
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of microporous layer – free hierarchical gas diffusion electrode as a low Pt-loading PEMFC cathode by direct growth of helical carbon nanofibers†
Robin Sandström a,
Joakim Eksponga,
Alagappan Annamalaia,
Tiva Sharifiab,
Alexey Klechikova and
Thomas Wågberg*a
a,
Joakim Eksponga,
Alagappan Annamalaia,
Tiva Sharifiab,
Alexey Klechikova and
Thomas Wågberg*a
aDepartment of Physics, Umeå University, Umeå 90187, Sweden. E-mail: Thomas.wagberg@umu.se
bDepartment of Materials Science and NanoEngineering, Rice University, Houston, TX, USA
First published on 12th December 2018
Abstract
Improving interfacial contact between each component in the proton exchange membrane fuel cell (PEMFC) can lead to a significant increase in power density and Pt utilization. In this work, the junction between the catalyst layer and gas diffusion layer (GDL) is greatly enhanced through direct attachment of helical carbon nanofibers, giving rise to a hierarchical structure within the electrical interconnections. The alternative novel GDL is produced by spraying a thin layer of Pd2C60 precursor on commercial carbon paper, followed by chemical vapor deposition growth resulting in a surface morphology of well-attached nanofibers surrounding the microfibers present in the commercial carbon paper. Subsequent solvothermal deposition of platinum nanoparticles allowed evaluation of its suitability as gas diffusion electrode in cathodic H2/O2 PEMFC environment. A combination of lowered charge transfer resistance and enhanced Pt-utilization is attributed to its unique wire-like appearance and its robust properties. The fabricated microporous layer – free GDL is suitable for relatively aggressive membrane electrode assembly fabrication procedures and is produced by industrially favorable techniques, rendering it capable of efficiently supporting small amounts of precious metal catalyst nanoparticles in various PEM applications.
1. Introduction
Development of sustainable energy sources is rapidly turning into an increasingly urgent task due to dire environmental problems, hence advances in hydrogen technologies are receiving substantial attention. Proton exchange membrane fuel cells (PEMFCs) offer an efficient and convenient solution for converting hydrogen into electricity at low operating temperature, and are considered as the primary candidate for replacing internal combustion engines in vehicles with a wide range of weight classes.1 In addition, PEM technology shows promise in a broader set of applications ranging from electrolysis2,3 to portable4–6 and stationary7 power. The high relative cost of precious metals does however still plague commercial viability in several such industries, motivating innovative solutions in enhanced catalyst utilization.All PEMFCs, held up by their carefully manufactured membrane electrode assemblies (MEA), comprise a set of key components including a (i) proton conducting solid membrane sandwiched between (ii) gas diffusion layers (GDL) coated with thin (iii) catalyst layers (CL). When the reactants are introduced to the cell, their diffusion into the CL has to be considered. In particular the I–V polarization profile often shows limited performance at higher current densities where the transport of reactants cannot keep up with the overall reaction rate.8 The primary tasks of the GDLs as a fuel cell component are thus, together with the fuel flow fields, to distribute the reactants in an efficient and homogeneous manner and provide an electrical connection to the current collector from the catalytically active sites. In addition, this component offers some structural integrity to the cell, helps to protect the catalyst layer from violent fuel flows and concurrently provides an optimized water management by tuning its hydrophobic attributes.9,10 Carbon papers (CP) or woven carbon cloths, with or without a microporous teflonized layer (MPL), are proven to be particularly suitable as GDLs. The CL, consisting of precious noble metal based catalyst particles decorated on some high surface area carbon support, is traditionally applied via an ink directly onto either the membrane or GDL. However, as this process results in a microporous blend of Nafion® polymer and catalyst, the electrical contact between GDL and active sites is not optimal.11,12 Moreover, the use of MPLs in H2/O2 PEMFCs may further contribute to increased cell resistance.
In order to decrease contact resistance contributing to ohmic potential drops, innovative procedures employing growth of alternative catalyst support directly on the GDL, has been scarcely proposed in literature. The resulting hierarchical 3D structures assure electrical contact of attached nanoparticles with the GDL, ultimately fulfilling one of the requirements of the three phase boundary and thereby enhancing catalyst utilization. Moreover, depending on the hygroscopic properties of the resulting 3D layer, the as-grown nanostructures has been proposed as a hydrophobic micro-porous layer used for water management, hence excluding the need of traditionally used insulating PTFE binder.13 Catalyst support types such as carbon nanotubes (CNTs)14 as well as various carbon nanofibers (CNFs)15–18 have shown promising performances in similar hierarchical configurations. Previous work has further demonstrated the possibility to grow modified CNTs directly on the relatively large microfibers contained within the carbon paper GDL, for alkaline water electrolysis and Li-ion battery purposes,19–21 showing that this 3D scheme is not restricted to solid polymer MEAs. However, concerns including low yield of support and difficulties in decoration of precious metal catalyst nanoparticles are yet to be fully addressed for PEMFC applications. Although successful decoration attempts have been performed based on methods such as electrodeposition, sputter-deposition and thermal evaporation,11,15 methods suitable for large scale production are also necessary to fully employ hierarchical supports in PEMFCs.
Carbon nanostructures such as CNFs and CNTs have shown to be interesting catalyst support candidates as they show superior conductive properties, high specific surface area and excellent durability.22–25 Compared to commercially available Vulcan XC-72 based catalysts, these alternative wire-like supports have also shown improved diffusion capabilities.26–28 High surface area helical carbon nanofibers (HCNFs), previously manufactured and extensively studied in literature,29–32 have already shown beneficial fuel cell performances as direct formic acid fuel cell anodes.33
Herein, we demonstrate a complete and novel protocol for the fabrication of gas diffusion electrodes (GDE) developed for production of HCNFs attached directly to carbon papers in a hierarchical manner. The method is invented with industrial large-scale production in mind and the final product offers support anchored directly to the surface of the diffusion layer with a corrosion resistant noble metal catalyst foundation (Pd). Platinum nanoparticles (NP) could be subsequently anchored, without functionalization, with a simple solvothermal method owing to the fibrous surface of the support containing plenty of attachment sites. Hydrogen fuel cell polarization curves show comparable cathodic performance as a MEA prepared with a conventional catalyst layer. Improvements in power output at elevated current densities is attributed to firm attachment of the support to the GDL leading to minimal diffusion of catalyst into the interior of the carbon paper during both hot-pressing procedures and natural cell compression when mounted in a cell fixture. Moreover, lowered charge transfer resistance and increased Pt-utilization are evidenced to be likely contributors to the desired fuel cell enhancements.
