
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence‘Phoenix polymers’: fire induced nanohardness in fibril-forming aromatic cyanate esters†
Lyndsey Mooring‡
a,
Scott Thompson§
a,
Stephen A. Halla,
Silvia Panib,
Peter Ziouposc,
Martin Swand,
Corinne Stoned,
Brendan J. Howlin a and
Ian Hamerton*e
a and
Ian Hamerton*e
aDepartment of Chemistry, Guildford, Surrey, GU2 7XH, UK
bDepartment of Physics, Faculty of Engineering and Physical Sciences, University of Surrey, Guildford, Surrey, GU2 7XH, UK
cCranfield Forensic Institute, CDS, Cranfield University, Shrivenham SN6 8LA, UK
dDstl, Porton Down, Salisbury, SP4 0JQ, UK
eBristol Composites Institute (ACCIS), Department of Aerospace Engineering, School of Civil, Aerospace, and Mechanical Engineering, Queen's Building, University of Bristol, University Walk, Bristol, BS8 1TR, UK. E-mail: ian.hamerton@bristol.ac.uk; Tel: +44(0) 0117 3314799
First published on 25th October 2018
Abstract
For the first time we present nanoindentation analysis of charred, cured aromatic cyanate esters, which exhibit outstanding mechanical properties when analysed under applied loads of 0.1–300 mN. Following charring (900 °C for 10 minutes to achieve graphitised structures), the samples display a remarkable combination of a modulus of elasticity of around 25 GPa and nanohardness of 300 kgf mm−2, making them some 30–40% stiffer than bone and practically as hard as tooth enamel. At the same time we find that under the same conditions the chars are highly resilient, displaying complete elastic recovery with very little plastic deformation. When cured in the presence of copper(II) acetylacetonate (200 ppm) in dodecylphenol (1% w/v active copper suspension) to form a polycyanurate, compound (2) forms a dense, consolidated structure compared with compound (1) under the same conditions. At high magnification, the presence of a nanoscale, fibrillar structure is observed, accounting for the high resilience.
Introduction
It has been reported1 that approximately 20% of the 1153 fatalities on U.S. transport airlines between 1981–1990 were caused by fire, with the vast majority resulting in post-crash fire accidents; 40% of the passengers who survive the impact of an aircraft accident subsequently die in these post-crash fires.2 The development of structural materials with improved fire resistance relative to commodity plastics is key to retard fire, increase the time available for passengers to escape the aircraft interior and thus reduce the loss of life. The European Union (EU) has restricted the use of brominated diphenyl oxide flame retardants because highly toxic and potentially carcinogenic brominated furans and dioxins may form during combustion.3 The World Health Organisation (WHO) and the US Environmental Protection Agency (EPA) also recommend exposure limit and risk assessment of dioxins and similar compounds.4,5 It is essential that new flame-retardant systems are developed to meet the constantly changing demand of new regulations, standards and test methods. This is often achieved by introducing highly aromatic or hetero-aromatic materials such as thermoset polymer composites that form intumescent chars during the combustion process, with the polymer swelling and becoming porous to protect the underlying structure.6 Thermoset polymers have an established history in civil aviation, in applications involving decorative panels, secondary composite structures and adhesives – around 90% of the interior furnishings of a typical civil airliner will contain thermoset composites.7 Difunctional cyanate esters8 constitute a family of addition cured high performance, thermosetting polymers, which occupy a niche intermediate between high glass transition temperature (Tg), tetrafunctional epoxy resins9 and bismaleimides (BMIs).10 Cured cyanate esters are increasingly used in high temperature applications as they combine high glass transition temperatures (e.g. dry Tg values of 270–300 °C, coupled with strain at break of over 5%), low dielectric loss properties (dielectric constant of 2.7, loss tangent of 0.003 at 25 °C and 1 MHz) with the potential to be toughened with thermoplastics (HexPly 954-2A, GIC = 250 J m−2)11,12 or elastomers (HexPly 953-3, GIC = 450 J m−2)13 for engineering applications. In this form they typically find application as matrices in advanced composites (either in combination with epoxy resins in aerospace applications14,15). While they are not currently widely used in civil aviation (unlike their phenolic-triazine counterparts), they are often combined with BMIs as dielectric polymers in the microelectronics industry.16 The ability of polymers containing the chloral moiety (derived from bisphenol C) to withstand high temperatures and to convert significant quantities of their structures into carbonaceous char makes them of particular interest when considering high temperature engineering applications. In the current study, we explore the effects of thermal conditioning on the development of char structure in two cyanate esters and the mechanical integrity of these structures after the potential effects of fire.Experimental section
Materials
