
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal stability of PMMA–LDH nanocomposites: decoupling the physical barrier, radical trapping, and charring contributions using XAS/WAXS/Raman time-resolved experiments†
H. W. P. Carvalho‡
 ac,
F. Lerouxb,
V. Brioisc,
C. V. Santillia and
S. H. Pulcinelli*a
ac,
F. Lerouxb,
V. Brioisc,
C. V. Santillia and
S. H. Pulcinelli*a
aUniversidade Estadual Paulista (UNESP), Instituto de Química, Rua Prof. Francisco Degni 55, 14800-060, Araraquara, SP, Brazil. E-mail: sandrap@iq.unesp.br
bICCF, Université Clermont Auvergne, UMR CNRS 6296, SIGMA Clermont, 24 av. des Landais, 63171 Aubière Cedex, France
cSynchrotron SOLEIL, L'Orme des Merisiers, Saint Aubin, BP48, 91192 Gif-sur-Yvette Cedex, France
First published on 9th October 2018
Abstract
In-depth understanding of the thermal stability of polymer–clay nanocomposites requires the use of advanced time-resolved techniques combined with multivariate data analysis, as well as the preparation of layered nanofillers with well-defined composition. The layered double hydroxide (LDH) compounds Zn2Al(OH)6·nH2O, Zn2Al0.75Fe0.25(OH)6·nH2O, ZnCuAl(OH)6·nH2O, and ZnCuAl0.5Fe0.5(OH)6·nH2O were prepared, each designed to specifically identify the physical barrier, radical trapping, and char formation contributions to the thermal stability of the PMMA–LDH nanocomposites. The unique combination of conventional methods (TG, DSC, and Raman spectroscopy) and synchrotron radiation techniques (XAS and WAXS), applied during PMMA–LDH heating, revealed the synergetic (of iron) and antagonist (of copper) effects of the LDH layers transformations on the three main endothermic steps of mass loss of the polymer. The diffusion barrier effect was proved by the downshift of the PMMA thermal decomposition temperature caused by the decrease of the LDH edifice thermostability when divalent cations were substituted in the LDH (passing from PMMA–Zn2Al(OH)6·nH2O to PMMA–ZnCuAl(OH)6·nH2O). For PMMA–Zn2Al0.75Fe0.25(OH)6·nH2O, a cooperative contribution of iron reduction, stabilisation of layered edifice, and radical trapping effects was observed for the thermal stability of the nanocomposite. LDH also acted as a diffusion barrier to the efflux and evaporation of depolymerized species, favouring the charring which exerts an additional contribution to thermal stability of the PMMA–LDH nanocomposites.
Introduction
Nanocomposites based on a polymeric matrix and inorganic nanoparticles as the dispersed phase have become essential materials in daily life, finding a broad range of applications due to their outstanding properties including mass transport and heat diffusion barrier effects, flame retardancy, and scratch/wear resistance.1–4 Nanocomposites offer improved properties, compared to conventional microcomposites. For example, they can be important in fire prevention5 because the polymer decomposition temperature can be increased by the addition of highly dispersed nanofillers such as oxide nanoparticles and clays.1–9 Clays are especially attractive as nanofillers, since they are abundant in nature or are readily synthesized, which reduces production costs.Several mechanisms have been invoked to explain the improved thermal stability of clay–polymer nanocomposites. It is well recognized that the high aspect ratio of clays can contribute to their diffusion barrier effects, preventing the entry of gaseous oxygen into the bulk polymer and the escape of volatile components.10 Moreover, the nanoconfinement created by clay layers can induce radical recombination and consequently delay depolymerization.11 The presence in clays of 3d transition metal cations, such as Fe and Cu, can enhance the thermal stability of polymers.12–16 Several interpretations can be found for the role played by the 3d cations. The presence of iron in clays favours the so-called radical trapping mechanism, as proposed by Wilkie et al.,14 but there is a lack of consensus in the literature concerning the role of copper. The observation of Cu(II), Cu(I), and Cu(0) species at different temperatures of thermal decomposition of polymer–clay materials has led to the proposal of a similar radical trapping mechanism for Cu.15 The same authors have proposed that Cu(I) ions could favour the catalytic formation of new polymer chains by polymerization involving combinations of methyl methacrylate (MMA) radicals.16 Finally, copper species formed during degradation are believed to be catalysts for the formation of cross-linked char, which acts as a new physical barrier.17 The radical trapping mechanism associated with the presence of structural iron in clays was recently demonstrated by the monitoring of thermal degradation of poly(methyl methacrylate) (PMMA) – montmorillonite (MMT) nanocomposites using in situ time-resolved XAS.18 It was shown that structural Fe(III) atoms in the octahedral layer of natural MMT were reduced to Fe(II) due to trapping of radicals formed during the thermal decomposition of PMMA.18 This iron reduction was not observed during heating of the pristine MMT.
Our recent study of thermal decomposition of clay-like materials belonging to the layered double hydroxide (LDH) family (without polymer) clearly revealed that for Zn–Cu–Al LDH, the formation of copper species with different oxidation states readily occurred.19 This indicated that the change of Cu oxidation state might not necessarily be associated with the trapping of radicals. Moreover, the dispersion of 3d metals introduced as adsorbed additives on the clay surface, or segregated into the bulk phase, has been suggested to provide an explanation for the differences in thermal stability of nanocomposites with similar 3d cation loadings.14,20,21
The aim of the present work was to disentangle the contributions of the different effects suggested to explain the improved thermal stability of synthetic PMMA–LDH nanocomposite materials. For this purpose, PMMA–LDH nanocomposites were prepared with different chemical compositions, with Zn(II), Cu(II), and Fe(III) in the octahedral positions of positive layers, defined by the general formula [M(1−x)2+Mx3+(OH)2]layer[DSx−·zH2O]interlayer, with dodecylsulphate (DS, CH3(CH2)11OSO3) anions embedded in the interlayer spaces of the lamellar edifice. In a previous study, based on an in-depth analysis of the local and medium range order evolution of ZnCuAl and ZnFeAl LDH during thermal treatment at T < 450 °C, it was demonstrated that the evolution of the chemical environment of the transition metal was highly dependent on the LDH chemical composition, and that the layer degradation passed through several intermediate phases until formation of the crystallized oxide phases.19 In the present work, a similar approach was used in order to establish a relationship between the speciation of 3d cations in the LDH guest matrix, embedded in PMMA, and the thermal properties of the resulting nanocomposites. The 3d cations were selected in order to elucidate the contributions of the different mechanisms (physical barrier, radical trapping, and char formation) to improved thermal stability of the nanocomposites.
Different to previous works reported in the literature on this subject,11–17 an elegant combination of complementary time resolved techniques was used for in situ investigation of the thermal decomposition of both organic (PMMA) and inorganic (LDH) phases of the nanocomposites. Thermogravimetry (TG) and differential scanning calorimetry (DSC) provided information about the thermal stability of PMMA. X-ray absorption spectroscopy (XAS) at the Cu, Zn, and Fe K-edges enabled tracking of the local atomic order of the cations in the LDH and decomposed phases during thermal decomposition of the nanocomposite. Raman spectroscopy provided information about the thermal degradation of the organic matrix and the formation of coke deposits. Wide angle X-ray scattering (WAXS) supplied additional information concerning mesoscopic changes of the LDH structure during the decomposition.
Experimental
Preparation of the nanocomposites
The LDH phases were prepared by co-precipitation of the corresponding metal-chloride salts with aqueous NaOH solution, as described previously.19 The resulting LDH–Cl phases were submitted to ionic exchange against sodium dodecyl sulphate (SDS) anions. For this purpose, 20 g of the precipitated pasty LDH–Cl were dispersed in 100 mL of deionized water, followed by addition of a quantity of SDS corresponding to twice the calculated anionic exchange capacity for each LDH–Cl (Table S1, ESI†). The slurry was stirred for 24 h under a flow of N2, before being centrifuged, washed four times with deionized water, and dried at 70 °C for 24 h. The resulting materials were denoted LDH–DS. Next, 800 mg of the LDH–DS were dispersed in 15 mL of tetrahydrofuran (THF), under stirring for 2 h in a 20 kHz ultrasonic bath. In parallel, 0.75 mol of methyl methacrylate (MMA) and 205 mg of benzoyl peroxide were added to 160 mL of THF. The solution was kept under reflux at 70 °C for 15 h, with increasing viscosity of the liquid being an indication of polymerization. Finally, the LDH–DS dispersion was mixed with 20 mL of the polymeric solution and kept under reflux at 70 °C for 24 h, before being deposited over polytetrafluoroethylene substrates, forming thick films. The films were dried first at room temperature (RT) for 24 h and then at 70 °C for a further 24 h. The films were easily removed from the substrates, forming self-supported structures. The resulting nanocomposites contained 17% (w/w) of LDH–DS. The PMMA–LDH nanocomposites were named according to the nominal cationic chemical composition of the LDH, as follows:| ZA = [Zn0.66Al0.33(OH)2.00][Cl0.33]·nH2O, |
| ZCA-1 = [Zn0.33Cu0.33Al0.33(OH)2.00][Cl0.33]·nH2O, |
| ZCA-0.14 = [Zn0.58Cu0.08Al0.33(OH)2.00][Cl0.33]·nH2O, |
| ZAF = [Zn0.66Al0.25Fe0.08(OH)2.00][Cl0.33]·nH2O, and |
| ZCAF = [Zn0.33Cu0.33Al0.165Fe0.165(OH)2.00][Cl0.33]·nH2O. |
In the case of LDH containing zinc, copper, and aluminium, the value added after ZCA refers to the copper/zinc ratio.