2. Experimental methods
2.1. Synthesis procedure
The full fabrication procedure for fabricating the platinum decorated hierarchical HCNF gas diffusion electrode (denoted Pt–HCNFs/CP), followed by MEA fabrication, is shown schematically in Fig. 1. Specifically, individual 15 cm2 pieces of carbon paper (Sigracet®, type 34 AA) were decorated with a thin layer of Pd2C60 by first spraying with a handheld airbrush (Iwata® Eclipse), under 3 bar N2 atmosphere, a mixture of 20 mg tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3, Aldrich) and 16 mg C60 (MER corp.) dispersed in 5 ml toluene by 10 min sonication. Care was taken to keep a spraying distance of ca. 10 cm while painting in a serpentine motion with a sufficient speed as to avoid too wet droplets upon impact producing unwanted “craters”. The painted carbon paper was then dried on a hotplate (60 °C) for 1 h followed by annealing at 200 °C in air for 2 h to remove the dba ligand. The total Pd loading was estimated, by measuring the weight of a large area CP before and after spray coating, to ∼0.02 mg (Pd) cm−2. Growth of HCNFs was performed by chemical vapour deposition (CVD) similar to earlier described procedure.30 First, the growth chamber containing the prepared substrate was purged by an Ar flow (80 ml min−1) for 20 min followed by a heat ramp to 550 °C over the course of 40 min. After an additional 20 min under Ar, the substrate was pretreated with ammonia (25 ml min−1) and 5% H2 in Ar (80 ml min−1) for 20 min. Finally, acetylene was introduced (20 ml min−1) for the growth of HCNFs during 1 h before cooling the oven in an Ar flow. The produced hierarchical GDL signified by a pitch black thin layer on one side of the paper is henceforth denoted HCNFs/CP.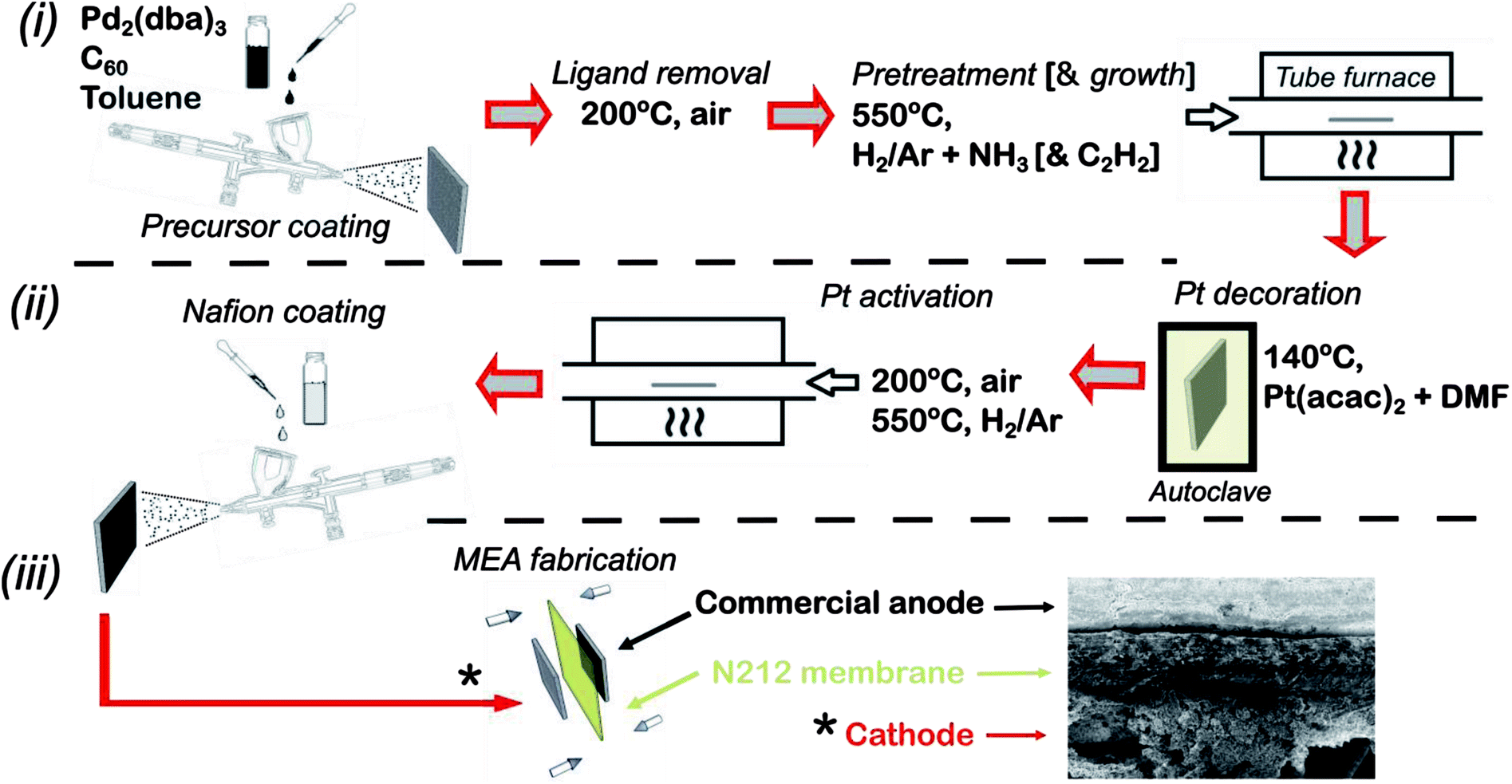 | ||
| Fig. 1 Schematic illustration of full fabrication procedure including MEA fabrication. The following are the key stages: deposition of Pd2C60 followed by growth of HCNFs (i), decoration of Pt NPs with a Nafion layer forming the catalyst layer (ii) and finally MEA fabrication through hot-pressing (iii). | ||
Decoration of Pt was performed in a Teflon lined stainless steel autoclave completely filled (to the top) with a mixture of anhydrous dimethylformamide (DMF) and Pt(acac)2 (2 mg ml−1). Two equally sized HCNFs/CP were then placed back-to-back and submerged in the yellow solution. The sealed autoclave was heated to 140 °C for 24 h after which the sample was thoroughly washed with distilled water (H2O(DI)) and ethanol respectively and subsequently dried on a 60 °C hotplate. After a brief annealing step in air for 10 min (200 °C), the sample was finally heated from room temperature to 550 °C in Ar atmosphere over 40 min and maintained for 15 min under 5% H2 in Ar. This resulted in Pt–HCNFs/CP with a Pt loading of 0.05 mg cm−2.