The dicyanate ester monomer 2,2-bis(4-cyanatophenyl)propane (1) (Fig. 1) was supplied by Lonza AG (Visp, Switzerland) (as PRIMASET™ BADCy) and, having confirmed its purity using 1H NMR and elemental analysis, was used as received without further purification. In the interests of brevity, the analytical data (from FTIR ATR, 1H and 13C NMR spectroscopy) are given as ESI.† The synthesis of l,l-dichloro-2,2-(4-cyanatophenyl)ethylidene was based on a well-used route, originally reported by Grigat and Pütter17 Copper(II) acetylacetonate, Cu(acac)2, (97%) and dodecylphenol (mixture of isomers) (used in the co-catalyst package) and cyanogen bromide (97%) were obtained from Sigma Aldrich. Bisphenol C (l,l-dichloro-2,2-(4-hydroxyphenyl)ethylidene, 97%) was obtained from AOKChem, Shanghai. All reagents were analysed to determine purity and used as received without further purification.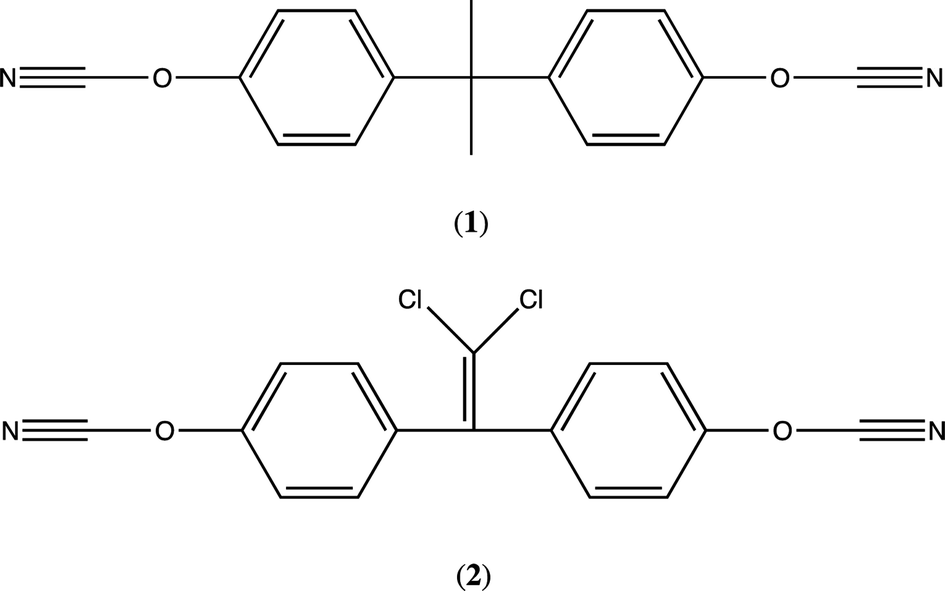 | ||
| Fig. 1 Chemical structures of the monomers studied in this work. | ||
Characterisation of l,l-dichloro-2,2-(4-cyanatophenyl)ethylidene, (2)
The crude product (yields 85–90%) was recrystallized from a dichloromethane/hexane mixture (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30). 1H NMR analysis was performed following recrystallization before samples were combined to ensure consistency of batch quality (examples of spectra are presented in the following sections). Calculated for C16H8Cl2N2O2, wt%: C 58.00, H 2.42, N 8.46, found: C 57.86, H 2.20, N 8.03; IR (ATR, cm−1): 2273, 2235 (OCN stretch), 3000–3100 (Ar–H stretch), 1595 (Ar, C
:30). 1H NMR analysis was performed following recrystallization before samples were combined to ensure consistency of batch quality (examples of spectra are presented in the following sections). Calculated for C16H8Cl2N2O2, wt%: C 58.00, H 2.42, N 8.46, found: C 57.86, H 2.20, N 8.03; IR (ATR, cm−1): 2273, 2235 (OCN stretch), 3000–3100 (Ar–H stretch), 1595 (Ar, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch), 975 (C–Cl stretch), 900–1000 (out-of-plane C–Cl deformation), 835 (out-of-plane C–H deformation characteristic of para-disubstituted phenyl), 600–800 (C–Cl stretch), and 656 (in-plane C–H deformation characteristic of para-substituted phenyl). 1H NMR δH (500 MHz, CDCl3, ppm from TMS): 7.39 (4H, ortho- to the alkene); 7.32 (4H, ortho- to the cyanate); 13C NMR δC (75 MHz, D6-DMSO, ppm from TMS): 152.32 (2 × C2); 137.52 (C6); 136.95 (C6); 131.65 (2 × C5); 122.10 (C7); 115.61 (2 × C4); 108.28 (2 × C1); 77.00 (C from CDCl3 solvent).
C stretch), 975 (C–Cl stretch), 900–1000 (out-of-plane C–Cl deformation), 835 (out-of-plane C–H deformation characteristic of para-disubstituted phenyl), 600–800 (C–Cl stretch), and 656 (in-plane C–H deformation characteristic of para-substituted phenyl). 1H NMR δH (500 MHz, CDCl3, ppm from TMS): 7.39 (4H, ortho- to the alkene); 7.32 (4H, ortho- to the cyanate); 13C NMR δC (75 MHz, D6-DMSO, ppm from TMS): 152.32 (2 × C2); 137.52 (C6); 136.95 (C6); 131.65 (2 × C5); 122.10 (C7); 115.61 (2 × C4); 108.28 (2 × C1); 77.00 (C from CDCl3 solvent).
Blending and cure of polymer samples for thermo-mechanical analyses
To provide sufficient material to examine the char in detail, circular disks, or “pucks”, of the cured cyanate esters were prepared by polymerising the monomers (in the presence or absence of catalyst) in a silicone mould prepared to the specific dimensions required by a published test method.18 This allowed for dimensional measurement as well as weight change to be recorded and attributed to thermal degradation. For catalysed samples, a CuII(acac)2·C18H30O initiator suspension (1% w/v) was made up by mixing CuII(acac)2 with dodecylphenol, heating to 80 °C for 30 minutes then raising the temperature to 120 °C for a further 30 minutes, stirring throughout. Good homogenisation was achieved and CuII(acac)2 (200 ppm) was added to each monomer and heated to 90 °C whilst stirring to yield a homogenous suspension with uniformly dispersed initiator after 15 minutes. The silicone mould was placed in a fan-assisted oven heating at 2 K per minute to 200 °C (1 hour isothermal), the monomer/initiator blend was then added to the mould and allowed to melt as the mould was filled. It was found that the silicone mould could not be held for long periods of time above 230 °C without failure. Consequently, the samples were vitrified in the mould at a lower temperature (e.g. (1) 230 °C for 60 minutes; (2) 220 °C for 90 minutes). The samples were de-moulded before cure and post cure was conducted. This had the advantage that larger batches of puck samples could be cured fully at the same time, negating differences in thermal history. Vitrified samples were heated at 2 K per minute to 260 °C (3 hours isothermal) then post cured by heating at 2 K per minute to 280 °C (1 hour isothermal) followed by a gradual cool (3 K per minute) to room temperature.Instrumentation
1H spectra were obtained at 298 K using a Bruker AV-300 spectrometer operating at 300 MHz and a Bruker DRX-500 spectrometer operating at 500 MHz. 13C spectra were obtained by Bruker AV-300 spectrometer operating at 75 MHz. Depending on the solubility, CDCl3 and D6-DMSO were used to prepare samples using TMS as an internal standard.Fourier transform infrared (FTIR) spectra were recorded using a Perkin Elmer 2000 FT-IR spectrometer interfaced with a computer running PE-Spectrum v5.3 software. Liquid samples were presented on KBr disks and 16 scans were recorded over the range 4000–400 cm−1 with a resolution of 4 cm−1 at intervals of 1 cm−1. Solid samples were placed on the ATR accessory equipped with single reflection, diamond ATR crystal. 16 scans were recorded (4000–550 cm−1, with a resolution of 4 cm−1 at intervals of 1 cm−1).
