (NG) by a modified Hummers method.22 NG (1 g) and potassium permanganate (5 g) were added in a mixture of sulfuric acid (98%, 81 mL) and phosphoric acid (≥85 wt%, 9 mL), producing a slight exothermic reaction, which increased the temperature to 30–40 °C. The reaction was then heated to 50 °C without subsequent aging and stirred for 12 h. The reaction was then cooled to room temperature and hydrogen peroxide (30%, 2 mL) was added. The next processes, namely, filtration through qualitative filter paper (GEB Co.), washing of remaining solid materials in succession with pure water (200 mL) and hydrochloric acid (10%, 200 mL), centrifugation (6000 rpm for 4 h) of the filtrate by a centrifuge (TG-16-WS XiangYi centrifuge Instruments Co.) and decantation of the supernatant, were performed. The as-synthesized GO was vacuum-dried overnight at room temperature to yield the desired product.Preparation of RGOOP-7/EGM sheets
For the reduction process, dry GO (500 mg) was dispersed in a ratio of 3:7 of OP-7/EGM (60:140 mL) to obtain a solution with high uniformity and good dispersion. The solution was sonicated in a bath-type sonicator (KQ-50, Kunshan Ultrasonic Instruments Co.) for 3 h at a power level of 50 W. Hydrazine hydrate (1 mL) was added, and the mixture was heated at 80 °C for 3 h with constant stirring. The finished product was separated by filtration, washed five times with anhydrous ethanol (400 mL), and vacuum-dried overnight at room temperature to obtain RGOOP-7/EGM powders. Then, 30 mg of the dried RGOOP-7/EGM powder, prepared according to the previous process, was transferred into a tablet mold with an inner diameter of 10 mm and then pressurized (15 MPa) by a hydraulic machine (MLN, QJD2518003). RGOOP-7/EGM sheets can be obtained after holding the pressure for 10 min, followed by demolding.
Preparation of RGOH2O sheets
For the reduction process, dry GO (500 mg) was dispersed in pure water and sonicated for 3 h at a power level of 50 W. Then, N2H4·H2O (1 mL) was added and the mixture was heated at 80 °C for 3 h with constant stirring. The finished product was separated by filtration and vacuum-dried overnight at room temperature to obtain RGOH2O powders. RGOH2O sheets can be obtained after holding the pressure (15 MPa) for 10 min, followed by demolding.Characterization of GO, RGOOP-7/EGM and RGOH2O sheets
Fourier transform infrared (FTIR) spectra were recorded on an FTIR spectrometer (Thermo Scientific Nicolet 6700, U.S.) to identify the functional groups. Raman spectra were collected via a confocal microscopic Raman spectrometer (Renishaw InVia Raman microscope, Britain) using a ×50 objective lens at room temperature, with a 530 nm laser beam and 1800 lines per mm grating. The UV-vis absorption spectra were recorded by a UV-vis spectrometer (Lambda 750 S, U.S.). Measurement of the interlayer distance of the GOs and RGOs was conducted on XRD (D8 Focus 3 KW, Bruker AXS, Germany) with Cu-Kα radiation (λ = 1.5406 Å). Elemental composition analyses were performed via X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi) with a monochromatic Al-Kα X-ray source at 100 W. The scanned energy has pass energies of 140.00 eV, and the high-resolution scans have a flux of 26.00 eV. The microstructure and crystal structure were observed by field emission transmission electron microscopy (FETEM, JEM-2100F, Japan) and field emission scanning electron microscopy (FESEM, Zeiss Ultra Plus, Germany). Furthermore, selected area electron diffraction (SAED) was performed using FETEM. Sheet resistance (Rs, Ω sq−1) of the RGO sheets was measured by a four-point probe method (RTS-8, China), and the corresponding volume conductivity (σ, S m−1) can be converted using the formula: σ = 1/(Rs × t), where t indicates the thickness of the RGO sheet.Results and discussion
Pure water has been used by many groups as a reaction medium to reduce the GO. However, several chemical reduction reactions in pure water cannot bring about effective excellent performance of the resultant GO. The underlying correlation with reaction medium has not been clearly observed. For this purpose, we compared graphene obtained by reduction in pure water and that in OP-7/EGM mixed medium. Fig. 2a shows the Fourier transform infrared spectra, confirming the different functional groups between GO, RGOH2O and RGOOP-7/EGM. The spectrum of GO shows too many peaks: wide and strong O–H stretching vibrations (3410 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations (1724 cm−1), CC from sp2 bonds (1628 cm−1), C–OH stretching vibrations (1220 cm−1), C–O–C stretching vibrations (1050 cm−1) in epoxy, and epoxy stretching vibrations (853 cm−1). The CO, C–OH and epoxy stretching vibrations evidently disappeared, indicating that the GO has been reduced. On comparing the spectrum of RGOOP-7/EGM with that of RGOH2O, we see that O–H and C–O–C stretching vibrations were hardly visible. This indicated that most of the oxygen groups on GO have been removed. This result shows that the edge and center of GO reduced completely under OP-7/EGM mixed reaction medium.