2.2. Physical characterization
Thermogravimetric analysis (TGA) was performed with a TGA/DSC 1 under an airflow of 30 ml min−1 with a heating rate of 5 K min−1 in the temperature range of 30 °C to 950 °C. Blank background curves were recorded, prior to each measurement, with the same crucibles as the one used for the sample for additional accuracy. X-ray diffraction (XRD) patterns were recorded by a Panalytical X'Pert3 Powder diffractometer with a CuKα (λ = 1.5406 Å) source with a 2θ scan range of 20–75°. A Renishaw InVia Raman spectrometer with a laser excitation wavelength of 514 nm was used for Raman analysis.Scanning electron microscopy (SEM) imaging was carried out with a Zeiss Merlin field emission SEM operating with an accelerating voltage of 4 keV and a probe current of 120 pA with images recorded by an in-lens positioned detector. High resolution and low resolution transmission electron microscopy (TEM/HRTEM) analysis took place in a JEOL 2100F microscope with an accelerating voltage of 200 keV. TEM grid preparation was performed by scratching off a small amount of material from the CP and dispersing it in ethanol by sonication prior to dropcasting a holey carbon Cu grid.
For the purpose of surface area estimation through the Brunauer–Emmett–Teller (BET) method, HCNFs/CP samples of three 15 cm2 pieces were fabricated with HCNFs coated on both sides. A reference with uncoated CPs with identical geometric areas, taken through the same treatment steps with excluded catalysts, was used in order to estimate HCNF loading. The measurements took place in a Quantachrome Nova 1200e at liquid nitrogen temperature where nitrogen adsorption isotherms were acquired using a Micromeritics ASAP 2460 analyzer supplied with a turbopump. Prior to measurements, samples were degassed under 150 °C vacuum conditions.
2.3. Electrochemical characterization and MEA fabrication
Ex situ electrochemical impedance spectroscopy (EIS) and electrochemical active surface area measurements (ECSA) were performed on custom made circular-cut 0.2 cm2 pieces of Pt–HCNFs/CP and Pt–Vulcan/C (20 wt%, Premetek) with an equivalent Pt loading as reference. The GDL electrodes were attached with tin-coated copper wires and glued with epoxy onto a glass slide. ECSA measurements took place by recording hydrogen under potential deposition curves (HUPD). Due to the sensitivity toward dissolution and corrosion of various materials within the fabricated electrode, a 0.5 M phosphate buffer solution (PBS) with a pH of 6.8 (−0.62 to 0.4 V vs. Ag/AgCl, 3 M KCl) was used as electrolyte and recorded with a potentiostat (Autolab PGSTAT302N). EIS spectra were measured in a 5 mM potassium hexacyanoferrate solution, with an Ag/AgCl reference (3 M). The EIS frequency was scanned with a BioLogic potentiostat with integrated FRA module and a range of 10 kHz to 0.1 Hz at an applied potential of +200 mV (Fe2+/Fe3+ redox couple) with a relatively high 100 mV AC amplitude due to issues regarding high measurement noise at lower currents.Fabrication of 5 cm2 membrane electrode assemblies (MEAs) were made by combining the fabricated electrode with a commercial anode produced by commercially available Pt – Vulcan (Premetek, 20 wt%) decorated with a loading of 0.1 mg (Pt) cm−2 on a type 10BC GDL (Sigracet®). Catalyst decoration took place by spraying a dispersion containing 4 mg ml−1 catalyst and 33 wt% Nafion (DuPont, Ion-Power 5 wt%) in 90% 2-propanol. Cathodes were produced by first applying a layer of Nafion on the surface (2 mg cm−2) through spray coating a mixture of Nafion, distilled water and 2-propanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 volumetric ratio) followed by drying at 60 °C overnight (hotplate). Membranes (Type N212) were first pretreated by heating for 1 h at 80 °C consequently in 3% H2O2, H2ODI, 0.5 M H2SO4 and H2ODI respectively and finally stored in H2ODI until usage. MEAs were finally produced by hot-pressing with a pretreated N212 membrane for 2 min at 130 °C at 60 kg cm−2 pressure with a Stahls Hotronix® 6” × 6” heat press. The MEAs were then loaded into a cell fixture (quickCONNECT, Baltic FuelCells GmbH) with serpentine flow fields connected to a Scribner 850e fuel cell test system. The fuel cell was operated at 60 °C and 95% RH on both anode and cathode with gas flows of 100 ml min−1. Break-in procedures took place by operating the cell at constant potential of 0.55 V until reaching stable performance typically within 12 h. Polarization curves were recorded at 0.1 A step size acquired after 1 min per step. In situ ECSA was measured with an integrated potentiostat (Scribner 885 Fuel Cell potentiostat), by first saturating the cathode in N2 atmosphere followed by potential sweeps between 0.05 to 0.8 V vs. counter/reference electrode (CE/RE). The scan rate was held to 40–50 mV s−1 while maintaining an H2 flow of 100 ml min−1 and repeated until reaching stable HUPD area.
:1:1 volumetric ratio) followed by drying at 60 °C overnight (hotplate). Membranes (Type N212) were first pretreated by heating for 1 h at 80 °C consequently in 3% H2O2, H2ODI, 0.5 M H2SO4 and H2ODI respectively and finally stored in H2ODI until usage. MEAs were finally produced by hot-pressing with a pretreated N212 membrane for 2 min at 130 °C at 60 kg cm−2 pressure with a Stahls Hotronix® 6” × 6” heat press. The MEAs were then loaded into a cell fixture (quickCONNECT, Baltic FuelCells GmbH) with serpentine flow fields connected to a Scribner 850e fuel cell test system. The fuel cell was operated at 60 °C and 95% RH on both anode and cathode with gas flows of 100 ml min−1. Break-in procedures took place by operating the cell at constant potential of 0.55 V until reaching stable performance typically within 12 h. Polarization curves were recorded at 0.1 A step size acquired after 1 min per step. In situ ECSA was measured with an integrated potentiostat (Scribner 885 Fuel Cell potentiostat), by first saturating the cathode in N2 atmosphere followed by potential sweeps between 0.05 to 0.8 V vs. counter/reference electrode (CE/RE). The scan rate was held to 40–50 mV s−1 while maintaining an H2 flow of 100 ml min−1 and repeated until reaching stable HUPD area.