DSC thermograms were obtained in triplicate using a TA Instruments DSC Q1000 Thermal analyser. All samples (3 ± 1 mg) were analysed in hermetically-sealed, aluminium pans under nitrogen (40 mL min−1) using one of three programmes: (i) samples were heated at 10 K min−1 from 20 °C to 350 °C; (ii) samples were heated rapidly to 225, 250 or 275 °C and held isothermally (120 minutes); (iii) samples were heated at 10 K min−1 from 20 °C to 350 °C, cooled to 20 °C at 10 K min−1 and rescanned at 10 K min−1 from 20 °C to 350 °C to determine the Tg. It was found that the repeatability of the DSC profiles was initially relatively poor for samples of monomer (2) with catalyst package added, which was found to be due to the uneven re-crystallisation of the sample after formulation, as evidenced by heterogeneous distribution of pigmentation arising from the complex in the bulk sample. The use of a ball mill (15 min) on re-crystallised samples of (2) led to improved reproducibility in the DSC scans.
Scanning electron microscopy was performed using an FEI ESEM Quanta 200 Environmental Scanning Electron Microscope (http://www.fei.com/products/sem/quanta-sem/) using an accelerating voltage of 20 kV. To view the samples (and avoid electrostatic charging from the resin) the SEM was used in environmental mode (ESEM), where water vapour in injected into the specimen chamber to a pressure of 1 mBar, to neutralise charge build-up (and get a clear image). Samples were simply mounted on the stub and placed in the specimen chamber (gold coating was not required).
Charred polycyanurate samples were analysed using the CSM-Nano Hardness Testing system, to yield modulus and hardness values for the charred samples of (1) (catalysed), (2) (uncatalysed), and (2) (catalysed). A Berkovich indenter was used at loads typically between 0.1 mN and 300 mN under dynamic or creep loading protocols.
X-Ray CT was carried out on replicate samples of (1) (catalysed), (2) (uncatalysed), and (2) (catalysed) following the “puck” method described above. The images were acquired using a microCT unit based on a Mo-anode X-ray source (Oxford instruments) operated at 50 kVp and 1 mA, a flat panel detector with a 50 μm pixel pitch and CsI scintillator (Hamamatsu C7942) read by means of a frame grabber card with a rate of 2 frames per s, and a set of rotation-translation stages (Micos VT-80 and DT-80 with a SMC Corvus Eco controller). A set of 720 projections was acquired across 360°. Each projection was obtained by integrating 20 frames. CT slices were reconstructed using a filtered back projection algorithm with a Shepp–Logan filter.
TGA thermograms were obtained using a TA Instruments TGA Q500 Thermal Analyser in a platinum pan in flowing nitrogen. During the project the Dstl TGA heating profile (or “Puck” method) was adopted.18 The following outlines the test regime written for our instrument to follow the Dstl method: data storage was engaged (‘On’), the mass flow was set to 100 mL min−1 and the TGA equilibrated at 25 °C and held isothermally (5 min). The mass flow was adjusted to 30 mL min−1 and the sample was heated from 20 °C to 100 °C at a rate of 20 K min−1 (later reduced to 10 K min−1 following experimentation). Samples were then held isothermally (10 min) before being heated at 20 K min−1 (later 10 K min−1) to 900 °C before being held isothermally (10 minutes); data storage was then switched off. The explosive nature of the out-gassing from the cured samples at 400 °C led to pan shake, occasional erratic weight measurement; high quantities of degradation products coating the internal mechanism of the TGA instrument. In extreme cases, the violent loss of volatile products led to samples being ejected from the platinum sample holder into the furnace. Several methods were employed to reduce the impact of these issues, including a reduction in the experimental heating rate from 20 K min−1 to 10 K min−1; milling of sample to 1 mm thickness to produce samples with even surface morphology, as well as reducing the quantity of sample. These changes did improve the sample success rate for the TGA analysis and enabled the generation of reasonable (and reproducible) TGA traces.