Characterization of the PMMA–LDH nano composites
Thermogravimetry (TG) and differential scanning calorimetry (DSC). Thermogravimetric analyses of ∼5 mg portions of the nanocomposites were performed using an SDT Q600 system (TA Instruments), under 100 mL min−1 flows of N2 and air, with heating at a rate of 10 °C min−1. DSC measurements were carried out using a DSC 2910 analyser (TA Instruments), under a flow of N2 at 100 mL min−1, with heating at a rate of 10 °C min−1. About 10 mg of sample were placed in aluminium crucibles, in which holes were drilled to allow gas exchange and maintain atmospheric pressure inside. The total heat flux was normalized by the total sample mass, so the results could be expressed as J g−1. For each step of degradation of the PMMA and the nanocomposites, the area under the curve was measured and normalized by the corresponding weight loss obtained by TG. The measurements were repeated three times, with the calculated errors corresponding to the standard deviation for the three measurements.
WAXS measurements. Wide angle X-ray scattering (WAXS) patterns were recorded during thermal decomposition of the nanocomposites, at the SWING beamline of the SOLEIL synchrotron radiation facility. The white beam was monochromatised (λ = 0.8266 Å) by a Si double crystal and focused by a Kirk-Patrick–Baez mirror. The intensity of the scattered beam was measured with a two-dimensional CCD detector located at 0.51 m from the sample. RT measurements enabled evaluation of the efficiency of the anionic exchange on the pristine LDH–DS, by measuring the peaks related to the basal distance of the LDH. Time-resolved monitoring of the intensities and positions of these peaks during heating at 10 °C min−1 from RT to 450 °C (using a LINKAM THM 600R cell) provided insights about the thermal decomposition of the layered edifice.
Quick-EXAFS and Raman measurements. X-ray absorption spectroscopy (XAS) data were recorded at the Fe, Cu, and Zn K-edges during thermal decomposition of the PMMA–LDH nanocomposites (with heating at 10 °C min−1), at the SAMBA beamline of the SOLEIL synchrotron radiation facility, using the Quick-EXAFS mode.22 The set-up used for the time-resolved XAS monitoring of the PMMA–LDH thermal decomposition was identical to the one described fully in previous work concerning the thermal decomposition of LDH–Cl.19 Data recorded at the Zn and Cu K-edges were collected in a single experiment, while a second measurement was used for collection of Fe K-edge data for the PMMA–ZAF and PMMA–ZCAF nanocomposites. The time for collection of one Quick-EXAFS spectrum was 0.5 s, and 12 spectra were merged in order to improve the signal/noise ratio. During the thermal decomposition at temperatures from RT to 450 °C, 210 spectra were typically recorded at each edge, with one spectrum obtained every 2 °C. The Quick-EXAFS measurements were performed simultaneously with Raman spectroscopy analyses performed using an RXN1 spectrometer (Kaiser Optical Systems, Inc.) equipped with a solid-state laser diode operating at 532 nm. The laser beam was focused using a 150 mm long working distance objective lens with a magnification of 10×. The thermal decomposition was carried out using the oven described previously,23 which allowed the simultaneous use of both spectroscopic techniques. A 2 mm thick pellet of PMMA–LDH nanocomposite (120 to 160 mg) was loaded into a sample holder fitted with a mica window at the face irradiated by the laser.
Multivariate analysis of the time-resolved data sets acquired during thermal decomposition of the nanocomposites. As previously carried out for analysis of the thermal decomposition of pristine LDH–Cl phases,19 the chemical species involved in the PMMA–LDH decomposition were determined using a multivariate analysis of the XAS data. The principle of the multivariate curve regression with alternating least squares (MCR-ALS) fitting procedure was based on decomposition of the matrix data set D(ij) containing the i XAS spectra of the samples, recorded at time ti, for the j energy values, according to the relation:
| D = CST + E | (1) |
Results
The thermal decomposition of PMMA-based polymers is a complex process that involves the scission of bonds and the transport of volatile decomposed products out of the bulk material.27 Information concerning the effects of the LDH composition and the surrounding atmosphere (synthetic air and N2) on the thermal stability of the PMMA–LDH nanocomposite matrix was obtained by thermogravimetry (TG, Fig. 1) and differential scanning calorimetry (DSC, Fig. 2). In order to interpret the PMMA–LDH mass loss events, it is important to consider the TG curves obtained for pristine LDH–DS (Fig. S1, ESI†). These were characterized by a first 10% mass loss between RT and 110 °C, corresponding to the removal of intercalated and adsorbed water molecules, a second 30% mass loss at around 200 °C, corresponding to dehydroxylation of the LDH sheets, and a third 15% mass loss at around 450–600 °C, associated with sulphate decomposition. However, the total 40% mass loss in the temperature range from RT to 450 °C for pristine LDH–DS is strongly levelled out on the thermogravimetric curves of the nanocomposites, due to the low loading of LDH (17%, w/w).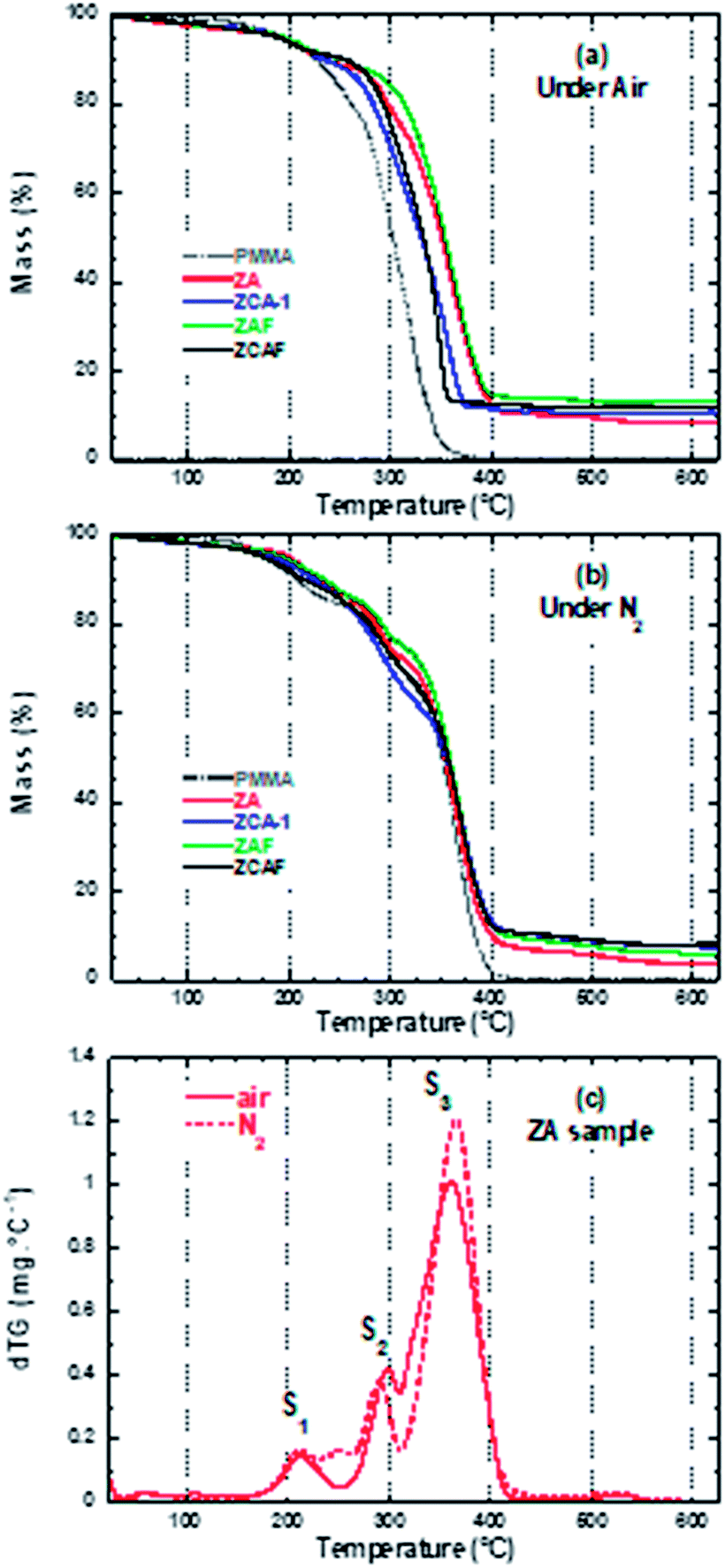 | ||
| Fig. 1 Thermogravimetric curves obtained under (a) air and (b) N2 atmospheres for the different nanocomposites and the PMMA matrix. (c) Comparison of dTG curves for ZA under air and N2 atmospheres. | ||