3. Results
3.1. Synthesis and morphological characteristics
Helical carbon nanofibers supported by conventional GDL substrates were attained according to the top row in the scheme (Fig. 1(i)). Contrary to numerous previously proposed routes, the catalyst meant for CNF growth was applied by spray coating a thin layer of the precursor compound dispersed in a spray-friendly volatile solvent (toluene). The combination of Pd2(dba)3 and C60 lead to a Pd2C60 phase (see Fig. S1†), which upon collapse in reductive 550 °C atmosphere results in highly active Pd particles capable of high yield CNF growth.30 As shown in Fig. 2a and b, the treatment of a commercial carbon paper by the fine airbrush mist of such a mixture yields CP-microfibers completely encapsulated in Pd2C60 polymer. Once collapsed, attached Pd NPs could be observed on both the graphitic binder and fibrous regions of the CP (Fig. 2c and d), yielding a homogeneously distributed, relatively thick and well-anchored HCNF layer upon growth (Fig. 2e and f). Ideally, the growth of a thicker HCNF layer dedicated for catalyst decoration, without compromising on its desirable firm attachment, could benefit the usage as a PEMFC catalyst layer. Unfortunately as shown in Fig. S2a and b,† increasing the Pd loading (roughly double) did not produce more HCNFs. Instead, large “blisters” and peeled-off layers were formed indicating the existence of a saturation limit for the HCNFs/CP growth; since an increasing growth time also did not result in any notable modifications. As larger HCNF surface coverage is clearly dependent on the CP-microfiber density, an intermediate growth of larger diameter CNFs may be desirable such as the approach proposed by Kundu et al.,34 where, an electrospinning step was performed followed by CNF growth yielding a “tree-like” structure with enhanced coverage. Alternatively, commercially available carbon papers with more dense microfiber morphology may aid the support coverage. | ||
| Fig. 2 SEM micrographs of blank 34 AA type CP (a), Pd2C60 decorated CP (b). Pd NPs on CP obtained directly after pretreatment and hence the collapse of the Pd2C60 in reductive atmosphere (c and d), and the resulting HCNFs/CP obtained after CVD growth (e and f). | ||
As a proof-of-concept, subsequent Pt decoration on the modified GDL was achieved by a simple solvothermal treatment in dimethylformamide (DMF), acting as both solvent and reducing agent on the organometallic Pt(acac)2 precursor,35 followed by additional reduction at 550 °C in 5% H2 in Ar atmosphere. It should be noted that a similar solvothermal procedure could potentially be employed toward synthesis of Pt NPs with controlled shapes and/or alloying with one or several transition metals, as is also feasible in similar reaction conditions.36,37 Here however, the HCNFs provided excellent nucleation sites for the Pt species and did not form a colloidal Pt dispersion in the remaining solution as it maintained its transparent yellow tint. SEM analysis in Fig. 3 shows that the resulting Pt–HCNF layer did not detach even after the harsh treatment caused by the decoration steps, albeit somewhat collapsed into a more “compact” CL. In addition, a striking homogeneity in decoration with no large visible Pt clusters is seen. This is impressing considering that the reaction takes place with HCNFs in a non-dispersed state and the decoration is comparable, or better, than that of various solution based decoration processes on HCNFs/CNFs in previous literature.38–41 The Pt(acac)2 concentration was tuned to realize a total Pt loading of about 0.05 mg cm−2 (Fig. 4a). Note that the relative Pd loading (originating from the catalysts for HCNF growth) of about 0.01 mg cm−2 is substantially lower and can be considered a negligible areal cost in most MEAs.