Results and discussion
The cured cyanate esters in the form of ‘pucks’ were charred in a systematic manner following a published method18 in which the heating rate and thermal exposure is clearly defined. Following a final isothermal exposure to 900 °C (10 minutes) in an air-circulating furnace, a series of charred pucks were produced from cyanate ester polymers that had been cured in the presence and absence of a co-catalyst package (designated ‘catalysed’ and ‘uncatalysed’ respectively). Nanoindentation measurements were performed on replicates of the charred samples and modulus and hardness values were determined under dynamic or creep loading protocols.19 Nano-hardness is generally defined as the resistance of a material to permanent or plastic deformation at the nano–micro level. In many materials, hardness is a measure of resistance to an indentation where a ball, diamond pyramid or cone is forced into the material being tested under an applied load. In the present study, a Berkovich indenter20 was used at loads of between 0.1 mN and 300 mN at several points (N = 4–6) on the surfaces of the pucks and the data are presented in Table 1 with the inclusion of data for poly(methyl methacrylate), PMMA, for comparison. The modulus of elasticity recorded for the charred cured cyanate ester samples is about 25 GPa, stiffer than bone by some 30–40%,21 comparable with graphite (20 GPa),22 and around five times that of liptinite and vitrinite coals (5–6 GPa).23 Perhaps even more remarkable is the hardness, which at 300 kgf mm−2, is significantly greater than graphite (7–11 kgf mm−2),24 anthracite coal (44–143 kgf mm−2),22 and almost hard as tooth enamel (a material that comprises some 98% ceramic25). The charred samples of the cyanate esters also exhibit extremely interesting elastic behaviour, which results in the samples displaying very low values (20–40 nm) for the permanent depth of indentation (hp) after removal of the force, indicating that the charred polymer reveals hardly any indentation and recovers completely. Furthermore, this is a magnitude lower than the PMMA standard, for which a value 650 nm was recorded, demonstrating significant deformation.| PMMA | (2) Uncatalysed | (1) Catalysed | (2) Catalysed | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a HV = Vickers hardness, EIT = modulus of elasticity, hp = permanent depth of indentation; CIT = indentation creep, RIT = indentation relaxation; nIT = closed force displacement loop. | ||||||||||||
| HV (O&P) | Mean | 14.9 | 276.7 | 308.5 | 325.0 | 331.8 | 342.1 | 260.4 | 304.8 | 327.2 | 307.7 | 262.2 |
| [kgf mm−2] | Std dev | 0.3 | 2.7 | 28.6 | 43.3 | 12.7 | 32.2 | 17.2 | 46.1 | 25.1 | 31.8 | 7.4 |
| N | 5 | 5 | 5 | 6 | 4 | 5 | 5 | 5 | 4 | 6 | 5 | |
| EIT (O&P) | Mean | 4.2 | 25.2 | 26.4 | 25.9 | 30.0 | 29.0 | 27.5 | 25.8 | 26.2 | 26.4 | 24.7 |
| [GPa] | Std dev | 0.1 | 0.3 | 1.0 | 0.8 | 0.2 | 1.3 | 0.9 | 1.0 | 0.6 | 1.1 | 0.9 |
| N | 5 | 5 | 5 | 6 | 4 | 5 | 5 | 5 | 4 | 6 | 5 | |
| hp (O&P) | Mean | 648.1 | 43.7 | 29.9 | 21.8 | 42.2 | 37.2 | 68.0 | 32.2 | 23.7 | 31.7 | 46.1 |
| [nm] | Std dev | 68.3 | 1.0 | 9.9 | 11.8 | 4.8 | 10.4 | 6.9 | 14.5 | 6.2 | 12.8 | 2.4 |
| N | 5 | 5 | 5 | 6 | 4 | 5 | 5 | 5 | 4 | 6 | 5 | |
| CIT | Mean | 5.8 | −0.1 | −0.7 | −1.5 | −0.5 | −0.8 | 1.5 | −1.3 | −1.3 | −1.0 | 0.0 |
| [%] | Std dev | 0.2 | 0.4 | 0.7 | 0.6 | 0.4 | 0.9 | 0.9 | 0.8 | 1.0 | 0.9 | 0.5 |
| N | 5 | 5 | 5 | 6 | 4 | 5 | 5 | 5 | 4 | 6 | 5 | |
| RIT | Mean | −0.05 | 0.04 | 0.05 | 0.05 | 0.02 | 0.07 | 0.01 | 0.06 | 0.01 | 0.06 | 0.07 |
| [%] | Std dev | 0.08 | 0.04 | 0.06 | 0.06 | 0.03 | 0.05 | 0.02 | 0.03 | 0.05 | 0.03 | 0.04 |
| N | 5 | 5 | 5 | 6 | 4 | 5 | 5 | 5 | 4 | 6 | 5 | |
| nIT | Mean | 36.1 | 93.2 | 99.6 | 102.6 | 93.0 | 96.3 | 79.5 | 101.5 | 105.0 | 100.1 | 93.1 |
| [%] | Std dev | 0.4 | 1.1 | 6.4 | 5.4 | 3.4 | 5.8 | 1.6 | 9.4 | 5.8 | 5.7 | 1.5 |
| N | 5 | 5 | 5 | 6 | 4 | 5 | 5 | 5 | 4 | 6 | 5 | |
At the same time the charred cured cyanate ester samples yield very high values for the closed force displacement loop (nIT is practically 100%) compared with PMMA (nIT = 36%). Furthermore, the closed force displacement loop is very tight with little hysteresis, indicating that there is complete elastic recovery with very little plastic deformation, or that a very small percentage of the work is deposited plastically into the material during deformation. The measured shift angle between force and displacement (tanδ) in a dynamic viscous analysis was also nearly zero, for the charred cyanates indicating that the materials display behaviour akin to a non-viscous solid.
Finally, both indentation creep (CIT) and indentation relaxation (RIT) values are also approximately zero for the charred cyanates. It is well known that extensive recovery takes place on most carbons26 and graphites27 with carbon filaments giving rise to figures of the order of 95%,28 although the detection and measurements of indentations on their uncoated surfaces is difficult and so relevant studies are comparatively rare. Hawthorne compared28 the microindentation hardness behaviour of carbon filaments, glassy carbons, and pyrolytic graphites and found that typically diminishing resistance to indenter penetration resulted from greater structural order, which would lead to greater resistance with lower order as is the case in our study.
A charred puck sample of polycyanurate (1) polymerized in the presence of catalyst was analysed by scanning electron microscopy (SEM) and is shown in Fig. 2 at a variety of magnifications. Fig. 2a shows a view of the edge of the charred puck from which the effects of outgassing are apparent e.g. the elliptical fissure (length 3 mm) in the centre of the image. The smaller (2 mm) fissure towards the upper surface in Fig. 2a is observed in greater detail in Fig. 2b, from which the variation in void sizes within the fissure can be seen. There is evidence of release of volatile matter and blistering in sample with the large area (2–3 mm diameter) on the top surface (Fig. 2c) with buckling to relieve the build up of stress within the puck. The observed morphology (Fig. 2c and d) is consistent with the effects of viscous flow during the loss of volatile matter followed by carbonization during the degradation process.