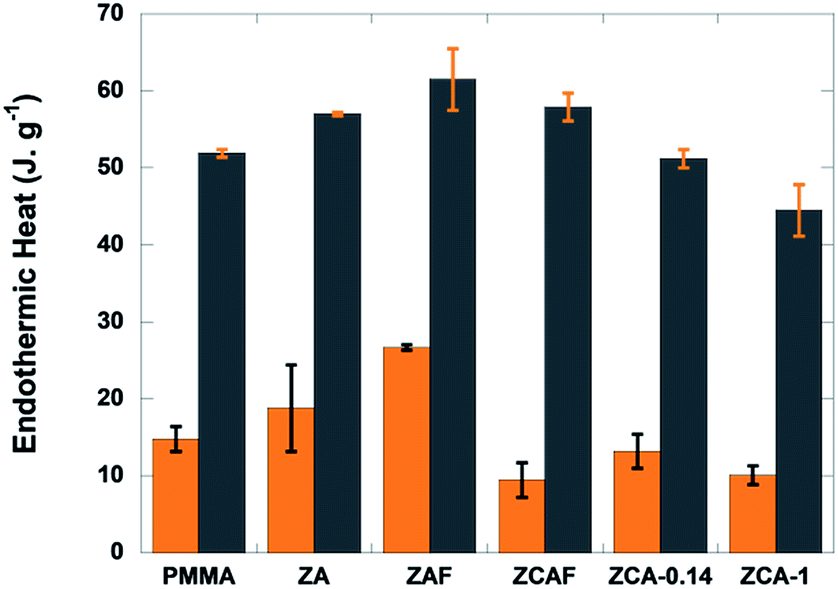 | ||
| Fig. 2 Heat absorbed by PMMA and the nanocomposites during the first (orange columns) and third (grey columns) decomposition steps. | ||
The decomposition of PMMA–LDH occurs in distinct steps (see the dTG curves in Fig. 1(c) and in Fig. 6(g)–(i)), as reported previously for the PMMA homopolymer27,28 and PMMA–montmorillonite nanocomposites.5,10,11,18 The first two steps with mass loss below 20%, denoted S1 and S2, were related to the initial depolymerisation involving head-to-head linkages and vinylidene end groups, respectively. The third and most important mass loss, denoted S3, was related to random unzipping depolymerisation. Under air atmosphere, all the nanocomposites presented higher thermal stability than the PMMA homopolymer (Fig. 1(a) and (b)). There were significant shifts towards higher temperatures for 50% mass loss (T0.5), with increases ranging from 27 to 51 °C, depending on the LDH composition (Table 1). The effect of the isovalent substitution of divalent and trivalent cations on thermal stability was more pronounced under air. The PMMA–ZCA material was less stable than the nanocomposite containing only Zn as the divalent cation (PMMA–ZA). The substitution of Al by Fe cations resulted in an increase of T0.5, compared to PMMA–ZA. Under nitrogen atmosphere (Fig. 1(b)), the onset of the acceleration of the first 20% of mass loss was delayed by this isovalent substitution, being shifted by 15–20 °C for PMMA–ZAF, compared to PMMA–ZA, with almost complete suppression of the S2 event in the case of PMMA–ZAF (Fig. 6(i)). Since the zinc and aluminium cations do not usually undergo redox reactions under these experimental conditions, the higher thermal stability of PMMA–ZA could be ascribed to a mass transport barrier effect of inorganic sheet labyrinth, which slows down the diffusion, permeation and releases of thermal decomposition products from the PMMA.29 Hence, the PMMA–ZA nanocomposite could be considered as a reference for the diffusion barrier stabilization mechanism.
| Sample | Under air | Under N2 | Residue expected (%) | ||||
|---|---|---|---|---|---|---|---|
| T0.5 (°C) | ΔT0.5 (°C) | Residue obtained (%) | T0.5 (°C) | ΔT0.5 (°C) | Residue obtained (%) | ||
| PMMA | 303 | 0 | 352 | 0 | 0 | ||
| PMMA–ZA | 351 | 48 | 10.1 | 355 | 3 | 7.6 | 7.9 |
| PMMA–ZCA-1 | 330 | 27 | 10.9 | 356 | 4 | 10.1 | 8.0 |
| PMMA–ZCAF | 333 | 30 | 12.0 | 357 | 5 | 10.1 | 9.0 |
| PMMA–ZAF | 354 | 51 | 13.8 | 359 | 7 | 9.0 | 8.6 |
The observed differences in thermal stability between PMMA–ZA and the other nanocomposites could be explained by the different structural and electronic characteristics of the LDH phase during thermo-oxidative depolymerisation of the matrix. This hypothesis was confirmed by the higher and similar T0.5 values (352–359 °C) observed for the TG curves obtained under N2 for all the PMMA–LDH and the PMMA homopolymer. This high thermal stability was consistent with an up to four-fold increase of the activation energy of the random scission depolymerisation reaction reported when the surrounding atmosphere was changed from oxidizing to non-oxidizing one.30 Accordingly, the observed atmosphere-dependent behaviour indicated that the LDH acted as an efficient O2 diffusion barrier, hindering random scission of the PMMA chains. Furthermore, the delay of the S1 thermal event for the nanocomposites under an N2 atmosphere indicated that the LDH not only acted as a physical barrier against the diffusion of O2, but was also involved in reactions affecting the chemical equilibrium of the polymeric chains decomposition. This hypothesis was also supported by the differences between the expected and measured residues at 450 °C, ascribed to char formation (Table 1), comparing the iron-containing nanocomposites to PMMA–ZA.
In order to elucidate the contributions of the copper and iron atoms to the non-diffusional process of PMMA–LDH thermal decomposition, a systematic calorimetric study was carried out under a non-oxidizing atmosphere (N2). Irrespective of the iron or copper content of the LDH, the three degradation steps (S1, S2, and S3) were endothermic events (Fig. S2, ESI†). At least two heat consumption events were involved during the first (S1) and third (S3) decomposition steps, one necessary for the unzipping reaction giving rise to monomer formation, and another one corresponding to the evaporation of MMA (Fig. 2). Comparing PMMA–ZA with PMMA–ZAF, the data evidenced that for both events, the isovalent substitution of aluminium by iron led to an increase of the endothermic heat. In contrast, the energy required for the degradation decreased as the Cu content increased from PMMA–ZA to PMMA–ZCA-0.14 and PMMA–ZCA-1. It is notable that the results obtained for the PMMA–ZCAF nanocomposites were consistent with the antagonistic roles played by the two cations. Although the iron content of ZCAF was twice that of the ZAF sample, the energy required for degradation of the nanocomposite containing Fe and Cu (PMMA–ZCAF) was lower than for PMMA–ZAF. The opposite behaviour could be explained by the occurrence of exothermic reactions such as char combustion or other oxidation reactions promoted by the reduction of copper cations.31–33 This hypothesis will be further discussed in light of the combined XAS and Raman monitoring of the thermal decomposition process.