 | ||
| Fig. 3 SEM of the Pt–HCNFs/CP sample. Overview of Pt–HCNFs wrapped around the CP microfibers (a and b) and close-up imaging the Pt decoration on a representative region of the nanofibers (c). | ||
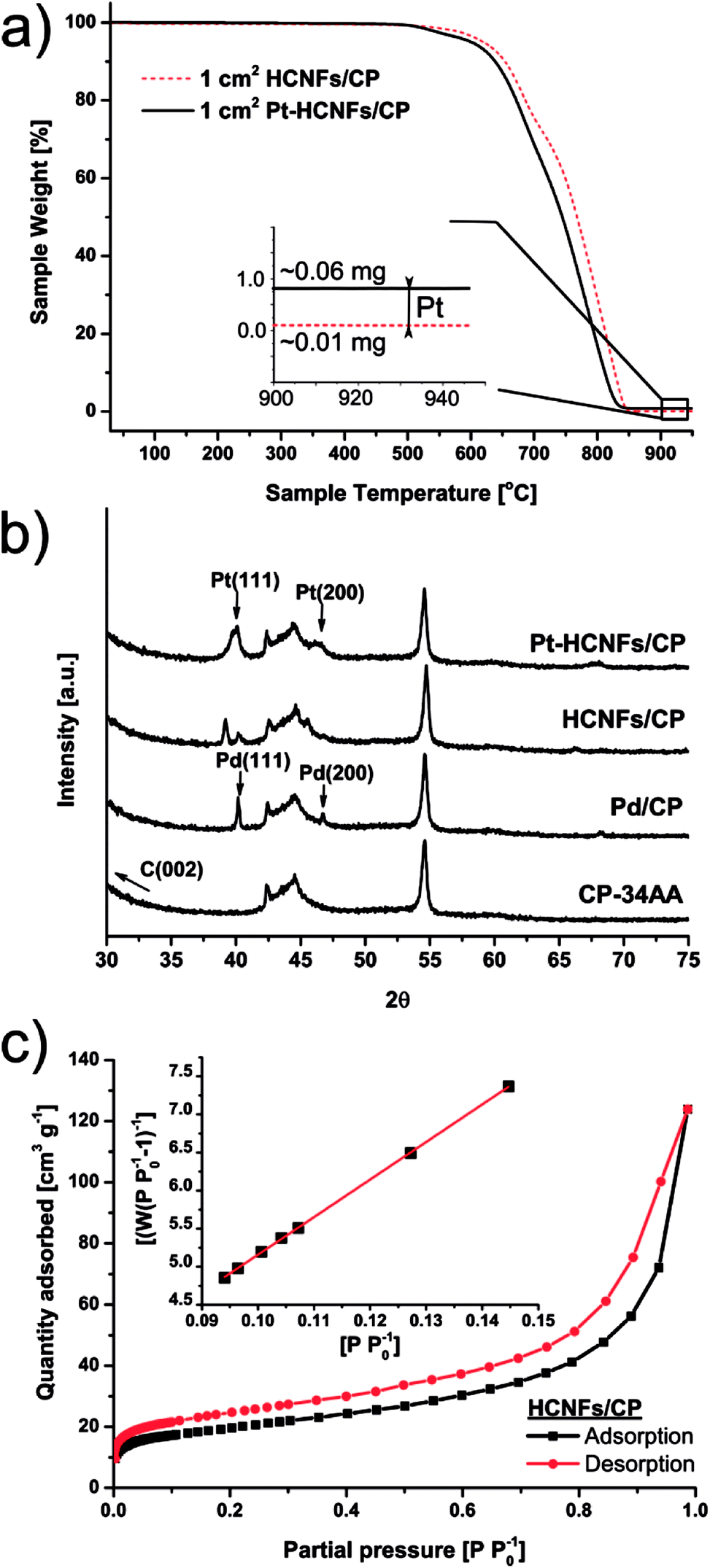 | ||
| Fig. 4 Thermogravimetric analysis of 1 cm2 area of samples before and after Pt decoration (a) recorded under airflow of 30 ml min−1 and a heating rate of 5 K min−1. X-ray diffraction patterns of various stages during the fabrication process (b) and BET of HCNFs/CP (c) where the inset displays the nitrogen adsorption plot. | ||
The emerging crystallites in each stage of the fabrication process were traced by X-ray diffraction as shown in Fig. 4b. Here, two sharp Pd fcc reflections above the noise level, revealing successful crystalline Pd NP formation, are clearly indexed on the pretreated Pd2C60 coated carbon paper by their characteristic Pd(111) (40.1°) and Pd(200) (46.7°) reflections. A large fraction of the intensities from both peaks are followed by a downshift to 39.1° and 45.6° respectively upon HCNF growth; likely signifying a lattice expansion caused by carbon diffusion into the Pd(0) structure leading to the formation of a Pd–C phase.42 Additional crystalline Pt reflections can also be identified in the final Pt–HCNFs/CP at about 40° and 47° for Pt(111) and Pt(200) respectively. As there are severe peak overlaps as well as larger Pt particles attached in the interior microporous regions (see backside of CP in Fig. S2d†), size estimation through the Scherrer approximation would unfortunately provide misleading average sizes.
During gaseous PEMFC operation, catalyst NPs further away from the membrane will likely never partake in the reaction due to the lower possibility of a connecting electrolyte bridge to the membrane. Hence the interior and backside unfortunately contain unemployed precious metal attached on the CP microfibers. We believe however that the significantly higher surface area of HCNFs compared to a blank carbon paper (below detection limit), as measured by BET shown in Fig. 4c, encourage the vast majority of the Pt mass to be deposited on the catalyst layer in close proximity to the membrane. By summarizing the BET analysis, the 70 m2 g−1 HCNF yield a total measured HCNFs/CP surface area of about 700 cm2 cmcp−2 with roughly 1 mg cmcp−2 of support, as estimated from the differential weight of equally sized blank CPs subjected to the same annealing atmospheres. Hence, the Pt-to-HCNF loading can, with caution, be assumed to average around 5 wt%. TEM evaluation, presented in Fig. 5a–c, provides further insight into the Pt characteristics. The particles show crystalline Pt lattice fringes, as shown by the highlighted Pt(111) face (also see SAED pattern in Fig. S3†). The exclusively spherically shaped NPs demonstrate a narrow size distribution of 4.9+/−1.0 nm.
 | ||
| Fig. 5 TEM (a) and HRTEM (b) of Pt NPs on a HCNF section from the Pt–HCNFs/CP sample where the Pt (111) plane is highlighted. The histogram (c) was produced from a large number of TEM images similar to the one presented in (a). | ||
