
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiffusion of methyl oleate in hierarchical micro-/mesoporous TS-1-based catalysts probed by PFG NMR spectroscopy†
Muslim Dvoyashkin *a,
Nicole Wildea,
Jürgen Haaseb and
Roger Gläsera
*a,
Nicole Wildea,
Jürgen Haaseb and
Roger Gläsera
aInstitute of Chemical Technology, Universität Leipzig, Linnéstr. 3, 04103 Leipzig, Germany. E-mail: muslim.dvoyashkin@uni-leipzig.de
bFelix-Bloch-Institut, Universität Leipzig, Linnéstr. 5, 04103 Leipzig, Germany
First published on 20th November 2018
Abstract
Pulsed field gradient (PFG) NMR is successfully applied to trace the diffusion of methyl oleate (MO) inside the mesopores of hierarchically structured titanium silicalite-1 (TS-1)-based catalysts. Introduction of mesoporosity by post-synthetic treatment of initially microporous TS-1 provides additional active surface to improve catalytic activity in the epoxidation of MO. The present study provides experimental evidence of the accessibility of mesopores for MO resulting from alkaline treatment of TS-1. The self-diffusion coefficients of MO inside the pores of hierarchically structured TS-1 catalysts are up to two orders of magnitude lower compared to the values in the bulk liquid phase. Additionally, the methodological capability of PFG NMR for measuring self-diffusion coefficients of long-chain hydrocarbons (up to C19) confined to narrow mesopores of catalytically active is demonstrated for the first time.
In micro- and mesoporous materials, transport of fluids with molecular dimensions comparable with those of the pores often determines the overall rate of processes such as adsorption or catalysis as a result of diffusion limitations.1 While the vast majority of self-diffusion studies are focused on transport characteristics of low molecular-weight species, in particular short-chain hydrocarbons,2,3 far less diffusion data are reported for higher molecular-weight compounds with dimensions of up to several nanometers, such as proteins, fats and oils or long-chain hydrocarbons.
A specific example for the need of understanding the transport of long-chain molecules in nanopores is the heterogeneously catalyzed epoxidation of fatty acid methyl esters, such as, e.g., methyl oleate (MO), in the presence of microporous molecular sieves4,5 for the production of high value-added epoxides. Olefin epoxidation over titanium-containing molecular sieves is already industrially applied for lower olefins such as propene.6 Epoxidations over TS-1 can be performed at ambient temperatures and pressures using hydrogen peroxide as the sole oxidant, thus forming only water as a byproduct.4 However, efficient utilization of this microporous catalyst is often limited due to the pore width of ∼0.58 nm,7 and, thus, strong limitations of MO (∼2.5 nm along the C–C segments, ∼0.5 nm for the cross section) diffusion inside the TS-1 pore system.
It is known that zeolites and related microporous materials possessing hierarchical pore systems, containing a secondary porosity with pore widths larger than the micropores can facilitates intra-crystalline transport.1,8 In such materials, the presence of pores and channels larger than the size of micropores might help to overcome the mass-transfer limitations often associated with catalytic conversions over conventional zeolites.9 Tailoring the pore space towards maximum catalytic activity, selectivity and stability is a challenging task requiring extensive knowledge of both textural properties of the catalysts and the diffusion characteristics of the confined molecular species, i.e., reactants and products.10 While transport mechanisms of lower molecular-weight adsorbates in multiple micro-/mesoporous materials were thoroughly investigated using pulsed field gradient (PFG) NMR,11 only a single conference contribution was reported on diffusion of MO in a mesoporous silicate, i.e., SBA-16,12 and just one proceeding report on PFG NMR study of n-hexadecane confined to MCM-41.13 No studies exist on the diffusion of hydrocarbons comparable in molecular size with MO in catalytically active materials. This is most probably due to the combination of two factors, i.e., short spin–spin relaxation times (from few to few tens of ms) and the need for ultra-high gradients (∼few tens of T m−1) required for detection of slowly diffusing molecules.