The RT WAXS pattern for the PMMA–ZCA nanocomposite (Fig. 3(a)) displayed well-defined peaks at around 0.22 and 0.44 Å−1, corresponding to the (003) and (006) diffraction signals characteristic of lamellar stacking of the LDH sheets. A significant shift was observed of the basal distance determined from the position of the (003) peak for PMMA–ZCA (d00l = 29.12 Å), compared to the values reported previously19 for pristine LDH–Cl (7.66 Å) and pristine LDH–DS (26.35 Å). This increase of the basal distance resulted first from the exchange of Cl anions by DS oriented interdigitated and more or less perpendicular to the LDH sheets,17,34 and then during preparation of the nanocomposite from the ingress (migration) of the PMMA chains into the interlamellar galleries still occupied by the hydrophobic tails of tethered DS anions. The basal distances for the pristine LDH samples and the nanocomposites are shown in Table S1 (ESI†). Similar behaviours were obtained, irrespective of the nanocomposite composition. The interlayer distance was greater for PMMA–ZCA, compared to the other nanocomposites, which could be ascribed to the corrugation of the LDH lamellae induced by the Jahn–Teller distortion around the Cu cations introduced into the regular brucite-like sheets.35
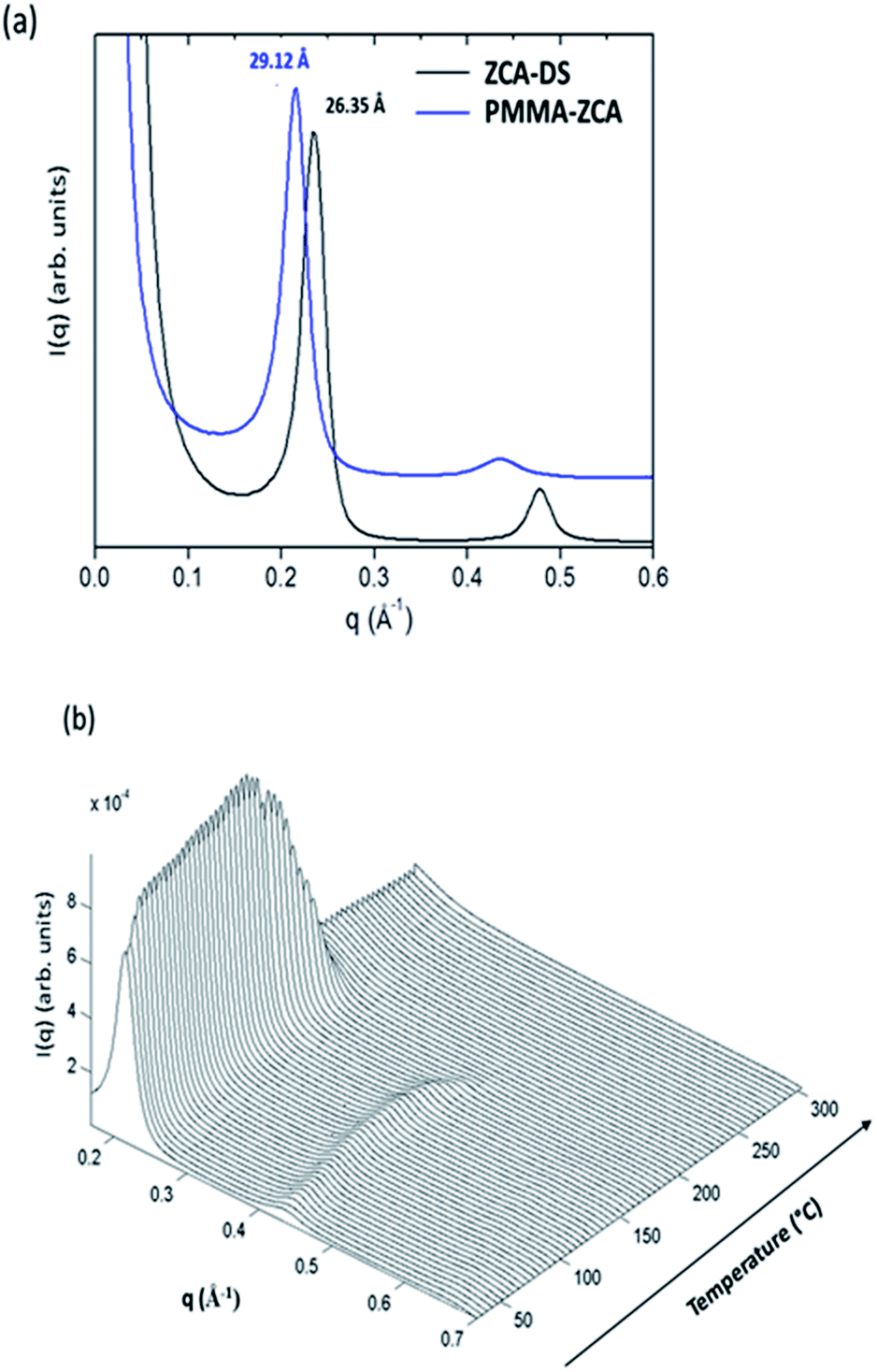 | ||
| Fig. 3 WAXS patterns recorded for the ZCA-based systems: (a) comparison at RT of the patterns for the ZCA–DS and PMMA–ZCA samples; (b) evolution of the WAXS patterns of PMMA–ZCA during thermal treatment under air atmosphere. | ||
The effect of heating on the medium range order of LDH embedded in the PMMA matrix was evaluated from the time-resolved WAXS patterns, presented in Fig. 3(b) for the PMMA–ZCA and in Fig. S3 (ESI†) for the other nanocomposite compositions. Both the positions and intensities of the (003), (006), and (009) diffraction signals evolved during heating. For PMMA–ZCA, the loss of signals above 190 °C was an indication of destruction of the periodic stacking of the LDH sheets. The onset of the collapse of the periodic structure was dependent on the LDH chemical composition (see Fig. 6(g)–(i)).
In order to correlate the thermal stability of the different nanocomposites with the transformation of the organic matrix and the inorganic LDH filler, the thermal decomposition was monitored simultaneously using the Raman and XAS spectroscopic techniques.
The energy position of the rising edge of the measured spectra at the Zn K-edge (Fig. 4(a)) was characteristic of divalent Zn species, irrespective of the nanocomposite composition, confirming that Zn species were not involved in redox reactions during nanocomposite heating. The thermally-induced local order transformations around Zn of the LDH embedded in the PMMA matrix were different to those reported for the pristine LDH–Cl.19 The LDH–Cl first evolved towards a dehydrated phase, which was further converted to nano-ZnAl2O4 and nano-ZnO phases. Here, the PMMA–LDH–DS phases were first transformed into an intermediate species presenting a common XANES spectrum (Fig. S4, ESI†). The EXAFS spectrum of this intermediate species could be satisfactorily reproduced considering a first coordination shell with ∼4 oxygen atoms at a distance of 1.95 ± 0.01 Å. Comparison of the Zn K-edge spectra of the nanocomposites at the end of the heating, 450 °C, (Fig. S5(a), ESI†), revealed a dependency of the spectral profiles on the LDH composition, which could be explained by different proportions of Zn-based phases. In fact, the XAS spectrum obtained for PMMA–ZAF at 450 °C was very similar to that of a nano-ZnO phase, whereas the spectrum obtained for PMMA–ZCA was characteristic of nano-ZnAl2O4 (Fig. S5(b), ESI†).19
 | ||
| Fig. 4 Evolution of Quick-EXAFS spectra during heating, evidencing the modifications of local range order around the 3d cations forming the LDH sheet (a) at the Zn K-edge and (b) at the Cu K-edge for the PMMA–ZCA sample, and (c) at the Fe K-edge for the PMMA–ZAF sample. The insert compares the spectra obtained at RT and 410 °C. | ||
The evolution observed at the Cu K-edge (Fig. 4(b)) displayed strong modifications, with the apparent low-energy shift of the rising edge being characteristic of redox transformations, first towards Cu(I) species, and then towards metallic Cu(0) species. It is notable that similar changes were observed for thermal decomposition of the pristine ZCA–Cl,19 confirming the well-known tendency of Cu phases to participate in redox reactions under mild heating.36 Taking into account this behaviour in absence of polymer, at first glance the direct relation between Cu reduction observed for nanocomposites and trapping of radicals formed during PMMA depolymerisation is not straightforward. This finding for the copper LDH samples did not hold true for the nanocomposites containing iron in the LDH structure, since the Fe K-edge energy position for PMMA–ZAF (Fig. 4(c)) was shifted towards lower energy when the temperature was increased. This is illustrated in the insert of Fig. 4(c), comparing the nanocomposite measurements at RT and at 410 °C. A similar energy shift was not observed for the pristine ZAF–Cl,19 suggesting that the redox reactions of iron species were coupled to radical formation during the PMMA depolymerisation process. ESI (Fig. S6–S8†) gathers the Quick-EXAFS measurements recorded during the thermal decomposition at the Zn K-edge for the PMMA–ZA, PMMA–ZAF, and PMMA–ZCAF nanocomposites, Cu K-edge and Fe K-edge for the PMMA–ZCAF nanocomposite.
Raman spectroscopy analyses, performed simultaneously with the XAS measurements, enabled time-resolved monitoring of PMMA degradation (Fig. 5(a)) and the associated char formation (Fig. 5(b)). For this purpose, the lines characteristic of the C–H vibrational modes of the PMMA polymeric backbone (at 2800–3100 cm−1) and the D and G lines characteristic of char deposits (at 1300–1500 cm−1) were considered. The G line located near 1590 cm−1 is attributed to graphitic-type carbon, while the D line located at 1320 cm−1 is associated with in-plane graphitic imperfections.37 The Raman spectrum of the char residue, with a dominant contribution of a narrow G line, was characteristic of graphite-like chars.13,37 The presence of such graphitic layers provides an additional physical barrier to mass transport.13 The Raman signal of the PMMA matrix was lost at around 200 °C (Fig. 5(a)), when signals of the char residues started to appear (Fig. 5(b) and 6(e)). This evidenced that the graphitic char deposit hindered monitoring of the decomposition of the bulk PMMA. Furthermore, monitoring of char formation was impossible for ZA and ZAF, due to the strong fluorescence backgrounds of the samples prepared without copper. Consequently, the evolution of the area of the G lines, as a function of temperature, would only be correlated to the copper speciation determined from XAS and the thermal stability of PMMA. These correlations are in deep discussed in the next section.