3.2. Ex situ electrochemistry and MEA evaluation
The intended application of the Pt–HCNFs/CP is as a gas diffusion electrode working in H2/O2 PEMFC conditions. For this reason a comparison with a conventional catalyst layer, on the same Sigracet® 34 AA type carbon paper substrate was performed. Thus, the reference CP was coated with a thin layer of 20 wt% Pt on Vulcan XC-72 (0.05 mg cm−2) containing 33 wt% Nafion (see overview SEM in Fig. S4†), as is a common blend for PEMFC cathodes.43 As illustrated in the Nyquist plot in Fig. 6a and its corresponding equivalent circuit model (see fitting details in Table S1† and corresponding Bode phase plot in Fig. S5.†), the high frequency resistance region shows an upshifted value for the commercial CL. In addition to solution resistance (RS), an increment of Fe2+/Fe3+ ionic transport resistance in this region is likely attributed to the electrolyte resistance (Rele) of the Nafion binder (in a non-H+ form) within the CL,44–47 which is absent in Pt–HCNFs/CP. More interestingly, the emerging low frequency response of Pt–HCNFs/CP displays a 38% reduction in charge transfer resistance (RCT) as compared to Pt–Vulcan/CP. The cause of this reduction can possibly be attributed to the wire-like support characteristics providing conductive pathways resulting in efficient charge transfer with minimum amounts of carbon–carbon junctions within the CL, contrary to its competitor. Here, a possible influence on RCT from a component related to diffusivity should not be excluded. However, such a component could not be reliably fitted into the equivalent circuit model, so the full influence of diffusion on the EIS remain unknown.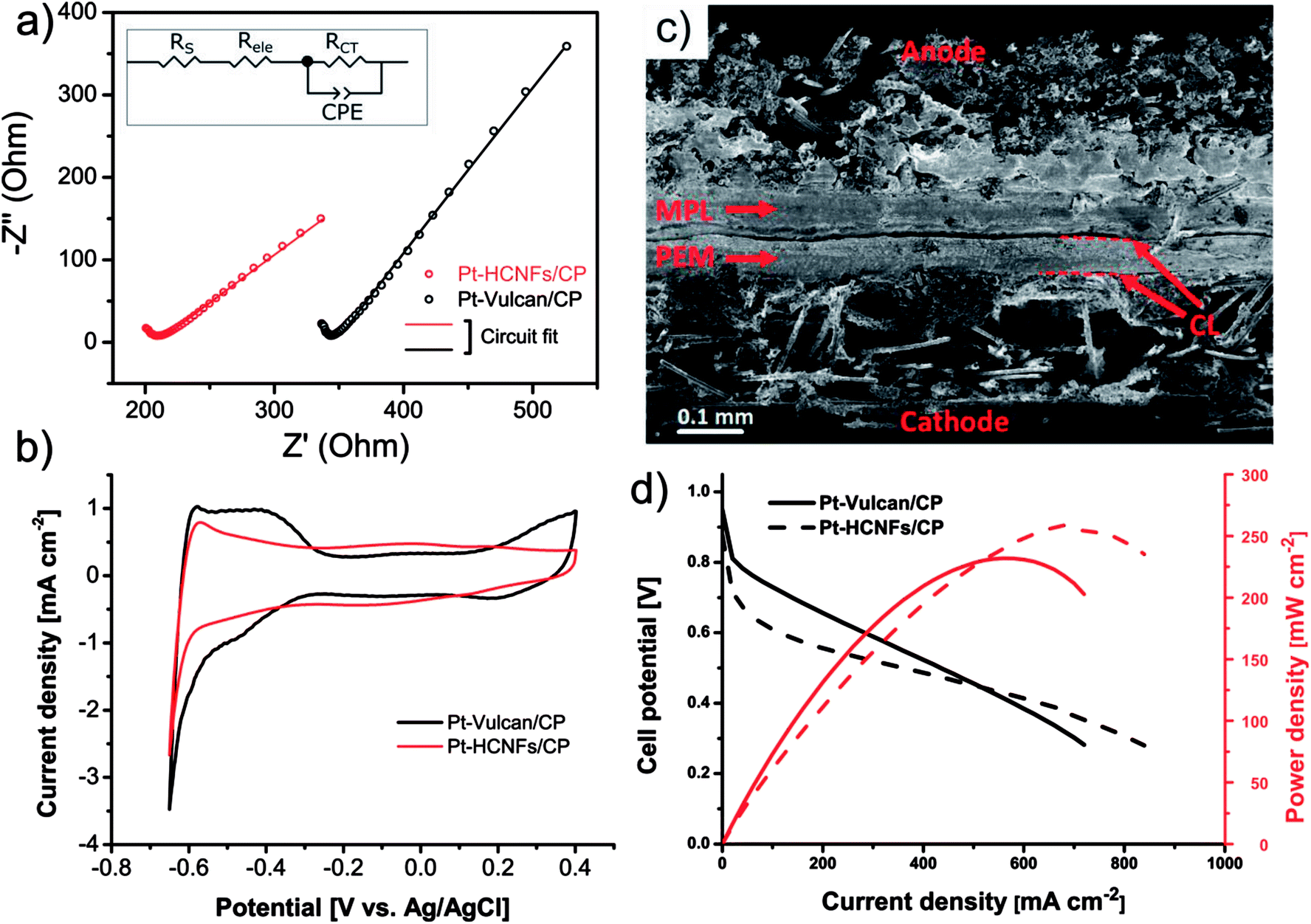 | ||
| Fig. 6 Nyquist plot resulting from electrochemical impedance spectroscopy performed on Pt–HCNFs/CP and the spray coated commercial Pt–Vulcan/CP catalyst layers; both with a CP type 34 AA as substrate (a). Ex situ ECSA as measured by HUPD in 6.8 pH phosphate buffer solution (b). Cross-section SEM of fabricated MEA where Pt–HCNFs/CP is used as a cathode and an anode consisting of commercial Pt–Vulcan (20 wt%) coated on type 10BC GDL (c). Corresponding H2/O2 fuel cell polarization curves recorded in 95% RH and 60 °C (d). | ||
Electrochemical active surface area measured (ex situ) by hydrogen underpotential deposition in PBS solution, completely flooding the porous structure of the GDL with electrolyte, is shown in Fig. 6b. Considering the significantly smaller NP size of the commercial Pt–Vulcan (XRD; ∼2 nm), Pt–HCNFs/CP of the same Pt loading is unsurprisingly showing lower ECSA (37.9 vs. 11.1 m2 g−1 respectively). In addition, the existence of NPs with thin coverage of amorphous carbon layers (visible in Fig. 5b) is a likely contributor to lowered ECSA. It must however be emphasized that the measurement took place directly on the GDE, containing a relatively thick CL attached to a carbon paper substrate. Likely, such non-ideal conditions may lead to a potentially underestimated intrinsic ECSA.48 However, since the catalyst layer of Pt–HCNFs/CP cannot be reliably extracted into a dispersible powder, an alternative such as the aforementioned method must be employed. Fig. S6† show that the ECSA of the commercial Pt–Vulcan powder measured on a conventional thin-film glassy carbon electrode in Ar saturated 0.1 M HClO4 electrolyte, suggest an underestimation of about 44% for this method.