In an attempt to improve the accessibility of the active Ti sites located within the framework, hierarchical micro-/mesoporous TS-1-based catalysts were prepared by post-synthetic treatment consisting of an alkaline treatment and a subsequent pseudomorphic transformation by recrystallization in the presence of a surfactant as a structure-directing agent.4 It has been demonstrated that such post-synthetic treatment results in a higher catalytic activity of the TS-1-based catalysts in MO epoxidation with aqueous hydrogen peroxide solution as compared to the purely microporous TS-1. It could, however, not been proven whether the enhanced accessibility of the active sites indeed leads to facilitated diffusion through the mesopores into the intra-crystalline space of the hierarchically structured TS-1 particles.
Therefore, the PFG NMR technique was applied in this study to directly assess the diffusion characteristics of MO inside the crystallites of three TS-1-based catalysts, i.e., a microporous TS-1 from microwave-assisted synthesis, and two micro-/mesoporous TS-1 catalysts from alkaline treatment and from alkaline treatment and a subsequent pseudomorphic transformation, respectively.
The three TS-1-based catalysts, i.e., the initial TS-1 (TS-1_ns70), and the post-synthetically treated micro-/mesoporous TS-1 catalysts from alkaline treatment (D_NH3/TPAOH) and from alkaline treatment and a subsequent pseudomorphic transformation (R_D_NH3/TPAOH) were prepared as described in ref. 4 Also, for the physicochemical properties as well as the catalytic behavior of the three TS-1-based catalysts in the liquid-phase epoxidation of FAME with hydrogen peroxide, the reader is referred to ref. 4.
Fig. 1 displays diffusion attenuation curves obtained for the three catalysts, i.e., the initial TS-1 (TS-1_ns70, Fig. 1A), and the two micro-/mesoporous materials obtained from alkaline treatment (D_NH3/TPAOH, Fig. 1B), and from alkaline treatment followed by pseudomorphic transformation (R_D_NH3/TPAOH, Fig. 1C) for diffusion times of 4, 18, 40 and 160 ms. All attenuation curves are expectedly not mono-exponential suggesting the presence of more than one molecular ensemble, possessing different mobilities. According to the procedure of the sample preparation, one shall expect a substantial amount of bulk excess of MO outside the pellets, and a fraction of MO that may occupy an inter-crystalline space between the individual crystallites within the pellet (see Fig. S1 of the ESI†). Contribution of these fractions is seen from the curves presented in Fig. 1A, and can satisfactorily be described using eqn (S1) of the ESI† by a sum of two exponents. The result of such fitting is given by a solid line and shows good agreement with the experimental data obtained for the diffusion times in the range from 8 to 160 ms. All results of the fitting are summarized in Table 1.