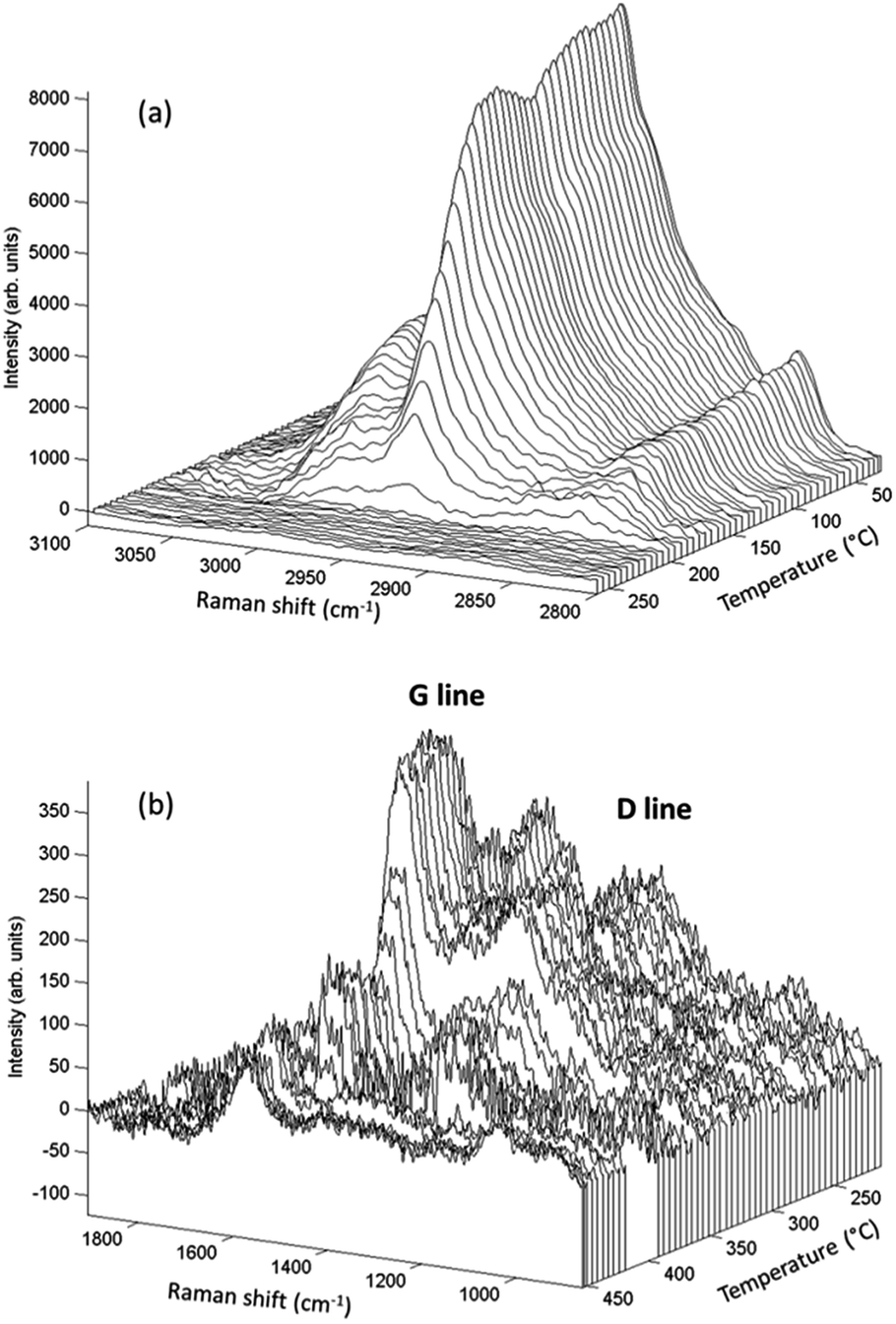 | ||
| Fig. 5 Raman spectra for PMMA–ZCA showing (a) decomposition of the polymeric matrix, and (b) coke formation following decomposition. | ||
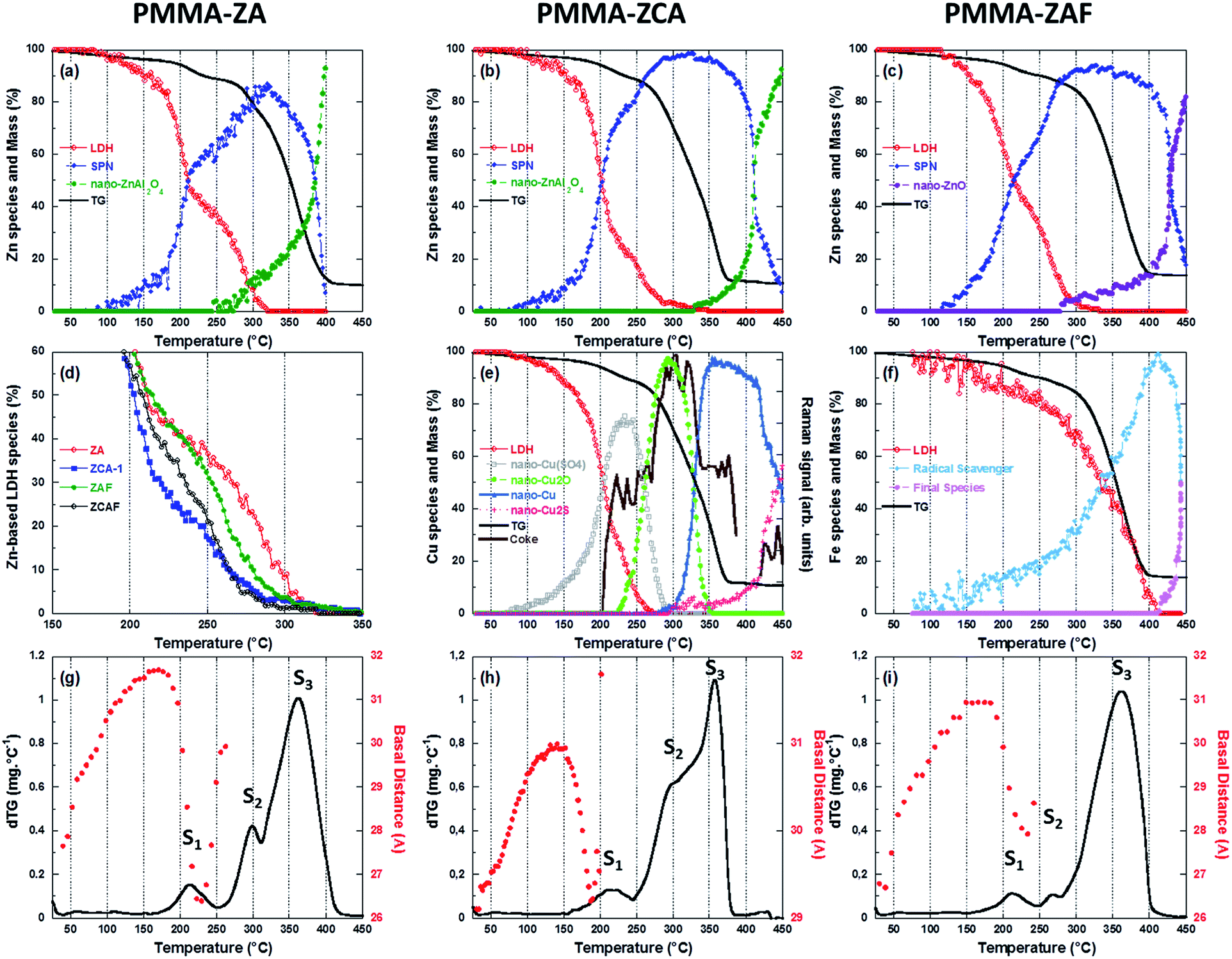 | ||
| Fig. 6 Comparison of the thermogravimetric curves under air atmosphere for the different nanocomposites with the concentration profiles determined by MCR-ALS analysis of the Quick-EXAFS data. Coke formation was determined by Raman monitoring and WAXS data were acquired during monitoring of the thermal decomposition. (a)–(c) Speciation from Zn K-edge XAS data for (a) PMMA–ZA, (b) PMMA–ZCA-1, and (c) PMMA–ZAF. (d) Comparison of the concentration profiles of the LDH species embedded in the polymeric matrix. (e) Speciation from Cu K-edge results for PMMA–ZCA and (f) speciation from Fe K-edge results for PMMA–ZAF. (g)–(i) Evolution of the basal distance of LDH stacking determined from WAXS for (g) PMMA–ZA, (h) PMMA–ZCA, and (i) PMMA–ZAF. | ||
Discussion
The modifications observed by Quick-EXAFS (Fig. 4 and ESI†) resulted from complex chemical transformations around each 3d cation of the LDH sheet, driven by the cooperative effects of the heating and the depolymerization of PMMA with formation of radicals. The metallic species involved could be elucidated from chemometric analysis based on multivariate curve resolution with alternating least squares fitting (MCR-ALS),38–40 which was applied for the determination of Zn, Cu, and Fe species, as detailed in the ESI.†In order to gain insight on the respective role of physical barrier, played by the LDH sheets and char residues, and of the redox reactions of the 3d cations building those sheets for improving the thermal resistance of PMMA, the thermal properties of the nanocomposites monitored by TG will be then discussed in light of the MCR-ALS speciation and structural results gained by WAXS and Raman, as displayed Fig. 6.