O stretching vibrations (1724 cm−1), CC from sp2 bonds (1628 cm−1), C–OH stretching vibrations (1220 cm−1), C–O–C stretching vibrations (1050 cm−1) in epoxy, and epoxy stretching vibrations (853 cm−1). The CO, C–OH and epoxy stretching vibrations evidently disappeared, indicating that the GO has been reduced. On comparing the spectrum of RGOOP-7/EGM with that of RGOH2O, we see that O–H and C–O–C stretching vibrations were hardly visible. This indicated that most of the oxygen groups on GO have been removed. This result shows that the edge and center of GO reduced completely under OP-7/EGM mixed reaction medium.
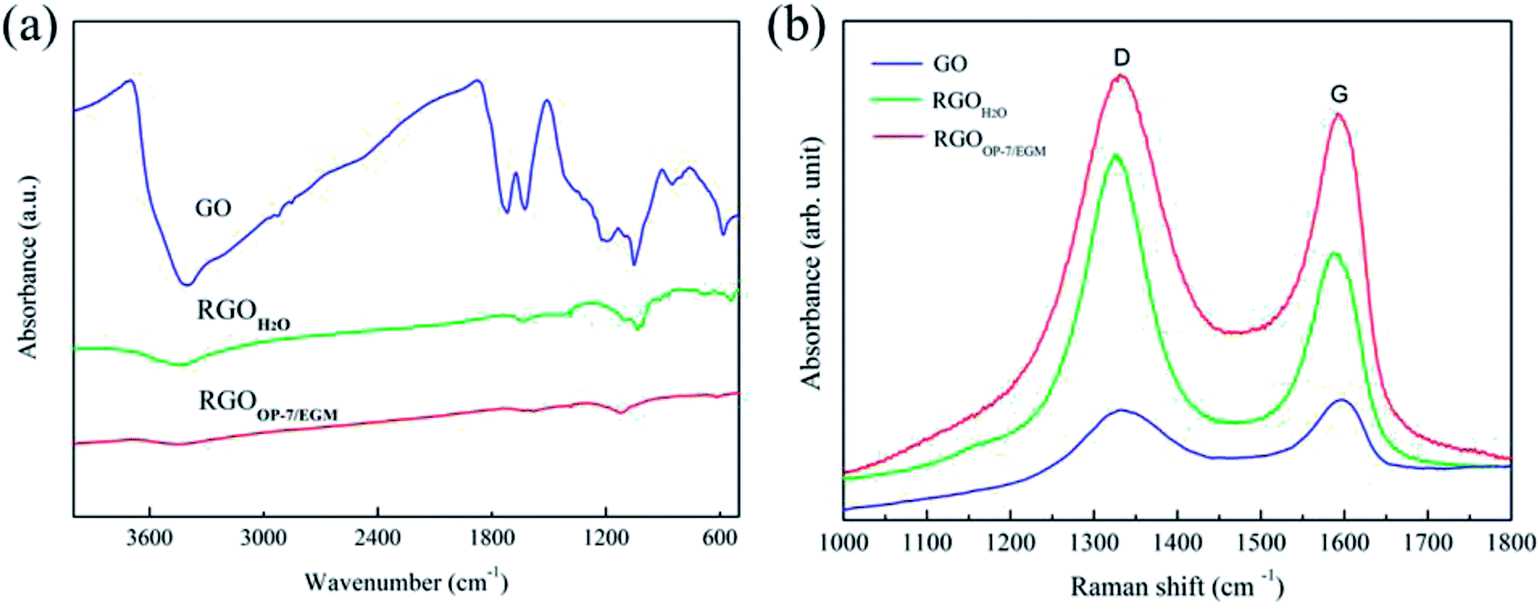 | ||
| Fig. 2 Fourier transform infrared spectra of GO, RGOH2O and RGOOP-7/EGM (a) and Raman spectra of GO, RGOH2O and RGOOP-7/EGM (b). | ||
The information of the defects was reflected in the ratio of the intensities of D and G bands (D:G) in the micro-Raman spectra, as shown in Fig. 2b. The G band reflects the symmetry and crystallinity of the material, while the D band is often called the defect band. It can be seen that the intensity ratio (ID/IG) of D band and G band of GO is lower than the ID/IG of RGOH2O and RGOOP-7/EGM. This is due to the presence of unpaired defects that remained after the removal of a large number of oxygen-containing functional groups.39 Moreover, the ID/IG of RGOH2O increased more significantly than RGOOP-7/EGM, indicating that RGOOP-7/EGM has fewer defects after reduction in the mixed reaction medium. This finding shows that OP-7 plays a crucial barrier role in preventing aggregation, which is consistent with previous infrared spectra results. Table 1 lists the physical values of the Raman spectra.