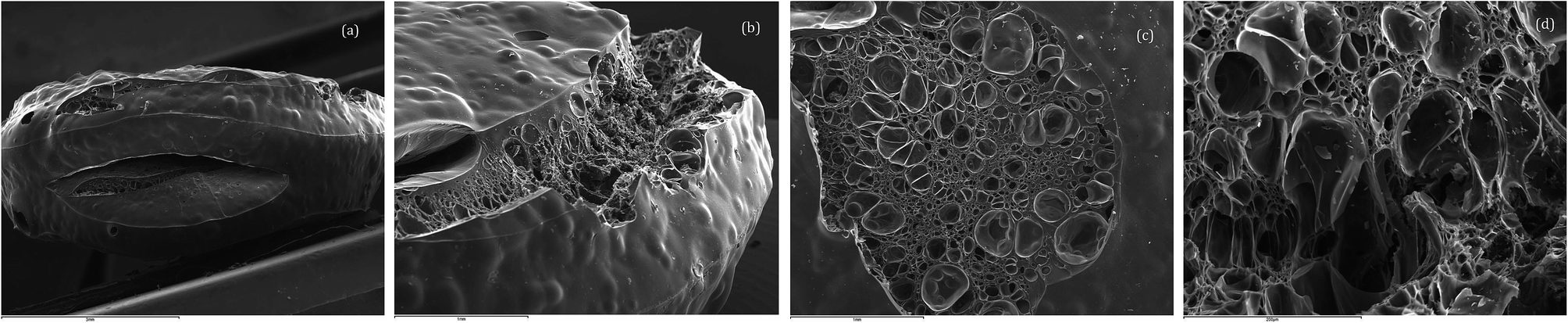 | ||
| Fig. 2 SEM images of charred TGA puck comprising (1) viewed at various magnifications. | ||
In contrast, the charred TGA puck arising from the catalysed chloral containing cyanate ester (2) displays a less distorted surface, although some surface fracture is evident (Fig. 3a), with far more compact cellular structure (Fig. 3b and c). Indeed under extreme magnification the presence of fine fibrils is apparent (Fig. 3d), presumably allowing greater flexibility and resilience in the charred structure.
 | ||
| Fig. 3 SEM images of charred TGA puck comprising (2), polymerised in the presence of catalyst, viewed at various magnifications. | ||
X-ray computed tomography (X-ray CT) was performed on replicates of the charred samples of both (1) (Fig. 4a) and (2) (Fig. 4b,c) to investigate the internal structure, from which it can been seen that the polycyanurates behave very differently as a result of the high temperature exposure experiment. Pucks from both compounds are presented on the same scale (in which the pixel size is 50 μm and the circular feature visible in Fig. 4a and c is a locating device unrelated to the samples) and display the plan view of three or four puck samples that have been positioned to show the depth of their side walls.
 | ||
| Fig. 4 X-Ray CT images of charred TGA pucks comprising (a) (1) (with catalyst), (b) (2) (without catalyst), (c) (2) (with catalyst). | ||
It is immediately apparent that the thermal treatment of (1) (polymerisation followed by char) led to the production of a distribution of voids in the body of the pucks, and this is consistent with the SEM images presented earlier. Furthermore, this results in a distorted puck with an irregular cross section. The charred samples of the uncatalysed polycyanurate of (2) (Fig. 4b) are more densely packed with extensive fracturing visible. However, one surface of each puck tends to retain a flat form and evolved volatiles have escaped from the opposite surface. However, in contrast, the charred samples of the polycyanurate of (2) that have been polymerised in the presence of catalyst (Fig. 4c) are still quite regular and recognizable as puck shaped disks. Some internal fracturing is apparent as thin cracks in these chars, but the samples remain well consolidated and the dimensions are little changed despite the exposure to the extreme temperatures.
The clear implication is that the introduction of a catalyst during the polymerization of (2) leads to a more regular, consolidated and integral char. It should be note that it was not possible to produce cured samples of (1) in the absence of catalyst at the designated cure schedule for comparison (the poor reactivity of the pure monomer rendering this impractical).
The choice of the l,l-dichloro-2,2-ethylidene (chloral) moiety in this work was based on its impressive reported char yield. Ramirez et al.29 have employed the group contribution method first reported by van Krevelen30 to calculate the char-forming tendency (CFT) for both the cyanate esters examined in this study. The CFT is defined as the amount of char (carbon equivalents) per structural unit and the incorporation of oxygen and nitrogen into the char as calculated from molar group calculations has been compared with elemental analyses of the polycyanurate chars recovered after flaming combustion at several hundreds of degrees celsius in a fire calorimeter. Thus, the elemental analysis of (2) yields an empirical formula of C16H8O2N2Cl2 versus an empirical measurement of C16H3O1.3N1.3 for the char. Heat release values for bisphenol C based polymers (i.e. containing the chloral bridge) are typically an order of magnitude lower in flaming combustion compared with bisphenol A analogues.31 For example, for the compounds studied here heat release data have been recorded as 283 J g−1 K−1 and 17.6 kJ g−1 for (1) compared with 13.0 J g−1 K−1 and 3.2 kJ g−1 for (2).32
When analysed using TGA in nitrogen (Fig. 5) the profiles of the polycyanurates resulting from catalysed cure revealed similar profiles and it has been reported29 that the thermal stability of aromatic polycyanurates is essentially independent of monomer chemical structure. Small differences were observed during the initial phase of the degradation (300–400 °C) where the loss of 10% of the mass of (2) precedes a significant mass loss, whereas (1) undergoes a single loss of 40% in the same temperature regime. This is consistent with reports that chloral-containing polymers undergo initial mass loss at lower temperatures than the corresponding isopropyl-bridged analogues.31 In the higher temperature regime (420–500 °C), the loss of mass is slower in (2) and does not appear to be broken down into more discrete steps. This major mass loss at about 450 °C is observed in all polycyanurates and has been found to be due to decyclisation of the triazine ring, liberating volatile decomposition products. Ramirez et al.29 attributed this step to decyclisation of the cyanurate to the corresponding isocyanurate followed by reaction with moisture to form carbamates. The latter underwent further hydrolysis to yield carbamic acid, that decomposes spontaneously to form carbon dioxide and ammonia. We recorded char yields at 800 °C of 38% in the case of (1) and 54% for (2), which are in line with the higher proportion of aromatic content in the chloral polymer and agree well with those found by Ramirez (39% for (1) and 56% for (2) 56% respectively).29 Despite the clear differences observed in the morphologies of the chars formed, the catalysed and thermally cured examples of each polycyanurate show similar char values.