Contribution of the physical barrier effect
Since the PMMA–ZA sample was considered an archetype of the physical barrier contribution to improvement of the thermal stability of the PMMA–LDH material, discussion was first focused on the transformations occurring around the Zn atoms (Fig. 6(a)–(c)). Irrespective of the type of LDH nanocomposite, the structural transformation around Zn during the thermal decomposition was quite similar, with three species involved. Two of these species were common to all the LDH, while the third was dependent on the metal substitution. The PMMA–ZA and PMMA–ZCA samples showed formation of mainly the nano-ZnAl2O4 phase, while for PMMA–ZAF, nano-ZnO was the main product obtained at the end of heating (Fig. S5, ESI†). The LDH embedded in the polymeric network was transformed into a first intermediate species, isolated by MCR-ALS and characterized in EXAFS by a first coordination shell of ∼4 oxygen atoms at 1.95 ± 0.01 Å and a second neighbors contribution located at around 3.0 Å in the no-phase corrected Fourier transform, which was well reproduced by Zn atoms located at 3.39 Å (Fig. S4, ESI†). Such a Zn contribution is usually encountered in ZnAl2O4.41 This finding, together with the similarity between the triangular shapes of the XANES spectrum and the spectrum for ZnAl2O4,19 led to this first intermediate species being identified as a spinel pre-nucleus (SPN) phase.Acceleration of transformation of the LDH phase to the SPN phase occurred at around 180 °C, irrespective to the LDH composition of the nanocomposites. This behaviour coincided with the onset of shrinkage of the basal distance of the lamellar edifice measured by WAXS (Fig. 6(g)–(i)). It is notable that at ∼200 °C, ∼50% of the LDH sheets remained embedded in the polymeric matrix (Fig. 6(d)). The periodic stacking was retained up to 200 °C for PMMA–ZCA, 240 °C for PMMA–ZAF, and 260 °C for PMMA–ZA (Fig. 6(g)–(i)). Comparison of these temperatures with the temperatures at which 10% of the LDH structure was still observed, as shown in Fig. 6(d) (260 °C for PMMA–ZCA, 275 °C for PMMA–ZAF, and 300 °C for PMMA–ZA), indicated that the LDH sheets persisted in the matrix, although the periodic stacking was lost. In the case of the PMMA–ZA sample, acceleration of the mass loss associated with the onset of the S3 event (Fig. 6(g)) occurred when 10% of the Zn species were still embedded in the LDH sheets. Considering the initial LDH loading (17%, w/w), this finding evidenced that stabilization of PMMA thermal decomposition, due to the physical barrier effect of the layered morphology of the fillers, was effective for LDH loadings higher than 1.7% (w/w). This low loading value was consistent with the classical literature42 reporting the gas diffusion barrier effect of clay in polymers. Despite similar transformation rates, the shift at higher temperature of the decrease of SPN amount with respect to the mass loss curve was a strong indication that neither the SPN species nor the nano-ZnAl2O4 or nano-ZnO products derived from it made any significant contributions to the physical barrier effect. This feature was in agreement with the transformation of two-dimensional (2D) nanofillers into zero-dimensional (0D) nanoparticles.42 From these comparisons between the thermal decomposition of PMMA and thermal transformation of LDH, it could be concluded that the persistence under heating of small amounts of layered nanofillers (1.7%) resulted in a high gas diffusion barrier efficiency. A fine tuning of the PMMA thermal stability was achieved, with an increase of the 2D to 0D transition temperature of ZA-based LDH.
In fact the isovalent substitution of either aluminium or zinc by iron or copper, respectively, resulted in opposite effects in terms of thermal stabilization of the nanocomposites. Comparison of the dTG curves (Fig. 6(g)–(i)) indicated a strong contribution of the S2 event to the stabilization effect. In comparison with PMMA–ZA, the higher stabilization observed for PMMA–ZAF was associated with a low contribution of the S2 event to the mass loss (Fig. 6(i)), while the destabilization observed for ZAC was related to an increase of the S2 contribution that led to merging of the S2 and S3 events (Fig. 6(h)). It is important to keep in mind that the S2 contribution was related to the degradation initiated at the vinylidene end groups, resulting in the formation of polymer radicals.29 It therefore seemed that the chemical nature of the LDH cations embedded in the PMMA matrix significantly contributed to the quantity of radicals formed as a result of the S2 event. This cooperative chemical effect will be discussed in the two next sections.
Contribution of the radical trapping
The S2 step occurred between 250 and 300 °C, a temperature range in which comparable percentages of Zn atoms were embedded in the LDH sheets of PMMA–ZA and PMMA–ZAF (Fig. 6(a) and (c)), so similar contributions of the physical barrier effect were expected for the two nanocomposites. However, the dTG curves evidenced that the isovalent substitution of aluminium by iron in PMMA–ZAF almost suppressed the S2 mass loss, indicating that the quantity of radicals participating in the PMMA decomposition was lower for PMMA–ZAF, compared to PMMA–ZA. This feature could be clearly understood from the iron speciation of ZAF (Fig. 6(f) and S9, ESI†). The intermediate species identified as an Fe(II) phase, according to the 2.6 eV energy downshift of the rising edge, compared to the pristine PMMA–LDH (Fig. S9†),43 started to be formed at 150 °C, with the amount increasing at a constant rate up to 300 °C. At this temperature, which corresponded to the maximum mass loss rate of PMMA–ZA in the S2 event (Fig. 6(g)), there was a remarkable acceleration of formation of the intermediate Fe(II) species (Fig. 6(f)). This provided clear evidence that for PMMA–ZAF, the radicals formed during the S2 event were scavenged by the Fe(III) cations embedded in the LDH sheet, yielding the iron(II) intermediate species and delaying the depolymerisation by radical propagation.Other evidence of this radical trapping phenomenon was provided by the superpositioning of the TG curve and the evolution of the position of the rising edge of the Fe K-edge experimental data (Fig. 7). The shift of the rising edge at lower energy was due to transformation of the LDH phase containing Fe(III) cations into the intermediate Fe(II)-based species (Fig. 6(f)). It is interesting to note that when the PMMA decomposition was over, there was a reverse energy shift towards the Fe(III) threshold energy position (Fig. 7), leading to the formation of a new Fe(III) species that was identified as ZnFe2O4, with a possible minor contribution of γ-Fe2O3. This set of time-resolved results obtained using independent techniques strongly suggested that the additional mechanism that acted to delay PMMA depolymerisation, in the case of the ZAF sample, was the redox reaction involving iron cations. In addition, the concentration profile of the Fe(III) atoms embedded in the LDH sheets (Fig. 6(f)) was clearly shifted to higher temperature, by almost 100 °C, compared to Zn in the LDH sheets of the ZAF nanocomposite (Fig. 6(c)). This could be explained by the coexistence of iron(III) and iron(II) within the LDH sheets, which was facilitated by the departure of zinc atoms of the layers to form the Zn-based SPN intermediate (Fig. 6(c)).
 | ||
| Fig. 7 Comparison of the Fe K-edge threshold energy, determined at a normalized absorbance coefficient value of 0.5, with the thermogravimetric curve for PMMA–ZAF. | ||
The Fe(III) in PMMA–ZAF and PMMA–ZCAF, as well as the Fe(II) intermediate species, displayed Fourier transforms of EXAFS signals with clear second neighbour contributions, as shown in Fig. 8. For both species, these contributions could be fitted by Zn atoms at characteristic distances for the LDH structure, with values of ∼3.07 Å for PMMA–ZAF and ∼3.00 Å for the Fe(II) intermediate species (Fig. S10, ESI†). Following reduction of Fe(III) to Fe(II), the positive charge of the layer was assumed to be maintained by the presence of Al(III) ions within the sheet. Notably, the PMMA–ZAF nanocomposite was the only sample for which nano-ZnO was the main product formed by transformation of the SPN species; the other nanocomposites showed the formation of spinel-like Zn-based species as the main products. This was a strong indication that aluminium cations remained in the layer structure during the redox reaction, stabilizing the lamellar edifice. The persistence of the characteristic LDH local order around iron at high temperature could also lead to increased PMMA–ZAF stabilization by the combined effects of the physical barrier and the higher glass transition temperature of PMMA chains confined on the LDH surface.44
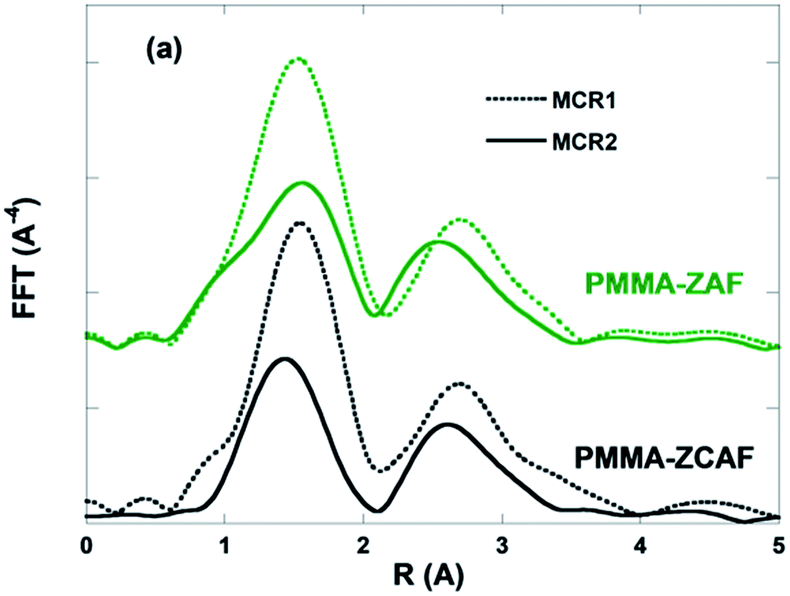 | ||
| Fig. 8 Fourier transforms of the Fe K-edge EXAFS spectra associated with the first two MCR-ALS components extracted from the thermal decomposition of PMMA–ZAF and PMMA–ZCAF. | ||
These results allowed to conclude that the addition of Fe(III) in the LDH structure produces synergetic contributions to the thermal stability of PMMA–ZAF nanocomposite due to the following cooperative effects: (i) the diffusion barrier caused by the LDH labyrinth, (ii) the trapping of radicals caused by depolymerisation leading to reduction of Fe(III) to Fe(II), and (iii) the preservation of the lamellar LDH edifice during the Zn(II) exudation due to the reduction of Fe(III) into Fe(II).