| Raman | |||
|---|---|---|---|
| D band (cm−1) | G band (cm−1) | ID/IG (%) | |
| Graphite | — | — | — |
| GO | 1357 | 1597 | 0.97 |
| RGOH2O | 1332 | 1587 | 1.37 |
| RGOOP-7/EGM | 1354 | 1591 | 1.01 |
The restoration of CC bonds during reduction was characterized by UV-vis spectrometry. As shown in Fig. 3, GO has a strong absorption peak at 235 nm and a distinct shoulder at 300 nm. The absorption peak of GO at around 235 nm gradually red-shifted towards 268 nm after reduction of GO. Furthermore, the absorbance in the whole spectral region increased, indicating that the CC bonds have been restored. The absorption peak shifted to 275 nm, and the absorption was saturated after reducing in mixed medium, indicating that the reduction had been completed.
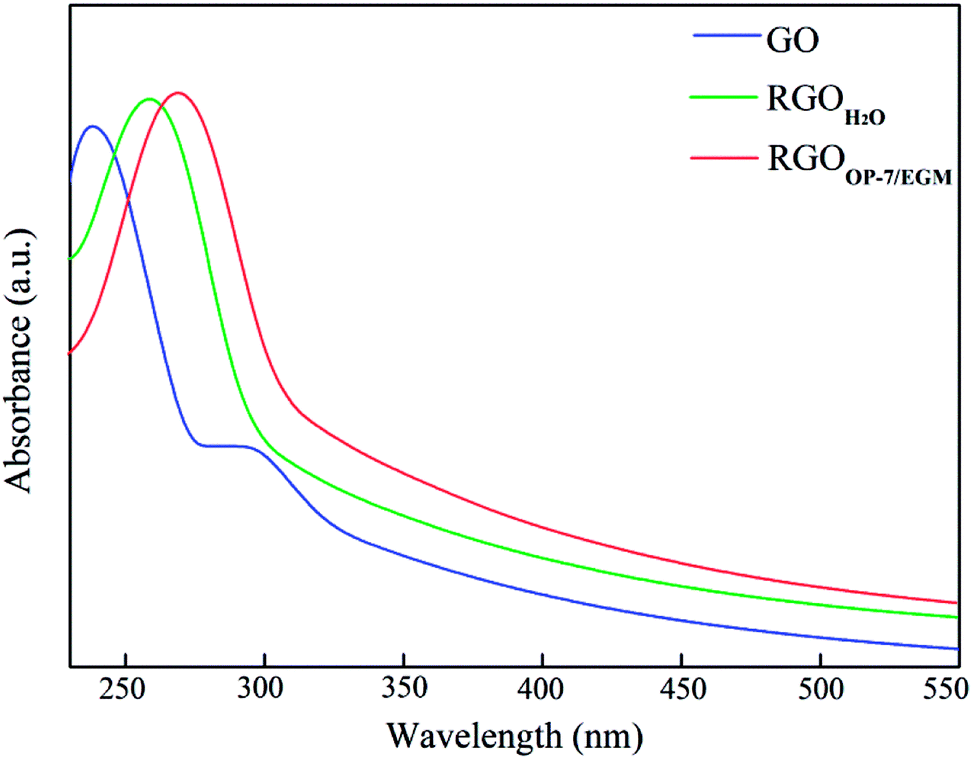 | ||
| Fig. 3 UV-vis absorption spectra of GO, RGOH2O and RGOOP-7/EGM. | ||
The powder XRD patterns of graphite (G) and GO were compared with those of the as-prepared RGOs, as shown in Fig. 4a. RGOOP-7 and RGOEGM were prepared by reducing GO in pure OP-7 and the pure solution of EGM, respectively; the preparation routes of RGOOP-7 and RGOEGM are shown in Fig. S1.† The peak position for G was 2θ = 26.60° (d-spacing ∼ 3.35 Å). After the oxidation reaction, GO exhibited a large interlayer distance (d-spacing = ∼8.66 Å, 2θ = 10.20°) because of the information of hydroxyl, carboxyl and epoxy groups. There was a significant decrease in the average interlayer distance of RGOs after reduction reaction, as described in Fig. 4b. Furthermore, Fig. 4a shows that RGOH2O has a distinct sharp peak, and its interlayer distance decreases to 3.54 Å (2θ = 24.92°), indicating that the RGOH2O has an ordered layer structure because of aggregation during the reduction phase. Although OP-7 can effectively prevent the aggregation of graphene, GO tends to agglomerate in a high concentration of OP-7, resulting in a low reduction degree of GO. The XRD spectrum of RGOOP-7 also displays an evident peak at 2θ = 24.80° (d-spacing = ∼3.59 Å), which is similar to the peak position of RGOH2O, indicating that RGOOP-7 still has a certain layer structure. In addition, the XRD spectrum of RGOEGM has no visible sharp peak, but an inconspicuous broad peak. The interlayer distance of RGOEGM was 3.94 Å (2θ = 22.55°), which was higher than that of RGOOP-7 because the solution of EGM can effectively disperse GO. However, the layered structure still exists in RGOEGM because the dispersion of graphene in EGM was not so ideal. If we only simply satisfy the good dispersion of GO or prevent graphene aggregation, it will be difficult to prepare graphene with good properties. Based on a comparison with interlayer spacing of graphene previously prepared by chemical reduction methods, we found that the spacing of the graphene layers was between 3.57 Å and 3.90 Å, while they all had a distinct sharp diffraction peak.27–30,36 On the contrary, a dispersing and broad diffraction peak between 22.0° and 26.9° can be observed in the XRD pattern of RGOOP-7/EGM. This shows that RGOOP-7/EGM has no typical layered structure because of the barrier effect of OP-7 and the good dispersion action of EGM. As shown in Fig. S2,† we calculated that the distance between the donor hydrogen and the acceptor oxygen atoms bonded to different functional groups of two graphene sheets was between 3.63 Å and 4.05 Å. This calculated result was consistent with the layer spacing of graphene prepared by the previous chemical methods and indicated that the individual graphene layers fit with each other via hydrogen bonds mediated by oxygen-containing functional groups. The average interlayer distance of RGOOP-7/EGM was 4.21 Å, corresponding to the red dot in Fig. 4b, which was higher than 4.05 Å owing to the prevention of the formation of hydrogen bonds by OP-7. In general, the barrier effect of OP-7 and the dispersion effect of EGM are indispensable in the preparation of graphene.
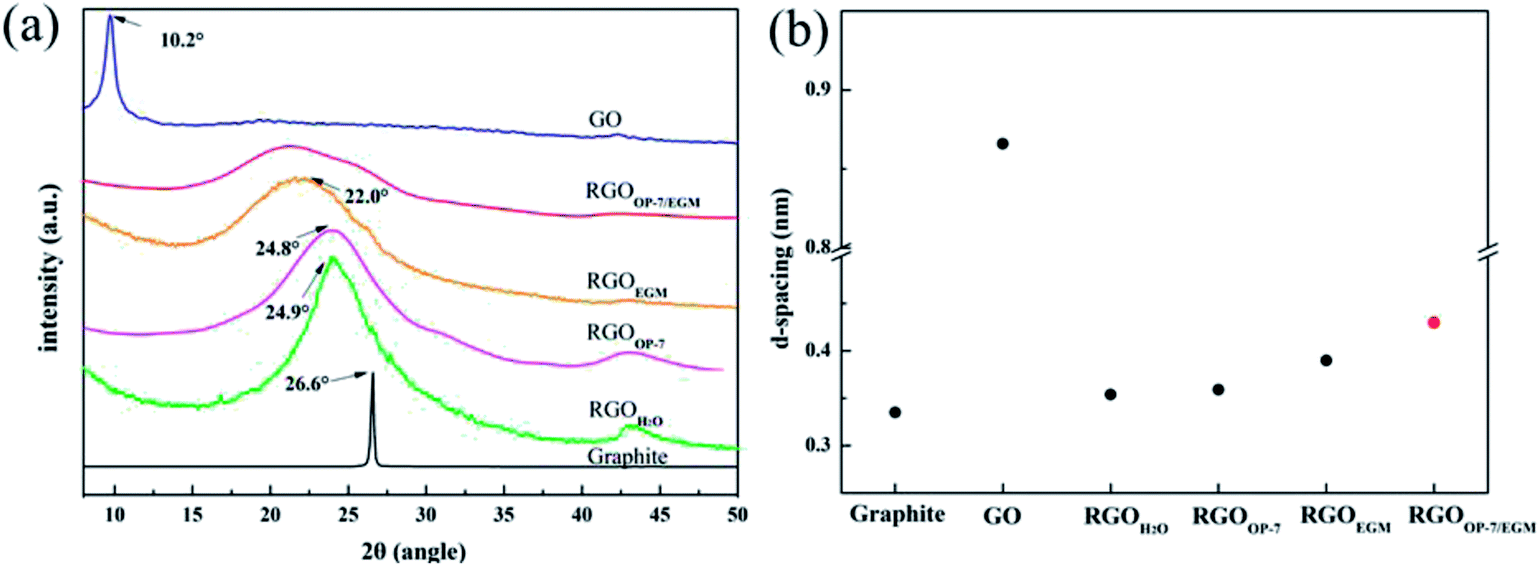 | ||