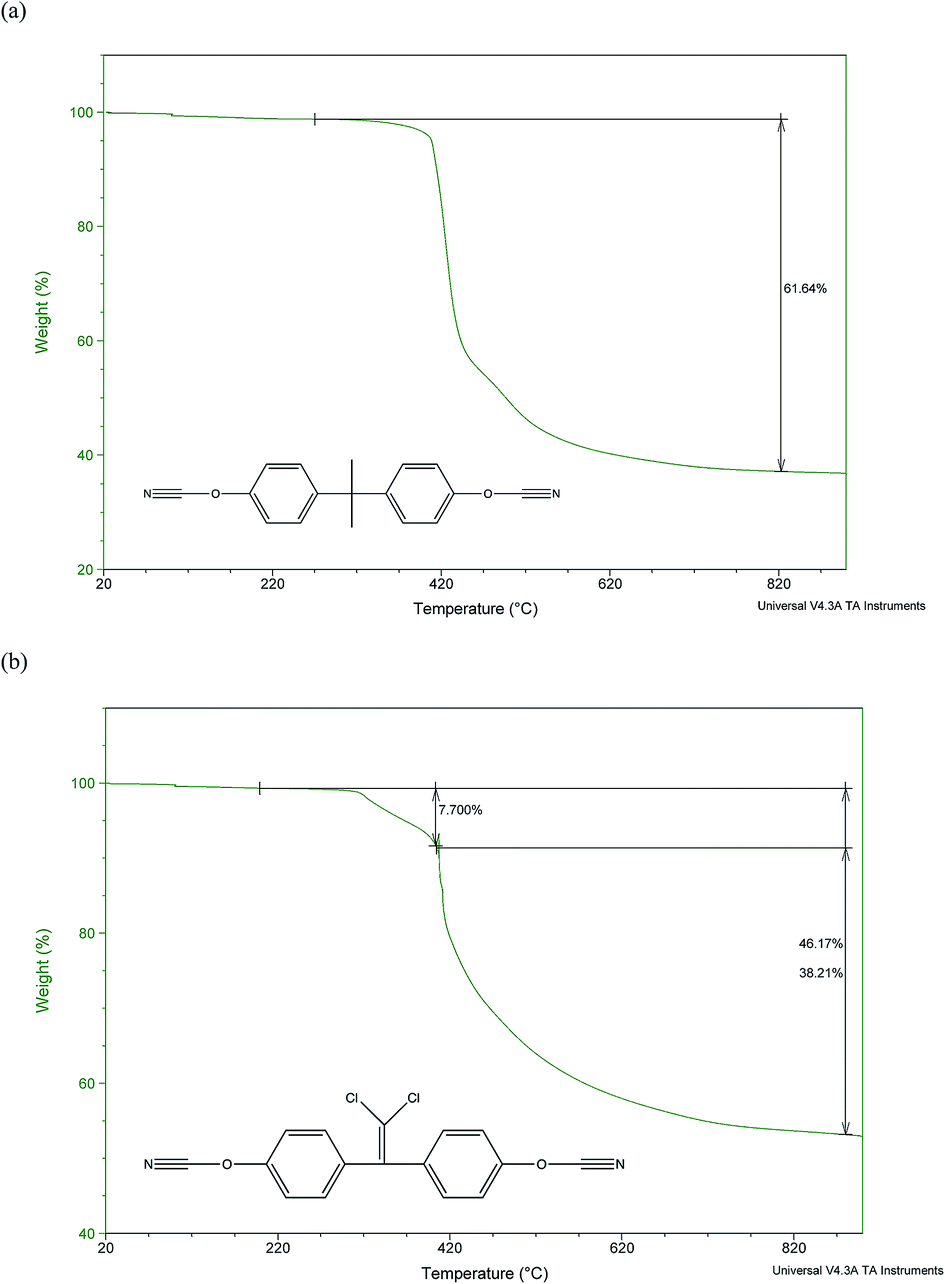 | ||
| Fig. 5 TGA data for (a) cured puck sample (1) and (b) cured puck sample (2). Note monomer structures shown inset and both monomers had been cured with catalyst. | ||
Ramirez has suggested31 the following degradation mechanism for polymers comprising the l,l-dichloro-2,2-(phenyl)ethylidene moiety (Fig. 6) wherein hydrogen chloride forms a major volatile yield along with the degradation products of the polymer-bridging group. The rearrangement through stilbenes and acetylenes is believed to be responsible for the high char yield when burned and quantum chemical calculations33 have supported the postulated mechanism.