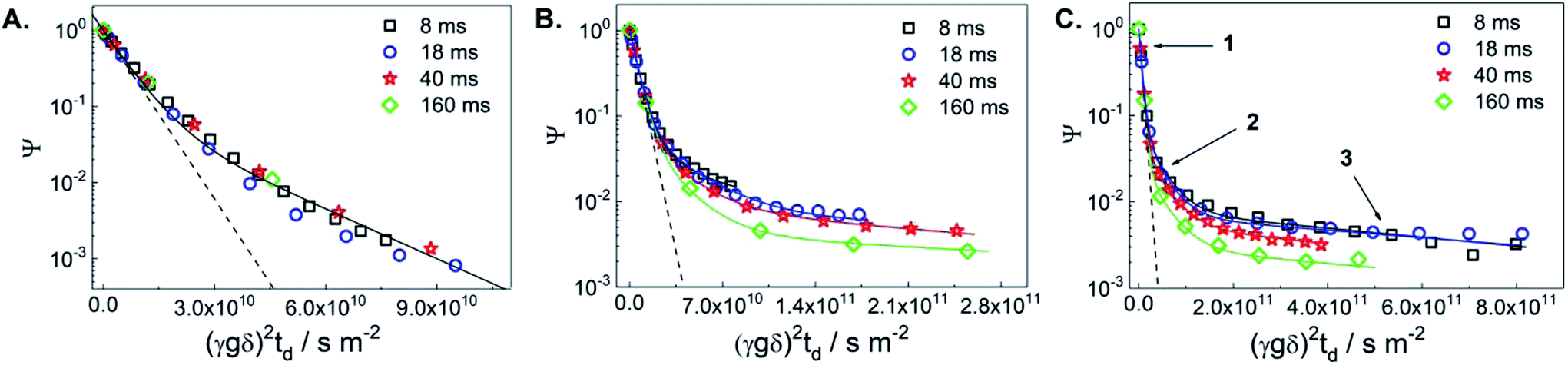 | ||
| Fig. 1 1H PFG NMR diffusion attenuation curves obtained for MO in the presence of TS-1_ns70 (A), D_NH3/TPAOH (B) and R_D_NH3/TPAOH (C) for 8 ms (squares), 18 ms (circles), 40 ms (stars) and 160 ms (diamonds) of diffusion time at 298 K. The solid lines represent the 2-exponential (A) and 3-exponential (B and C) fits of the data using eqn (S1) of the ESI.† The dashed lines represent the attenuation of the pure MO measured in a separate experiment. The arrows point to the different sections of the attenuation curves, in which different diffusion process dominate: (1) – bulk-like diffusion outside pores, (2) – long-range diffusion caused by exchange between the spaces inside and outside pores and/or diffusion in the inter-crystalline space, and (3) – diffusion within mesopores. | ||
| Catalyst | Δa/ms | D1b/10−10 m2 s−1 | RMSD/μm | D2/10−11 m2 s−1 | RMSD/μm | D3/10−12 m 2s−1 | RMSD/μm |
|---|---|---|---|---|---|---|---|
| a The diffusion time td = Δ − δ/3.b Parameter D1 = 1.7 × 10−10 m2 s−1 was fixed in the fitting procedure. | |||||||
| TS-1_ns70 | 8–160 | 1.7 | 2.8–12.8 | 5 ± 1 | 1.5–6.9 | n.d. | n.d. |
| D_NH3/TPAOH | 8 | 1.7 | 2.8 | 4 ± 1 | 1.4 ± 0.1 | 3 ± 1 | 0.4 ± 0.1 |
| 18 | 1.7 | 4.3 | 3 ± 1 | 1.8 ± 0.3 | 2 ± 1 | 0.5 ± 0.2 | |
| 40 | 1.7 | 6.4 | 4 ± 1 | 3.1 ± 0.4 | 3 ± 2 | 0.8 ± 0.3 | |
| 160 | 1.7 | 12.8 | 4.5 ± 2.5 | 6.6 ± 2.2 | 3 ± 2 | 1.7 ± 0.7 | |
| R_D_NH3/TPAOH | 8 | 1.7 | 2.8 | 2.8 ± 0.3 | 1.2 ± 0.1 | 1.2 ± 0.2 | 0.24 ± 0.02 |
| 18 | 1.7 | 4.3 | 2.9 ± 0.3 | 1.8 ± 0.1 | 1.2 ± 0.2 | 0.36 ± 0.03 | |
| 40 | 1.7 | 6.4 | 2.6 ± 0.8 | 2.5 ± 0.4 | 1.3 ± 0.7 | 0.56 ± 0.18 | |
| 160 | 1.7 | 12.8 | 3 ± 1 | 5.4 ± 1.0 | 1.1 ± 0.9 | 1.03 ± 0.59 | |
The self-diffusion coefficient representing the faster diffusing component is denoted as D1 and is equal to the diffusivity value of the bulk MO within the uncertainty of the experiment. The latter was measured in a separate diffusion experiment with only bulk MO inside the NMR tube at the same temperature (298 K) in which the self-diffusion coefficient D1 = 1.7 × 10−10 m2 s−1 was obtained. This value was fixed in the fitting of diffusion decays in Fig. 1. The decay that corresponds to this diffusivity is presented in Fig. 1(A–C) by dashed lines. The self-diffusion coefficient representing the slower diffusing component in Fig. 1A is denoted as D2 and is assigned to MO diffusing within the inter-crystalline space, i.e., in the voids between the crystallites of TS-1 generated during pelletization. The observed two-exponential decay suggests that MO located within the pellets diffuses slower resulting in an approx. 3-fold smaller self-diffusion coefficient (D2 = 5 × 10−11 m2 s−1) compared to D1.