| Fig. 4 XRD patterns of GO, Graphite (G) and RGOs (a) and d-spacing for the indicated sample types (b). | ||
The C/O ratio and information about various functional groups of GO, RGOH2O and RGOOP-7/EGM were investigated by X-ray photoemission spectroscopy (XPS). In Fig. 5a, the O 1s peak and C 1s peak positions are observed at 284.8 eV and 533.0 eV, respectively. Through the comparison of GO and RGOH2O, we found that the intensity of the O 1s peak of RGOH2O visibly decreased in the broad region. This implied that the C/O ratio of RGO increased significantly after reduction due to the removal of oxygen-containing groups. In addition, the intensity of the O 1s peak of RGOOP-7/EGM was slightly lower than that of RGO, suggesting that RGOOP-7/EGM was reduced more completely. Fig. 5b shows the C 1s spectrum of GO, which reveals that it consists of two main components arising from the CC/C–C (284.6 eV) and CO (carbonyl, ∼288.3 eV) groups and two minor components from the C–O (hydroxyl and epoxy, ∼286.5 eV) and O–CO (carboxyl, ∼290.3 eV) groups.40 After reduction by the traditional chemical method, the C 1s XPS spectrum of RGOH2O also displayed these peaks, but their intensities were much lower than those of GO (Fig. 5c), indicating that most of the oxygen functional groups were removed. Most of the CO bonds (carbonyl groups, ∼286.9 eV) were converted into C–O bonds (hydroxyl groups, ∼286.1 eV), and a number of carboxyl groups were removed. By using mixed reaction medium of OP-7 and EGM, all of the carbonyl groups of RGOOP-7/EGM were almost completely removed, and the C–O bonds were reduced, as shown in Fig. 5d. Compared with RGOH2O, the reduction degree of RGOOP-7/EGM is higher. Areas of the contributing peaks are listed in Table 2. The atomic ratio of C/O increased from 1.70 for GO to 9.04 for RGOOP-7/EGM, which was higher than the value for the RGOH2O obtained by reduction in pure water. The C–C groups of RGOOP-7/EGM compared with RGOH2O increased substantially, while the C–O and CO groups decreased significantly and the O–CO groups disappeared completely. These results show that RGOOP-7/EGM was reduced much better than RGOH2O, which is consistent with the XRD spectra due to the barrier effect of surfactant OP-7 and the great dispersion action of EGM.