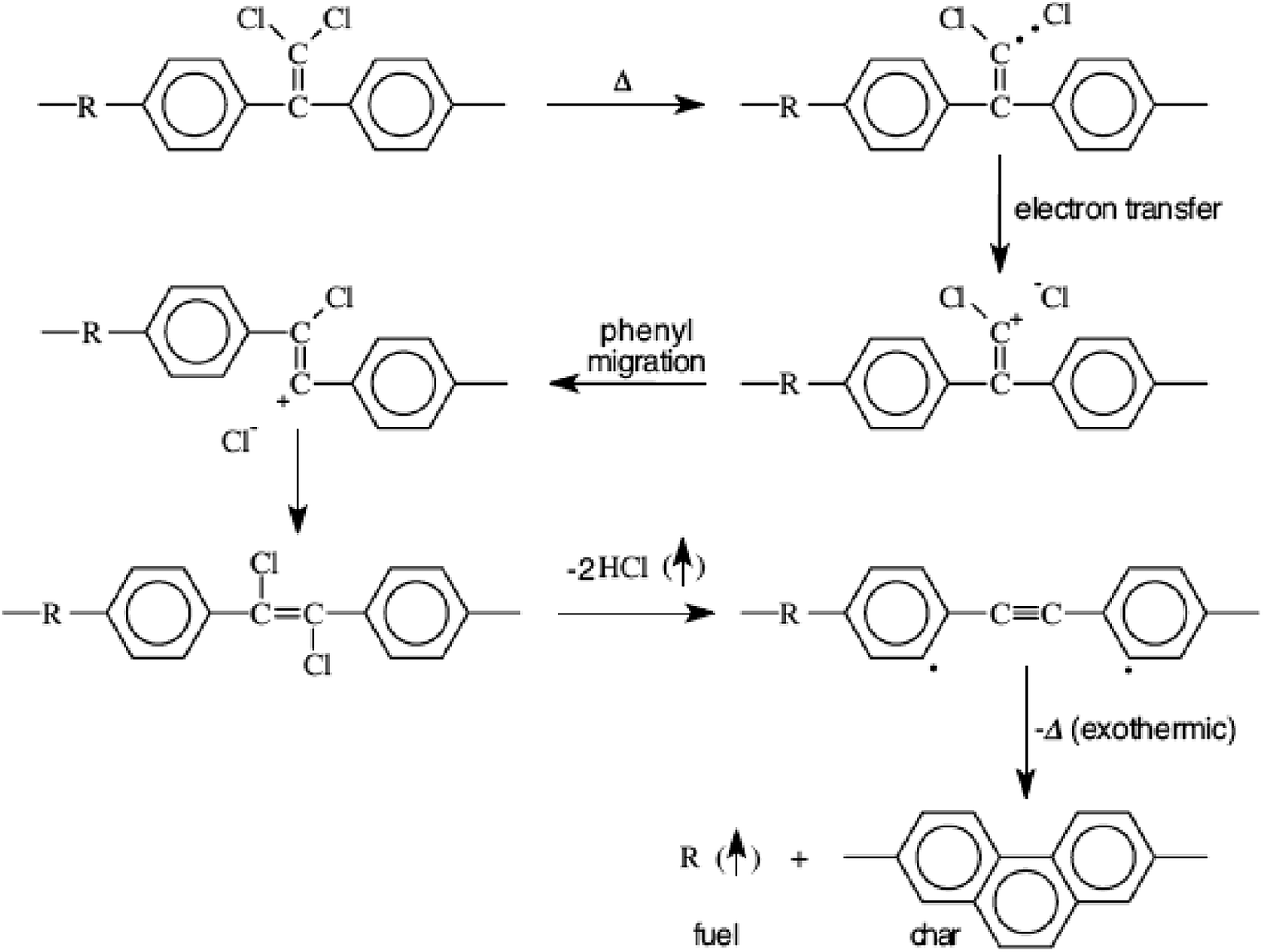 | ||
| Fig. 6 Schematic showing the postulated degradation mechanism of chloral based polymers.31,34 | ||
Conclusions
These exciting thermal and mechanical data demonstrate the potential of this family of fire resistant polymers to yield strong, resilient chars in the event of combustion offering the possibility of greater fire safety. After exposure to extreme temperatures (900 °C) the cured polymers both undergo charring, but display quite different structural behaviour. The polycyanurate of bisphenol A forms a porous char, with moderate structural integrity, but with evidence of severe blistering. The polycyanurate of bisphenol C yields a quite different result and is especially sensitive to the effects of additives. In the absence of catalysis the polycyanurate of bisphenol C forms a highly porous, but greatly degraded char. When a catalyst is employed, the charring of the same polymer yields a dense, mechanically strong, yet highly resilient structure. Work is currently underway to improve the processing characteristics of bisphenol C and its derivatives of through formulation and to introduce them into more representative structures. In due course, we will report whether these characteristics translate successfully into these advanced structural composites.Author contributions
IH and BJH proposed and supervised the project. IH, BJH, SAH, and MS designed the experiments. LM and ST carried out the experiments. IH, BJH, LM analysed data, IH, BJH, and CS wrote the manuscript. PZ advised on the nanoindentation measurements and SP advised on the X-ray CT measurements. All the authors participated in discussions of the research.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The Defence Science and Technology Laboratory (Dstl) generously funded this work through two research contracts to support LM (RD1109) and ST (RD1103A). The authors wish to thank Messrs Chris Burt and Peter Haynes, and Dr Jose Anguita (Advanced Technology Institute, Faculty of Engineering and Physical Sciences, University of Surrey) for assistance given during the course of this work.References
- R. E. Lyon, Fire Resistant Materials: Research Overview DOT/FAA/AR-97/99, FAA, 1997 Search PubMed.
- Special Study: U.S. Air Carrier Accidents Involving Fire, 1965-1974 and Factors Affecting the Statistics, Report NTSB-AAS-77-1, National Transportation Safety Board, February 17, 1977 Search PubMed.
- S.-Y. Lu and I. Hamerton, Recent developments in the chemistry of halogen-free flame retardant polymers, Prog. Polym. Sci., 2002, 27, 1661–1712 CrossRef CAS.
- G. J. Van Esch, Environmental health criteria 218—flame retardants: tris(2-butoxyethyl)phosphate, tris(2-ethylhexyl)-phosphate and tetrakis-(hydroxymethyl)phosphonium salts, WHO, Geneva, 2000 Search PubMed.
- U.S. Environmental Protection Agency, EPA/600, US GPO, Washington, 1994, vol. 1–3 Search PubMed.
- M. S. M. Alger, High-temperature and fire resistant polymers, in Specialty Polymers, ed. R.W. Dyson, Blackie Academic and Professional, Glasgow, ch. 3, pp. 189–227, (ISBN 978-0-85709-086-7) Search PubMed.
- I. Hamerton and L. Mooring, The use of thermosets in aerospace applications, in Thermosets Structure, Properties and Applications, ed. Qipeng Guo, Woodhead Publishing, Cambridge, 2012, ch. 7, pp. 189–227, (ISBN 978-0-85709-086-7) Search PubMed.