The root mean squared displacements (RMSDs) of the MO molecules can be estimated using the Einstein relation: 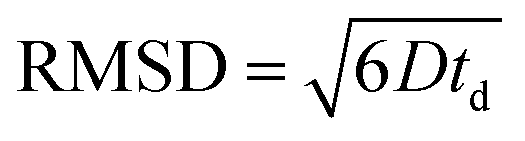 . The RMSD for the shortest observation time (8 ms) results in 1.5 μm for the MO located between the crystallites (Table 1). This value is 2–3 fold larger than the size of the crystallites, thus, resulting in the lower self-diffusion coefficient compared to the value in the bulk phase. Absence of a time dependence of the self-diffusion coefficient D2 can be caused by approximately similar size and tortuosity of the inter-crystalline space throughout the pellet. This does not change significantly after MO molecules “explore” more than a few crystallites during the observation time. At the same time, the RMSD is much smaller than the size of the pellets (∼100–200 μm) resulting in slow molecular exchange with the MO located outside the pellets, i.e., in the bulk liquid phase. As expected, due to the narrow micropore width of TS-1 (∼0.5 nm), the component corresponding to possible diffusion within micropores was below the detection limit.
. The RMSD for the shortest observation time (8 ms) results in 1.5 μm for the MO located between the crystallites (Table 1). This value is 2–3 fold larger than the size of the crystallites, thus, resulting in the lower self-diffusion coefficient compared to the value in the bulk phase. Absence of a time dependence of the self-diffusion coefficient D2 can be caused by approximately similar size and tortuosity of the inter-crystalline space throughout the pellet. This does not change significantly after MO molecules “explore” more than a few crystallites during the observation time. At the same time, the RMSD is much smaller than the size of the pellets (∼100–200 μm) resulting in slow molecular exchange with the MO located outside the pellets, i.e., in the bulk liquid phase. As expected, due to the narrow micropore width of TS-1 (∼0.5 nm), the component corresponding to possible diffusion within micropores was below the detection limit.
Fitting of diffusion data obtained for the micro-/mesoporous materials D_NH3/TPAOH and R_D_NH3/TPAOH as presented in Fig. 1B and C requires consideration of at least three components. In addition to the diffusion in the bulk phase (including the space between the pellets) D1 and diffusion in the inter-crystallite space D2 (within the pellets), it is reasonable to assign the third component (D3) to diffusion within the mesopores of the crystallites of the materials from post-synthetic treatment of the parent TS-1. The width of the mesopores in the alkaline treated material, 3–40 nm, is large enough to accommodate MO, having a largest dimension of ∼2.5 nm (ref. 14) and a certain degree of a chain flexibility. It has to be mentioned that, if MO has indeed access to the mesopores, for the treated samples the ensemble contributing to part of the diffusion decay resulting in D2 cannot be related to diffusion between the crystallites only. For the probed diffusion times, the resulting RMSDs (1.6–6.6 μm) are long enough for partial diffusion within the mesopores. This is the case when molecules spend part of the time inside mesopores and part of the time outside, i.e., they undergo an exchange between the inter-crystalline space and mesopores of the crystallites themselves. Under such conditions, the diffusion process resulting in D2 has to be treated as long-range diffusion.15
Diffusion of MO in the mesopores of the post-synthetically treated materials results in the intra-crystalline self-diffusion coefficients 3 × 10−12 m2 s−1 for the alkaline treated D_NH3/TPAOH and 1 × 10−12 m2 s−1 for the alkaline treated and pseudomorphically transformed R_D_NH3/TPAOH TS-1-based catalyst. These diffusion coefficients do not reveal any time dependence. Both values are almost two orders of magnitude lower than the diffusivity in the bulk liquid phase surrounding the pellets. This suggests that small mesopores within the TS-1 crystallites impose a strong confinement on the mobility of MO in the treated micro-/mesoporous materials. The RMSDs calculated for the smallest observation time used in the experiment (8 ms) results in ∼0.24 μm and ∼0.4 μm for the materials D_NH3/TPAOH and R_D_NH3/TPAOH, respectively. These values are smaller or comparable to the sizes of individual crystallites of TS-1 (∼0.5–1 μm) seen in the SEM images (see Fig. 6 of ref. 4). This suggests that the observed diffusivities D3 are predominantly determined by the diffusion within intra-crystalline mesopores. Analysis of the obtained RMSDs suggests absence of pore blockages for the MO diffusion on the length-scale comparable to the size of the crystallites. Thus, one may conclude that the network of mesopores within the TS-1 crystallites is accessible for MO on the millisecond time-scale. A threefold difference in the intra-crystalline diffusivities can be explained by the higher fraction of larger mesopores in the range 10–40 nm (0.11 cm3 g−1) in D_NH3/TPAOH than in R_D_NH3/TPAOH (0.05 cm3 g−1, see Table 1). This imposes less confinement on diffusion of adsorbed MO. It is in complete accordance with the higher fraction of smaller mesopores in the width range of 3–10 nm after pseudomorphic transformation in R_D_NH3/TPAOH compared to D_NH3/TPAOH.