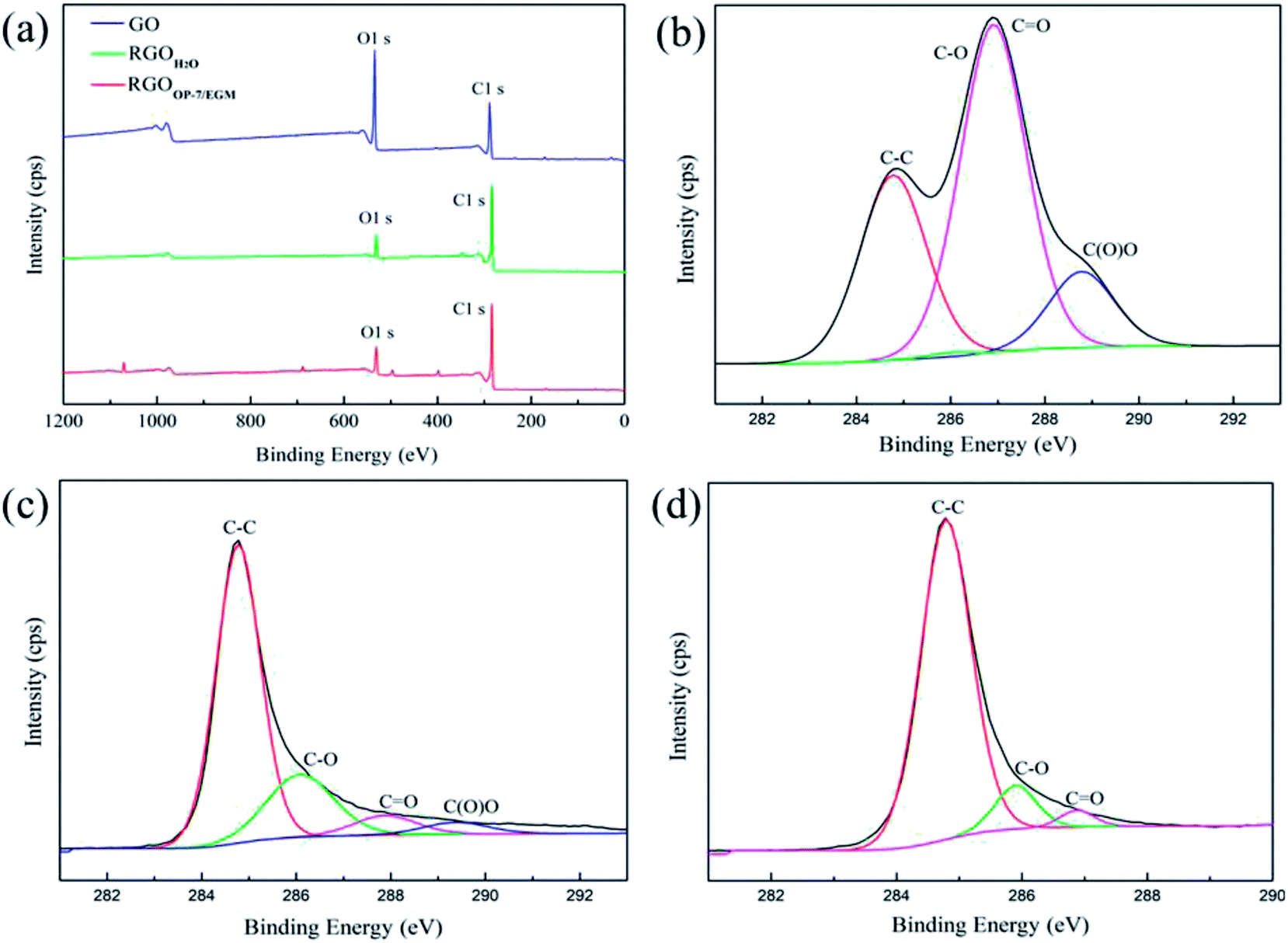 | ||
| Fig. 5 XPS spectra of GO, RGOH2O and RGOOP-7/EGM: wide region (a) and C 1s region of GO (b), RGOH2O (c) and RGOOP-7/EGM (d). | ||
| Fitting of the C 1s peak binding energy [eV] (relative atomic percentage [%]) | XPS | ||||
|---|---|---|---|---|---|
| C–C | C–O/C–O–C | CO |
O–CO |
C:O ratio |
|
| GO | 284.78 (31.55) | 286.01 (1.48) | 286.91 (55.33) | 288.78 (11.64) | 1.70 |
| RGOH2O | 284.77 (67.98) | 286.08 (21.74) | 287.88 (6.30) | 289.38 (3.98) | 6.25 |
| RGOOP-7/EGM | 284.79 (89.33) | 285.90 (8.62) | 286.88 (2.05) | — | 9.04 |
As shown in Fig. 6a, a field emission scanning electron microscopy (FESEM) image of the surface of the RGOH2O power sample exhibited many stacks due to aggregation. On the contrary, a clearly single-layer structure of the RGOOP-7/EGM sheet is observed in Fig. 6b. Fig. 6c shows the field emission transmission electron microscopy (FETEM) image of the RGOH2O sheets; we can find that the RGOH2O sheets overlapped and aggregated into a multilayer structure. Fig. 6d and e show the TEM images of the RGOOP-7/EGM platelets at different magnifications, respectively. Although a few folds appear on the RGOOP-7/EGM sheets, most of the graphene sheets were single-layer or few-layer owing to the complementary role of OP-7 and EGM. As can be seen in Fig. 6f, the selected area electron diffraction (SAED) pattern distinctly indicates the graphitic crystalline structure. The first ring came from the (1100) plane, and the bright spots consistent with the (1100) reflections retained the hexagonal symmetry of the [0001] diffraction pattern. Furthermore, we found that the relative strength of the inner and outer spot rings was close, corresponding to the structure of single-layer graphene or few layer graphene.