- Chemistry and Technology of Cyanate Ester Resins, ed. I. Hamerton, Blackie A&P, Glasgow, 1994 Search PubMed.
- I. Hamerton, Epoxy Resins, in The Encyclopedia of Chemical Processing, ed. S. Lee, Marcel Dekker, New York, 2005, pp. 911–928 Search PubMed.
- R. Iredale, C. Ward and I. Hamerton, Modern advances in bismaleimide resin technology: a 21st century perspective on the chemistry of addition polyimides, Prog. Polym. Sci., 2016, 69, 1–21 CrossRef.
- Q. S. Tao, W. J. Gan, Y. F. Yue, M. H. Wang, X. L. Tang and S. J. Li, Viscoelastic effects on the phase separation in thermoplastics modified cyanate ester resin, Polymer, 2004, 45, 3505–3510 CrossRef CAS.
- J. Suman, J. Kathi and S. Tammishetti, Thermoplastic modification of monomeric and partially polymerized Bisphenol A dicyanate ester, Eur. Polym. J., 2005, 41, 2963–2972 CrossRef CAS.
- C. P. R. Nair, D. Mathew and K. Ninan, Cyanate Ester Resins, Recent Developments, New Polymerization Techniques and Synthetic Methodologies, Springer, Berlin, 2001, pp. 1–99 Search PubMed.
- S. K. Karad, D. Attwood and F. R. Jones, Moisture absorption by cyanate ester modified epoxy resin matrices. Part III. Effect of blend composition, Composites, Part A, 2002, 33, 1665–1675 CrossRef.
- D. Leach and J. Boyd, Proc. 46th Int. SAMPE Symp. Exhib., SAMPE, Long Beach, 2001, pp. 1506–1514 Search PubMed.
- C. P. R. Nair, F. T. Vijayan and K. Krishnan, Sequential interpenetrating polymer networks from bisphenol A based cyanate ester and bimaleimide: Properties of the neat resin and composites, J. Appl. Polym. Sci., 1999, 74, 2737–2746 CrossRef CAS.
- E. Grigat and R. Pütter, Synthesis and reactions of cyanic esters, Angew. Chem., Int. Ed., 1967, 6(3), 206 CrossRef CAS.
- Dstl Test Method DSTL/DOC38187/1.0, 16th September 2009 Search PubMed.
- Indentation Software User's Manual, Version R0.1.0, December 2008, CSM Instruments, Rue de la Gare 4, CH-2034 Peseux, Switzerland, http://www.csm-instruments.com Search PubMed.
- J. Woirgard, J.-C. Dargenton and T. V. Audurier, A new technology for nanohardness measurements: principle and applications, Surf. Coat. Technol., 1998, 100–101, 103–109 CrossRef CAS.
- J. Y. Rho, P. Zioupos, J. D. Currey and G. M. Pharr, Microstructural elasticity and regional heterogeneity in human femoral bone of various ages examined by nano-indentation, J. Biomech., 2002, 35, 189–198 CrossRef CAS PubMed.
- GAB Neumann GmbH, Graphite Heat Exchangers and Equipment, https://www.slideshare.net/LoicBERNARD/gab-neumann-graphite-presentation, accessed 31 August 2018.
- S. A. Epshtein, F. M. Borodich and S. J. Bull, Evaluation of elastic modulus and hardness of highly inhomogeneous materials by indentation, Appl. Phys. A, 2015, 119, 325–335 CrossRef CAS.
- Physical properties of graphite,https://www.mindat.org/min-1740.html, accessed 31 August 2018.
- P. Zioupos and K. D. Rogers, Complementary physical and mechanical techniques to characterise tooth: A bone-like tissue, J. Bionic. Eng., 2006, 3, 19–31 CrossRef.
- S. Srinivasagopalan and D. B. Fischbach, 26th Pacific Coast Meeting, Amer. Ceram. Soc., San Francisco, October 1973, (Abstract: Ceram. Bull., 1973, 52, 9, 713) Search PubMed.
- J. C. Bokros, Chemistry and Physics of Carbon, 1996, ed. P. L. Walker Jr., Marcel Dekker, New York, vol. 5, pp. 1–108 Search PubMed.
- H. M. Hawthorne, The microindentation hardness behaviour of carbon filaments, glassy carbons, and pyrolytic graphites, Carbon, 1975, 13, 215–223 CrossRef CAS.
- M. L. Ramirez, R. Walters, R. E. Lyon and E. P. Savitski, Thermal decomposition of cyanate ester resins, Polym. Degrad. Stab., 2002, 78, 73–82 CrossRef CAS.
- D. W. Van Krevelen, Some basic aspects of flame resistance of polymeric materials, Polymer, 1975, 16, 615 CrossRef CAS.
- M. L. Ramirez, Thermal decomposition mechanism of 2,2-bis-(4-hydroxy-phenyl)-1,1-dichloroethylene based polymers DOT/FAA/AR-00/42, FAA, 2001 Search PubMed.
- J. R. Stewart, Synthesis and characterisation of chlorinated bisphenol A-based polymers and polycarbodiimides as inherently fire-safe polymers, DOT/FAA/AR-00/39, FAA, 2000 Search PubMed.
- S. I. Stoliarov and P. R. Westmoreland, Mechanism of the thermal decomposition of bisphenol C polycarbonate: nature of its fire resistance, Polymer, 2003, 44, 5469–5475 CrossRef CAS.
- M. L. Ramirez, Thermal Degradation Mechanism of Chloral-based Polymers, MSc thesis, University of Puerto Rico, Mayaguez, January 2000.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07449f |
| ‡ Current address: Basildon Chemical, Kimber Road, Abingdon, Oxfordshire, U.K. |
| § Current address: Hexcel Composites, Duxford, Cambridge, CB22 4QD, U.K. |
| This journal is © The Royal Society of Chemistry 2018 |