The fitting parameters pi presented in eqn (S1) of the ESI† reflect the contribution of a certain component possessing the diffusivity Di and are proportional to the number of molecular nuclei diffusing with Di during the observation time td. However, obtaining the exact mole fractions for each component would require knowledge of the nuclear spin relaxation times (T1, T2) of each of the three components. The procedure of the correct determination of the relaxation times affecting observed signal in the diffusion experiment is described in the ref. 16. Following this procedure, it was, unfortunately, not possible to extract corresponding relaxation times due to insufficient signal-to-noise levels. It is worth noting the molecular exchange between the intra- and inter-crystalline pore spaces cannot be completely ruled out, and thus might also affect the pi-values.
The diffusivities of the MO within the mesopores of D_NH3/TPAOH and R_NH3/TPAOH are of the same order of magnitude (see values of D3 presented in Table 1). From this, one may conclude that the active Ti sites on the (internal) surface of these hierarchical catalysts are similarly accessible for the MO molecules. However, the catalytic activity of the pseudomorphically transformed material (R_D_NH3/TPAOH) was found to be notably higher than for the catalyst from alkaline treatment only (D_NH3/TPAOH).4 The observed differences in activities, thus, might be caused by additional factors, e.g., by differences in surface hydrophilicity of the materials present after alkaline treatment and pseudomorphic transformation. Addressing this point would require additional studies in which, for example, the hydrophobicity of the mesopore surface of the treated samples will be probed by water sorption isotherms.
The present contribution represents first successful experimental results on molecular self-diffusion coefficients of MO confined to mesopores of catalytically active molecular sieves with a hierarchical micro-/mesopore system. As a case study, the self-diffusion processes of MO in pelletized TS-1-based catalysts containing mesopores of 3–40 nm pore width as a result of a post-synthetic treatment were investigated. Three different diffusion regimes in the treated catalysts were observed: (i) intra-crystalline diffusion within mesopores, (ii) long-range diffusion in the mesopore and in the inter-crystalline space of a pellet, and (iii) bulk-like diffusion in the voids between and outside the pellets. The intra-crystalline diffusivities are approximately two orders of magnitude lower compared to those of diffusion in the bulk phase at the same temperature. In the parent microporous TS-1, no diffusion of MO within the crystallites was observed (as expected). The experimental data suggest that the mesopore network of the hierarchical micro-/mesoporous TS-1-based catalysts is well interconnected and accessible for MO molecules at room temperature. This, however, does not rule out possible existence of isolated mesopores being not accessible for methyl oleate. In such case, introduction of mesopores might not facilitate the intra-crystalline transport, such as it has been reported in ref. 3. To clarify this, further microscopy studies are required. The obtained self-diffusion coefficients allow the estimation of the time required for MO to diffuse through the entire pellet at room temperature. Moreover, the diffusivities differ for the materials with different width fractions of intra-crystalline mesopores. Finally, the presented diffusion data demonstrate the feasibility of application of the PFG NMR technique for the direct determination of diffusion coefficients of high molecular-weight long-chain hydrocarbons and oxygen-functionalized derivatives, such as MO, in catalysts with hierarchically structured pore systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MD acknowledges the financial support within the Max-Buchner-Forschungsstiftung (DECHEMA) #3427 and DFG Research Grant DV 58/2-1.References
- J. Kärger, M. Göpel and R. Gläser, Nanotechnology in Catalysis, ed. M. van de Voorde and B. Sels, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, ch. 13, pp. 293–334 Search PubMed
.