 | ||
| Fig. 6 SEM images of RGOH2O (a) and RGOOP-7/EGM (b), TEM image of RGOH2O (c), TEM images of RGOOP-7/EGM (d) and (e) at different magnifications, SAED pattern of RGOOP-7/EGM platelet (f). | ||
Table 3 summarizes the electrical conductivity of RGOH2O, RGOOP-7, RGOEGM and RGOOP-7/EGM sheets dried at room temperature. The conductivity of RGOH2O was only 1050 S m−1 because hydrophobic graphene sheets were easily aggregating in an aqueous environment. The conductivity of RGOOP-7 was 1400 S m−1, which was similar to that of RGOH2O, because GO tended to aggregate in a high concentration of OP-7, resulting in a low reduction degree of GO. Although the conductivity of RGOEGM increased to 3770 S m−1, the poor dispersion of graphene in EGM led to non-ideal conductivity. With the use of the mixed reaction medium of OP-7 and EGM, the conductivity of RGOOP-7/EGM was 14000 S m−1, showing an over tenfold increase with respect to the RGOH2O value due to the interactions between the barrier effect of OP-7 and the good dispersion action of EGM. This result is consistent with the previous test results.
| Reduced graphene oxide | Drying temperature | Conductivity (S m−1) |
|---|---|---|
| RGOH2O | Room temperature | 1050 |
| RGOOP-7 | Room temperature | 1400 |
| RGOEGM | Room temperature | 3770 |
| RGOOP-7/EGM | Room temperature | 14000 |
Table 4 summarizes the electrical conductivity of the RGOOP-7/EGM power pellets and graphene power pellets or free-standing paper samples under different reduction conditions. In contrast to the electrical conductivity reported previously for modified graphene reduced by different chemical methods, our RGOOP-7/EGM power pellet had a high electrical conductivity (14000 S m−1), which was almost 70 times that of graphene reduced by hydrazine (200 S m−1), and achieved a higher value without treatment at high-temperatures. The conductivity of RGOHI-AcOH reduced by hydroiodic acid (HI) in acetic acid (AcOH) solvent is higher than that of RGOOP-7/EGM, the final conductivity of graphene may be different due to the differences in the reduction system. Our method of reduction in the mixed medium of OP-7 and EGM is a simple separation process, involves mild reaction conditions and short reduction time, and exhibits great reproducibility. The high electrical conductivity of RGOOP-7/EGM was attributed to the avoidance of aggregation because of the barrier effect of surfactant OP-7 and the good dispersion action of the organic solvent EGM. The electrical conductivity of RGOOP-7/EGM with 220 °C treatment reached 15800 S m−1, which was higher than that of the RGOOP-7/EGM sheet (14000 S m−1) obtained via room temperature treatment. We consider that the increase in electrical conductivity could be attributed to the escape and volatilization of some organic matter or adsorbate on the surface of RGOOP-7/EGM.
| Reduced graphene oxide | Reduction temperature (°C) | Reduction time (h) | Drying temperature (°C) | Conductivity (S m−1) |
|---|---|---|---|---|
| RGOOP-7/EGM | 80 | 3 | Room temperature | 14000 |
| 220 | 15800 |
|||
| Reduced graphene oxide by hydrazine33 | 80 | 24 | 60 | 200 |
| RGOHI-AcOH28 | 40 | 40 | Room temperature | 30400 |
| hKMG34 | 35 | 6 | Room temperature | 690 |
| RGH35 | 90 | 48 | −37 °C freeze-dried | 1351 |
| HRG36 | 80 | 12 | Room temperature | 1700 |
| 150 | 16000 |
|||
| CCG31,32 | 95 | 1 | Room temperature | 7200 |
| 220 | 11800 |
|||
| CCG2 (ref. 25) | 120 | 12 | Room temperature | 1600 |
Conclusions
In summary, we have analyzed the aggregation of graphene during the chemical reduction method and reported the method to prevent the aggregation of graphene. In addition, the potential reduction mechanism and the root cause for the aggregation of graphene have been addressed. RGOOP-7/EGM was reduced from GO with hydrazine hydrate in a mixed medium of OP-7 and EGM. EGM provided a good dispersion environment for GO, and OP-7 could effectively prevent superimposed aggregation. Furthermore, the XRD spectrum of RGOOP-7/EGM shows a dispersing diffraction peak, indicating that there was no typical layer structure. The RGOOP-7/EGM sheet obtained via room temperature treatment had an electrical conductivity value as high as 14000 S m−1.
Conflicts of interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work. There is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled, “Aggregation Prevention: Reduction of Graphene Oxide in Mixed Medium of Alkylphenol Polyoxyethylene (7) Ether and 2-Methoxyethanol”.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (51273155).References
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- S. Ramirez, K. Chan, R. Hernandez, E. Recinos, E. Hernandez, R. Salgado, A. G. Khitun, J. E. Garay and A. A. Balandin, Mater. Des., 2017, 118, 75–80 CrossRef CAS.