To investigate the effect of Cu(II) uptake within the LDH layer the spectra of the copper species determined by MCR-ALS during the thermal decomposition of PMMA–ZCA (Fig. S11, ESI†) were analysed. The decomposition of the PMMA–ZCA layered structure was shifted towards lower temperatures by 40–50 °C, compared to PMMA–ZA (Fig. 6(d)), hence substantially affecting the physical barrier effect for the former sample. This resulted in the early departure of copper from the LDH sheet to form the Cu(SO4)x phase (Fig. 6(e)). Concomitantly, the basal distance of the LDH (Fig. 6(h)) started to decrease at 150 °C, while the periodic stacking was lost at 210 °C. In addition, the copper speciation (Fig. 6(e)) showed successive reductions: (i) above 250 °C, coincident with S2 step, the sulphate Cu(II) species was converted to the nanometric Cu2O phase; (ii) above 300 °C the S3 PMMA decomposition step was concomitant with the formation of a nanometric Cu(0) phase; and (iii) above 400 °C, when the polymer was totally decomposed, the Cu(0) phase fraction decreased, with the formation of a monovalent Cu2S phase. Some of these phases have been reported from ex situ X-ray diffraction analyses of nanocomposite residues obtained at different heating temperatures,15,17 but contradictory interpretations were made concerning their roles in thermal stabilization, considering effects such as radical trapping, catalytic activation of MMA polymerization, and char formation. In the present case, different to the previously mentioned role of iron redox species, such successive redox reactions involving copper showed no contribution to improvement of the thermal stability of PMMA–ZCA.
The formation of these different Cu phases requires the exudation of copper from the LDH sheet, leading to destruction of the layered structure. This finding again highlights the key role played by the layered LDH edifice in stabilizing the nanocomposites when cation redox reactions occur. The tortuous paths created by the LDH layers hinder gas permeation through the PMMA matrix.28 In particular, the formation of peroxo radicals resulting from the reaction of O2 with radicals formed early during the decomposition (events S1 and S2) was hampered by the presence of the LDH layers. Consequently, efficient radical trapping only occurred when the physical barrier was preserved, as observed for PMMA–ZAF. Therefore, the dispersion of copper redox species in a matrix without layered structures did not contribute to radical trapping, since oxygen permeation led to the formation of stable peroxo radicals, as evidenced by the increasing contribution of S2 in the dTG thermogram for PMMA–ZCA (Fig. 6(h)).
In conclusion, comparison of the thermal stabilities of PMMA–ZAF and PMMA–ZAC with the redox speciation of iron and copper demonstrated that radical trapping was only efficient when it was assisted by the nanoconfinement of radicals caused by the presence/persistence of the lamellar nanostructure. This provides experimental evidence of the hypothesis of radical extinction facilitated by nanoconfinement.17
Contribution of the char deposits
It is well known that the formation of carbonaceous structures during the thermal degradation of clay–polymer nanocomposites depends on the nature of the clay and the presence of oxygen, even though carbonaceous materials are usually obtained by means of a carbonization reaction under an inert atmosphere.45 The acidic sites of the clays induce char formation,46,47 while the oxygen reacts with the polymeric surface species undergoing decomposition, forming carbonaceous structures. Several studies have reported that the formation of carbonaceous char can be responsible for improving the thermal stability of nanocomposites, due to the formation of a barrier against escape of volatile degradation products.13,48,49 Here, a higher proportion of char residues was observed for the ZAF (Table 1), and more generally for the iron-containing LDH nanocomposites. This was in good agreement with the efficiency of the gas diffusion barrier effect of the Fe-based LDH, throughout the entire temperature range of PMMA decomposition.In addition to the destabilization of the PMMA–ZCA layered edifice caused by the early departure of copper from the sheets, a second harmful effect on PMMA thermal stability was related to the influence of successive reduction of copper on the amount of graphitic coke deposits (Fig. 6(e)). It was first observed that at 200 °C, when about 50% of the LDH had been decomposed, the intensity of the G line of coke increased drastically, while at 225 °C, when the divalent CuSO4 species were reduced to monovalent Cu2O, the intensity of the Raman G line remained almost constant. This finding suggested that concurrent reactions competed in this temperature range, with the reduction of Cu(II) to Cu(I) favouring oxidation of the coke formed from PMMA degradation. When the protective barrier provided by the LDH sheets had disappeared (T > 275 °C), there was a new increase of the intensity of the coke G line, which coincided with the increase of the PMMA mass loss. At this stage, no change of copper oxidation was readily observed. Finally, at higher temperature, when Cu2O was reduced to Cu(0), a new decrease of the coke G line intensity occurred, again evidencing coupling with the oxidation of coke to CO2. In conclusion, the monitoring of Raman line intensity together with Cu speciation clearly demonstrated that the decreased char deposit induced by successive Cu reduction, coupled with destabilization of the LDH sheets, led to the nanocomposites containing Cu-based LDH presenting the lowest thermal stabilities (Table 1). These results revealed that redox reactions were associated with the oxidative coking and subsequent combustion, in partial agreement with the proposed catalytic role played by copper redox processes during char formation.17
Conclusions
In the present work, complementary X-ray absorption and scattering techniques, together with multivariate data analysis, enabled correlation between the thermal transformations (at long and short range order) of layered nanofillers of the LDH family and the mechanisms of decomposition/stabilization of the PMMA matrix.The mechanism of the gas diffusion barrier provided by the layered nanofiller was effectively demonstrated from the speciation profiles of Zn atoms in the layer of LDH of decreasing thermal stability (ZnCuAl(OH)6·nH2O < ZnCuAl0.5Fe0.5(OH)6·nH2O < Zn2Al(OH)6·nH2O < Zn2Al0.75Fe0.25(OH)6·nH2O), together with the temperature upshifts (compared to ZnAl(OH)6·nH2O; ΔT0.5 = 27, 30, 48, and 51 °C, respectively) of the main PMMA mass loss steps for these nanocomposites.
The contribution of the radical trapping mechanism could be confirmed by the coupled reaction between the 3d cations present in the LDH edifice and the PMMA, observed during thermal decomposition of the nanocomposites. This synergetic contribution involving the reversible reduction of Fe(III) to Fe(II), induced by the scavenging of PMMA radicals and the stabilization of layered edifice by charge compensation ensured by Fe(II) when Zn(II) are exuded from the LDH, was evidenced by independent time-resolved techniques, including superpositioning of the thermogravimetric curve and the evolution of the position of the rising K-edge of iron.
The radical trapping mechanism was only effective in the nanoconfined environment provided by the LDH layers. This finding indicated that the LDH layered edifice acted to restrict the movement of the PMMA chains, hindering radical propagation and increasing the probability of radical extinction.
The potential for char formation was unambiguously related to the thermal stability of the LDH and to the presence of oxidative species. On the one hand, higher thermal stability of the LDH lamellar structure was associated with a higher amount of char, demonstrating that the LDH also acted as a diffusion barrier to the efflux and evaporation of depolymerized species, which favoured charring. On the other hand, when redox reactions involving 3d cations occurred in a non-confined medium, such as the PMMA–ZCA nanocomposite, the coke was combusted and char deposition was minimized, hence contributing to decrease PMMA thermal stability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank SOLEIL for providing access to the SAMBA and SWING beamlines, and Dr Florian Meneau for assistance during the SAXS experiments. This work was partially supported by the Brazilian National Council for Scientific and Technological Development (CNPq/PVE, grant #401679/2013-6) and the São Paulo State Research Support Foundation (FAPESP, grant #2008/05475-9).References
- L. An, Y. Pan, X. Shen, H. Lu and Y. Yang, J. Mater. Chem., 2008, 18, 4928 RSC.