In addition to intimate CP|CL interfacial contact, an enhanced CL|PEM interface is a highly desirable trait that is also greatly benefiting from optimized MEA fabrication and novel innovations.49 Here, the fabrication of MEAs by deposition of a thin Nafion film followed by hot-pressing, resulted in firmly attached GDEs to the N212 membrane as illustrated in the cross-section SEM image in Fig. 6c where the dashed lines highlight the CL|PEM interface. Despite absence of a MPL, no part of the membrane was left visibly untouched by the catalyst whereas the deposited Nafion efficiently behaved as a “glue”, in line with earlier suggested MEA fabrication procedures where such methods resulted in enhanced interfacial contact.50 MEA performance in H2/O2 PEMFC conditions of Pt–HCNFs/CP and Pt–Vulcan/CP cathodes fabricated in the same manner can be seen in Fig. 6d. At higher current densities, the Pt–HCNFs/CP surpass the Pt–Vulcan/CP. At any point on the polarization curve, the potential drop can be considered as a superposition of several contributing factors. The higher power output pronounced at higher current densities can presumably be explained by the enhanced mass transport owing to the HCNFs unique porous micro structure.39 It should be noted that even though fuel cells are normally operated at lower potential than where there is a maximum power output, enhanced mass transport will still benefit the lower current-density regions to some degree. In addition, it is likely that Pt–HCNFs/CP had a lower loss of Pt during hot-pressing and natural cell compression as is expected for GLDs with absent or large-pore MPLs51–53 and lower ohmic drop caused by the superior CP|CL interface containing no parasitic ionomer blocking the e− path. The latter is, in addition to the EIS results, supported by simultaneous i-interrupt measurements recorded during the polarization curves (Fig. S7†). The higher overpotential of Pt–HCNFs/CP shows however that the produced Pt catalyst does not fully match the commercial alternative, revealing that there is still room for improvement in optimization of the ORR activity. However, by comparing the in situ ECSA measured by CV scans under an N2 saturated cathode to that of the ex situ measured ECSA values (see Fig. S8 and Table S1†), Pt surface area utilization factors of approximately 40% and 68% can be estimated for Pt–Vulcan/CP and Pt–HCNFs/CP respectively. While the commercial reference GDE performance is severely held back by the lack of an MPL, the Pt–HCNFs/CP approaches a precious metal utilization factor comparable to reported conventional state-of-the-art MEAs (∼75–97%).43 Here we note however that measured utilization factors in literature can vary greatly depending on MEA preparation procedure. As also shown by the real catalyst utilization factor calculated according to eqn S1,† taking into account total Pt loading, the commercial Pt–Vulcan/CP outperforms Pt–HCNFs/CP with 0.3% compared to 0.1% respectively. Together, the practical measured catalyst utilization factors thus highlight the conceptual benefit of the HCNFs/CP gas diffusion layer rather than intrinsic catalyst performance.
4. Conclusions
In summary, helical carbon nanofibers were grown by chemical vapour deposition directly on a commercial carbon paper diffusion layer, employing a fabrication procedure with a large degree of industrial benignity owing to the catalyst deposition step through a highly scalable spray-technique. The fibers are well anchored/entangled to the substrate as evidenced by Pt-deposition under harsh solvothermal growth conditions without any notable support detachment. A low Pd loading of around 0.01 mg cm−2 allows the modified GDL to be suitable for supporting precious metal catalysts or within similar applications where the Pd cost can be rendered negligible.The as-synthesized Pt catalyst served well as a proof-of-concept by successfully highlighting clear advantages the hierarchy GDL has to offer in H2/O2 PEMFC conditions by demonstrating a comparable maximum power output. The ability of the Pt–HCNFs/CP to compensate for the lower ECSA and higher overpotential can be ascribed to an increase in Pt utilization due to small loss of catalyst to the GDL interior during cell compression as well as a higher chance to achieve a three phase boundary credited to the almost guaranteed wire-like electrical connection to the current collector. Moreover, the lowered charge transfer resistance alleviates the ohmic polarization that is prominent at higher current densities.
The concept of hierarchy GDLs, such as the one reported in this work, could potentially be desirable in additional PEM systems ranging from anodes in fuel cells with liquid fuels to PEM electrolyzer cathodes where high hydrophilicity is desirable. In the case of high humidity H2/O2 PEMFCs, unwanted flooding issues may yet arise caused by its non-optimized hygroscopic properties for such environments. Thus, it is advisable to consider the long term operational stability for each application individually. The GDL nevertheless offer an innovative MPL-free alternative that may prove to be appropriate in certain carefully selected MEAs and operating conditions.
Conflicts of interest
There are no conflicts of interest.Acknowledgements
RS thanks the Kempe foundation for financial support. TW thanks Å gpanneföreningen (15-483), Energimyndigheten and Vetenskapsrådet for financial support. The authors acknowledge the Umeå Core Facility for Electron Microscopy (UCEM), the vibrational spectroscopy core facility (VISP) at Umeå University, the NanoLab facility at Umeå University, and the electron microscopy center (EMC) at the department of materials and environmental chemistry (MMK), Stockholm University.References
- F. T. Wagner, B. Lakshmanan and M. F. Mathias, J. Phys. Chem. Lett., 2010, 1, 2204–2219 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Merge and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- F. Barbir, Sol. Energy, 2005, 78, 661–669 CrossRef CAS.
- X. W. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124–132 CrossRef CAS.
- S. Wasmus and A. Kuver, J. Electroanal. Chem., 1999, 461, 14–31 CrossRef CAS.
- T. Iwasita, Electrochim. Acta, 2002, 47, 3663–3674 CrossRef CAS.
- M. T. Gencoglu and Z. Ural, Int. J. Hydrogen Energy, 2009, 34, 5242–5248 CrossRef CAS.
- E. Antolini, R. R. Passos and E. A. Ticianelli, J. Power Sources, 2002, 109, 477–482 CrossRef CAS.
- E. Sengul, S. Erkan, I. Eroglu and N. Bac, Chem. Eng. Commun., 2009, 196, 161–170 CrossRef CAS.
- A. Jayakumar, S. P. Sethu, M. Ramos, J. Robertson and A. Al-Jumaily, Ionics, 2015, 21, 1–18 CrossRef CAS.
- V. Kamavaram, V. Veedu and A. M. Kannan, J. Power Sources, 2009, 188, 51–56 CrossRef CAS.
- A. M. Kannan, P. Kanagala and V. Veedu, J. Power Sources, 2009, 192, 297–303 CrossRef CAS.
- S. Celebi, T. A. Nijhuis, J. van der Schaaf, F. A. de Bruijn and J. C. Schouten, Carbon, 2011, 49, 501–507 CrossRef CAS.
- C. Y. Du, B. R. Wang and X. Q. Cheng, J. Power Sources, 2009, 187, 505–508 CrossRef CAS.
- T. Bordjiba, M. Mohamedi and L. H. Dao, Nanotechnology, 2007, 18, 035202 CrossRef.
- K. K. Karuppanan, A. V. Raghu, M. K. Panthalingal, S. Ramanathan, T. Kumaresan and B. Pullithadathil, J. Mater. Chem. A, 2018, 6, 12768–12781 RSC.
- E. Salernitano, L. Giorgi and T. D. Makris, Int. J. Hydrogen Energy, 2014, 39, 15005–15016 CrossRef CAS.
- L. Giorgi, E. Salernitano, T. D. Makris, S. Gagliardi, V. Contini and M. De Francesco, Int. J. Hydrogen Energy, 2014, 39, 21601–21612 CrossRef CAS.
- T. Sharifi, E. Gracia-Espino, X. Jia, R. Sandstrom and T. Wagberg, ACS Appl. Mater. Interfaces, 2015, 7, 28148–28155 CrossRef CAS PubMed.