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS PubMed.
- R. Verma, S. Bhattacharya and S. Mahapatra, IEEE Trans. Electron Devices, 2013, 60, 2695–2698 Search PubMed.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 52–77 Search PubMed.
- H. Bai, C. Li and G. Shi, Adv. Mater., 2011, 23, 1089–1115 CrossRef CAS PubMed.
- B. Guo, L. Fang, B. Zhang and J. R. Gong, Insci. J., 2011, 40, 80–89 CrossRef.
- H. Spanggaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2004, 83, 125–146 CrossRef CAS.
- G. Wang, X. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049–2053 CrossRef CAS.
- W. Bower, W. Head, G. T. R. Droop, R. Zan, R. A. D. Pattrick, P. Wincott and S. J. Haigh, Mineral. Mag., 2015, 79, 337–344 CrossRef CAS.
- K. Kim, J. Park, C. Kim, W. Choi, Y. Seo, J. Ahn and I. Park, Micro Nano Lett., 2012, 7, 1133 CrossRef.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- D. Long, W. Li, L. Ling, J. Miyawaki, I. Mochida and S. Yoon, Langmuir, 2010, 26, 16096–16102 CrossRef CAS PubMed.
- S. Pei and H. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- S. Pei, J. Zhao, J. Du, W. Ren and H. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- R. Iguchi, T. Kawamura, Y. Suzuki, M. Inoue, Y. Kangawa and K. Kakimoto, Jpn. J. Appl. Phys., 2014, 53, 65601 CrossRef.
- Z. Juang, C. Wu, C. Lo, W. Chen, C. Huang, J. Hwang, F. Chen, K. Leou and C. Tsai, Carbon, 2009, 47, 2026–2031 CrossRef CAS.
- B. Hu, H. Ago, Y. Ito, K. Kawahara, M. Tsuji, E. Magome, K. Sumitani, N. Mizuta, K. Ikeda and S. Mizuno, Carbon, 2012, 50, 57–65 CrossRef CAS.
- M. Batzill, Surf. Sci. Rep., 2012, 67, 83–115 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- L. Tang, Y. Wang, Y. Li, H. Feng, J. Lu and J. Li, Adv. Funct. Mater., 2009, 19, 2782–2789 CrossRef CAS.
- A. F. Morpurgo, H. B. Heersche, L. M. K. Vandersypen, X. Liu and J. B. Oostinga, Nat. Mater., 2008, 7, 151–157 CrossRef PubMed.
- W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- S. Pei, J. Zhao, J. Du, W. Ren and H. Cheng, Carbon, 2010, 48, 4466–4474 CrossRef CAS.
- H. Shin, K. K. Kim, A. Benayad, S. Yoon, H. K. Park, I. Jung, M. H. Jin, H. Jeong, J. M. Kim, J. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987–1992 CrossRef CAS.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 1–6 CrossRef PubMed.
- Z. Fan, K. Wang, T. Wei, J. Yan, L. Song and B. Shao, Carbon, 2010, 48, 1686–1689 CrossRef CAS.
- V. H. Pham, T. V. Cuong, T. D. Nguyen-Phan, H. D. Pham, E. J. Kim, S. H. Hur, E. W. Shin, S. Kim and J. S. Chung, Chem. Commun., 2010, 46, 4375–4377 RSC.
- D. Li, M. B. Mã Ller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- H. Chen, M. B. Müller, K. J. Gilmore, G. G. Wallace and D. Li, Adv. Mater., 2008, 20, 3557–3561 CrossRef CAS.
- C. Bao, L. Song, W. Xing, B. Yuan, C. A. Wilkie, J. Huang, Y. Guo and Y. Hu, J. Mater. Chem., 2012, 22, 688–696 Search PubMed.
- S. Park, J. An, R. D. Piner, I. Jung, D. Yang, A. Velamakanni, S. T. Nguyen and R. S. Ruoff, Chem. Mater., 2008, 20, 6592–6594 CrossRef CAS.
- V. H. Luan, H. N. Tien, L. T. Hoa, N. T. M. Hien, E. Oh, J. Chung, E. J. Kim, W. M. Choi, B. Kong and S. H. Hur, J. Mater. Chem. A, 2013, 1, 208–211 RSC.
- S. Park, J. An, I. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2009, 114, 832–842 CrossRef.
- Y. Zhou, Q. Bao, L. A. L. Tang, Y. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950–2956 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07263a |
| This journal is © The Royal Society of Chemistry 2018 |