- M. Kotal and A. K. Bhowmick, Prog. Polym. Sci., 2015, 51, 127 CrossRef CAS.
- M. Naffakh, A. M. Díez-Pascual, C. Marco, G. J. Ellis and M. A. Gómez-Fatou, Prog. Polym. Sci., 2013, 38, 1163 CrossRef CAS.
- G. Choudalakis and A. D. Gotsis, Eur. Polym. J., 2009, 45, 967 CrossRef CAS.
- L. Wang, X. He and C. A. Wilkie, Materials, 2010, 3, 4580 CrossRef CAS PubMed.
- F. Leroux and J. Besse, Chem. Mater., 2001, 13, 3507 CrossRef CAS.
- M. Zanetti, G. Camino, D. Canavese, A. B. Morgan, F. L. Lamelas and C. A. Wilkie, Chem. Mater., 2002, 14, 189 CrossRef CAS.
- J. M. Yeh, S. J. Liou, C. Y. Lai and P. C. Wu, Chem. Mater., 2001, 13, 1131 CrossRef CAS.
- J. Choi, J. Harcup, A. F. Yee, Q. Zhu and R. M. Laine, J. Am. Chem. Soc., 2001, 123, 11420 CrossRef CAS PubMed.
- J. P. DeRocher, T. Gettelfinger, J. Wang, E. E. Nuxoll and E. L. Cussler, J. Membr. Sci., 2005, 254, 21 CrossRef CAS.
- K. Chen, C. A. Wilkie and S. Vyazovkin, J. Phys. Chem. B, 2007, 111, 12685 CrossRef CAS PubMed.
- N. S. Allen, M. J. Harrison, M. Ledward and G. W. Follows, Polym. Degrad. Stab., 1989, 23, 165 CrossRef CAS.
- Y. Cai, F. Huang, Q. Wei, L. Song, Y. Hu, Y. Ye, Y. Xu and W. Gao, Polym. Degrad. Stab., 2008, 93, 2180 CrossRef CAS.
- J. Zhu, F. M. Uhl, A. B. Morgan and C. A. Wilkie, Chem. Mater., 2001, 13, 4649 CrossRef CAS.
- E. Kandare, D. Hall, D. D. Jiang and J. M. Hossenlopp, in ACS Symposium Series 922, Fire and Polymers IV: Materials and Concepts for Hazard Prevention, ed. Wilkie C. A. and Nelson G. L., American Chemical Society, Washington, DC, 2006, p. 131 Search PubMed.
- E. Kandare, H. Deng, D. Wang and J. M. Hossenlopp, Polym. Adv. Technol., 2006, 17, 312 CrossRef CAS.
- E. Kandare, G. Chigwada, D. Wang, C. A. Wilkie and J. M. Hossenlopp, Polym. Degrad. Stab., 2006, 91, 1781 CrossRef CAS.
- H. W. P. Carvalho, C. V. Santilli, V. Briois and S. H. Pulcinelli, RSC Adv., 2013, 3, 22830 RSC.
- H. W. P. Carvalho, S. H. Pulcinelli, C. V. Santilli, F. Leroux, F. Meneau and V. Briois, Chem. Mater., 2013, 25, 2855 CrossRef CAS.
- A. Laachachi, E. Leroy, M. Cochez, M. Ferriol and J. M. Lopez Cuesta, Polym. Degrad. Stab., 2005, 89, 344 CrossRef CAS.
- J. Hausner, B. Fischer, M. Stoter, A. Edenharter, J. Schmid, R. Kunz, S. Rosenfeldt, V. Altstadt and J. Breu, Polym. Degrad. Stab., 2016, 128, 141 CrossRef CAS.
- E. Fonda, A. Rochet, M. Ribbens, L. Barthe, S. Belin and V. Briois, J. Synchrotron Radiat., 2012, 19, 417 CrossRef CAS PubMed.
- C. La Fontaine, L. Barthe, A. Rochet and V. Briois, Catal. Today, 2013, 205, 148 CrossRef CAS.
- A. de Juan and R. Tauler, Anal. Chim. Acta, 2003, 500, 195 CrossRef CAS.
- A. de Juan, J. Jaumot and R. Tauler, Anal. Methods, 2014, 6, 4964 RSC.
- C. Ruckebusch and L. Blanchet, Anal. Chim. Acta, 2013, 765, 28 CrossRef CAS PubMed.
- L. E. Manring, Macromolecules, 1989, 22, 2673 CrossRef CAS.
- J. D. Petterson, S. Vyazovkin and C. A. Wight, J. Phys. Chem. B, 1999, 103, 8087 CrossRef.
- C. Bao, H. Zhang, C. A. Wilkie, S. Bi, X. T. Tang, J. Wu and J. Yang, Carbon, 2016, 107, 774 CrossRef CAS.
- T. Hirata, T. Kashiwagi and J. E. Brown, Macromolecules, 1985, 18, 1410 CrossRef CAS.
- S. Mahamulkar, K. Yin, P. K. Agrawal, R. J. Davis, C. W. Jones, A. Malek and H. Shibata, Ind. Eng. Chem. Res., 2016, 55, 9760 CrossRef CAS.
- H. W. Chen, C. Y. Wang, C. H. Yu, L. T. Tseng and P. H. Liao, Catal. Today, 2004, 97, 173 CrossRef CAS.
- X. Yu, F. Zhang and W. Chu, RSC Adv., 2016, 6, 70537 RSC.
- M. A. Drezdzon, Inorg. Chem., 1988, 27, 4628 CrossRef CAS.
- H. Roussel, V. Briois, E. Elkaim, A. de Roy and J. P. Besse, J. Phys. Chem. B, 2000, 104, 5915 CrossRef CAS.
- J. Y. Kim, J. A. Rodriguez, J. C. Hanson, A. I. Frenkel and P. L. Lee, J. Am. Chem. Soc., 2003, 125, 10684 CrossRef CAS PubMed.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Poschl, Carbon, 2005, 43, 1731 CrossRef CAS.
- W. H. Cassinelli, L. Martins, A. R. Passos, S. H. Pulcinelli, C. V. Santilli, A. Rochet and V. Briois, Catal. Today, 2014, 229, 114 CrossRef CAS.
- A. Voronov, A. Urakawa, W. Van Beek, N. E. Tsakoumis, H. Emerich and M. Rønning, Anal. Chim. Acta, 2014, 840, 20 CrossRef CAS PubMed.
- A. Rochet, B. Baubet, V. Moizan, C. Pichon and V. Briois, C. R. Chim., 2016, 19, 1337 CrossRef CAS.
- R. Revel, D. Bazin, E. Elkaim, Y. Kihn and H. Dexpert, J. Phys. Chem. B, 2000, 104, 9828 CrossRef CAS.
- K. Yano, A. Usuki, A. Okada, T. Kurauchi and O. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2493 CrossRef CAS.
- K. Okudera, A. Yoshiasa, K. I. Murai, M. Okube, T. Takeda and S. Kikkawa, J. Mineral. Petrol. Sci., 2012, 107, 127 CrossRef.
- X. H. Li, L. Chen, Q. L. Li, J. J. Zhang, Z. Su, X. Zhang, K. Zheng and X. Y. Tian, RSC Adv., 2016, 6, 101941 RSC.
- T. Stimpfling and F. Leroux, Chem. Mater., 2010, 22, 974 CrossRef CAS.
- A. R. Horrocks, Polym. Degrad. Stab., 1996, 54, 143 CrossRef CAS.
- C. A. Wilkie and A. B. Morgan, in Fire Retardance of Polymeric Materials, Taylor and Francis Group, New York, 2nd edn, 2010 Search PubMed.
- T. Agag and T. Takeichi, Polymer, 2000, 41, 7083 CrossRef CAS.
- A. Leszczyńska, J. Njuguna, K. Pielichowski and J. R. Banerjee, Thermochim. Acta, 2007, 453, 75 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: LDH basal distance measured by WAXS (Table S1); MCR-ALS analysis of time-resolved XAS data; scree plots associated with the XAS data (Fig. S0); TG and DSC plots (Fig. S1 & S2); time-resolved WAXS (Fig. S3); XANES and experimental and fitted EXAFS spectra (Fig. S4); experimental and MCR-ALS simulated XANES spectra (S5, S9 & S11); time-resolved Quick-EXAFS (Fig. S6–S8); fitted EXAFS spectra (S10). See DOI: 10.1039/c8ra07611a |
| ‡ Present address: Centro de Energia Nuclear na Agricultura, Universidade de São Paulo, P.O. Box 96, 13400-970, Piracicaba, SP, Brazil. |
| This journal is © The Royal Society of Chemistry 2018 |