- T. Sharifi, M. Valvo, E. Gracia-Espino, R. Sandstrom, K. Edstrom and T. Wagberg, J. Power Sources, 2015, 279, 581–592 CrossRef CAS.
- J. Ekspong, T. Sharifi, A. Shchukarev, A. Klechikov, T. Wagberg and E. Gracia-Espino, Adv. Funct. Mater., 2016, 26, 6766–6776 CrossRef CAS.
- S. Sharma and B. G. Pollet, J. Power Sources, 2012, 208, 96–119 CrossRef CAS.
- T. Tamaki, H. L. Wang, N. Oka, I. Honma, S. H. Yoon and T. Yamaguchi, Int. J. Hydrogen Energy, 2018, 43, 6406–6412 CrossRef CAS.
- Y. Wang, G. Li, J. H. Jin and S. L. Yang, Int. J. Hydrogen Energy, 2017, 42, 5938–5947 CrossRef CAS.
- A. Bharti and G. Cheruvally, J. Power Sources, 2017, 360, 196–205 CrossRef CAS.
- G. Alvarez, F. Alcaide, P. L. Cabot, M. J. Lazaro, E. Pastor and J. Solla-Gullon, Int. J. Hydrogen Energy, 2012, 37, 393–404 CrossRef CAS.
- S. Chung, D. Shin, M. Choun, J. Kim, S. Yang, M. Choi, J. W. Kim and J. Lee, J. Power Sources, 2018, 399, 350–356 CrossRef CAS.
- X. Wu, Z. Liu, J. H. Zeng, Z. H. Hou, W. Y. Zhou and S. J. Liao, ChemElectroChem, 2017, 4, 109–114 CrossRef CAS.
- X. E. Jia, G. Z. Hu, F. Nitze, H. R. Barzegar, T. Sharifi, C. W. Tai and T. Wagberg, ACS Appl. Mater. Interfaces, 2013, 5, 12017–12022 CrossRef CAS PubMed.
- F. Nitze, E. Abou-Hamad and T. Wagberg, Carbon, 2011, 49, 1101–1107 CrossRef CAS.
- Y. Qin, Z. K. Zhang and Z. L. Cui, Carbon, 2004, 42, 1917–1922 CrossRef CAS.
- X. A. Jian, M. Jiang, Z. W. Zhou, M. L. Yang, J. Lu, S. C. Hu, Y. Wang and D. Hui, Carbon, 2010, 48, 4535–4541 CrossRef CAS.
- F. Nitze, M. Mazurkiewicz, A. Malolepszy, A. Mikolajczuk, P. Kedzierzawski, C. W. Tai, G. Z. Hu, K. J. Kurzydlowski, L. Stobinski, A. Borodzinski and T. Wagberg, Electrochim. Acta, 2012, 63, 323–328 CrossRef CAS.
- S. Kundu, T. C. Nagaiah, X. X. Chen, W. Xia, M. Bron, W. Schuhmann and M. Muhler, Carbon, 2012, 50, 4534–4542 CrossRef CAS.
- M. K. Carpenter, T. E. Moylan, R. S. Kukreja, M. H. Atwan and M. M. Tessema, J. Am. Chem. Soc., 2012, 134, 8535–8542 CrossRef CAS.
- C. Gumeci, A. Marathe, R. L. Behrens, J. Chaudhuri and C. Korzeniewski, J. Phys. Chem. C, 2014, 118, 14433–14440 CrossRef CAS.
- J. P. Lai, W. X. Niu, R. Luque and G. B. Xu, Nano Today, 2015, 10, 240–267 CrossRef CAS.
- G. Z. Hu, F. Nitze, T. Sharifi, H. R. Barzegar and T. Wagberg, J. Mater. Chem., 2012, 22, 8541–8548 RSC.
- F. Nitze, R. Sandstrom, H. R. Barzegar, G. Z. Hu, M. Mazurkiewicz, A. Malolepszy, L. Stobinski and T. Wagberg, J. Mater. Chem. A, 2014, 2, 20973–20979 RSC.
- M. L. Toebes, M. K. van der Lee, L. M. Tang, M. H. H. in 't Veld, J. H. Bitter, A. J. van Dillen and K. P. de Jong, J. Phys. Chem. B, 2004, 108, 11611–11619 CrossRef CAS.
- S. L. Knupp, W. Z. Li, O. Paschos, T. M. Murray, J. Snyder and P. Haldar, Carbon, 2008, 46, 1276–1284 CrossRef CAS.
- H. Gabasch, E. Kleimenov, D. Teschner, S. Zafeiratos, M. Havecker, A. Knop-Gericke, R. Schlogl, D. Zemlyanov, B. Aszalos-Kiss, K. Hayek and B. Klotzer, J. Catal., 2006, 242, 340–348 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- K. Shinozaki, J. W. Zack, S. Pylypenko, B. S. Pivovar and S. S. Kocha, J. Electrochem. Soc., 2015, 162, F1384–F1396 CrossRef CAS.
- K. Hongsirikarn, J. G. Goodwin, S. Greenway and S. Creager, J. Power Sources, 2010, 195, 7213–7220 CrossRef CAS.
- T. Okada, Y. Ayato, M. Yuasa and I. Sekine, J. Phys. Chem. B, 1999, 103, 3315–3322 CrossRef CAS.
- M. J. Kelly, G. Fafilek, J. O. Besenhard, H. Kronberger and G. E. Nauer, J. Power Sources, 2005, 145, 249–252 CrossRef CAS.
- M. H. Shao, Q. W. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS.
- M. Breitwieser, M. Klingele, S. Vierrath, R. Zengerle and S. Thiele, Adv. Energy Mater., 2018, 8, 1701257 CrossRef.
- Z. X. Liang, T. S. Zhao and J. Prabhuram, Electrochim. Acta, 2006, 51, 6412–6418 CrossRef CAS.
- M. Han, J. H. Xu, S. H. Chan and S. P. Jiang, Electrochim. Acta, 2008, 53, 5361–5367 CrossRef CAS.
- C. J. Tseng and S. K. Lo, Energy Convers. Manage., 2010, 51, 677–684 CrossRef CAS.
- J. M. Song, S. Y. Cha and W. M. Lee, J. Power Sources, 2001, 94, 78–84 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07569g |
| This journal is © The Royal Society of Chemistry 2018 |