-
(a) A. Galarneau, F. Guenneau, A. Gedeon, D. Mereib, J. Rodriguez, F. Fajula and B. Coasne, J. Phys. Chem. C, 2016, 120, 1562–1569 CrossRef CAS
- P. Kortunov, S. Vasenkov, J. Kärger, R. Valiullin, P. Gottschalk, M. Fé Elía, M. Perez, M. Stöcker, B. Drescher, G. McElhiney, C. Berger, R. Gläser and J. Weitkamp, J. Am. Chem. Soc., 2005, 127, 13055–13059 CrossRef CAS PubMed
- N. Wilde, M. Pelz, S. G. Gebhardt and R. Gläser, Green Chem., 2015, 17, 3378–3389 RSC
-
(a) N. Wilde, J. Přech, M. Pelz, M. Kubů, J. Čejka and R. Gläser, Catal. Sci. Technol., 2016, 6, 7280–7288 RSC
- L. Y. Chen, G. K. Chuah and S. Jaenicke, J. Mol. Catal. A: Chem., 1998, 132, 281–292 CrossRef CAS
- A. Keshavaraja, V. Ramaswamy, H. S. Soni, A. V. Ramaswamy and P. Ratnasamy, J. Catal., 1995, 157, 501–511 CrossRef CAS
-
(a) D. Verboekend, N. Nuttens, R. Locus, J. van Aelst, P. Verolme, J. C. Groen, J. Perez-Ramirez and B. F. Sels, Chem. Soc. Rev., 2016, 45, 3331–3352 RSC
- S. Mitchell, A. B. Pinar, J. Kenvin, P. Crivelli, J. Kärger and J. Pérez-Ramírez, Nat. Commun., 2015, 6, 8633 CrossRef CAS PubMed
-
(a) S. M. Rao, E. Saraçi, R. Gläser and M.-O. Coppens, Chem. Eng. J., 2017, 329, 45–55 CrossRef CAS
- D. Schneider, D. Mehlhorn, P. Zeigermann, J. Kärger and R. Valiullin, Chem. Soc. Rev., 2016, 45, 3439–3467 RSC
- S. M. Wang, R. Kanungo, B. Nohair, S. Vasenkov and S. Kaliaguine, Diffusion fundamentals, 2011, 16, 1–2 Search PubMed
- F. Stallmach, A. Gräser, J. Kärger, C. Krause, M. Jeschke, U. Oberhagemann and S. Spange, Microporous Mesoporous Mater., 2001, 44–45, 745–753 CrossRef CAS
- P. Berman, N. Meiri, L. A. Colnago, T. B. Moraes, C. Linder, O. Levi, Y. Parmet, M. Saunders and Z. Wiesman, Biotechnol. Biofuels, 2015, 8, 12 CrossRef PubMed
-
(a) S. Vasenkov and J. Kärger, Magn. Reson. Imaging, 2005, 23, 139–145 CrossRef PubMed
- M. Dvoyashkin, J. Zang, G. I. Yucelen, A. Katihar, S. Nair, D. S. Sholl, C. R. Bowers and S. Vasenkov, J. Phys. Chem. C, 2012, 116, 21350–21355 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Materials preparation, post-synthetic treatment, textural properties, diffusion NMR experimental procedures. See DOI: 10.1039/c8ra07434h |
| This journal is © The Royal Society of Chemistry 2018 |