
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic synthesis and self-assembly of glycolipids: robust self-healing and wound closure performance of assembled soft materials†
Yadavali Siva Prasada,
Balasubramani Sarithaa,
Ayyapillai Tamizhanbana,
Krishnamoorthy Lalithaa,
Sakthivel Kabilana,
C. Uma Maheswari a,
Vellaisamy Sridharanb and
Subbiah Nagarajan*ac
a,
Vellaisamy Sridharanb and
Subbiah Nagarajan*ac
aDepartment of Chemistry, School of Chemical and Biotechnology, SASTRA Deemed University, Thanjavur – 613401, Tamil Nadu, India
bDepartment of Chemistry and Chemical Sciences, Central University of Jammu, Rahya-Suchani (Bagla), District-Samba, Jammu-181143, Jammu and Kashmir, India
cDepartment of Chemistry, National Institute of Technology Warangal, Warangal-506004, Telangana, India. E-mail: snagarajan@nitw.ac.in; snrajannpt@yahoo.co.in; Tel: +91-9940430715
First published on 5th November 2018
Abstract
In developing countries, wounds are a major health concern and pose a significant problem. Hence, the development of new materials that can act as scaffolds for in situ tissue regeneration and regrowth is necessary. In this report, we present a new class of injectable oleogel and composite gel derived from glycolipids that provide reversible interlinked 3D fiberous network architecture for effective wound closure by tissue regrowth and regeneration. Glycolipids were derived from α-chloralose and various vinyl esters using Novozyme 435, an immobilized lipase B from Candida antarctica as a catalyst, in good yield. These glycolipids undergo spontaneous self-assembly in paraffin oil to form an oleogel, in which curcumin was successfully incorporated to generate a composite gel. Morphological analysis of the oleogel and composite gel clearly revealed the formation of a 3D fiberous network. Rheological investigation revealed the thermal and mechanical processability of the oleogel and composite gel under various experimental conditions. Interestingly, the developed injectable oleogel and composite gel are able to accelerate the wound healing process by regulating the overlapping phases of inflammation, cell proliferation and extracellular matrix remodelling. Since chloralose displays anesthetic properties, this study will establish a new strategy to develop anesthetic wound healing oleogels in the future.
Introduction
Self-assembled supramolecular materials are unambiguously important components in everyday life and continue to be of great intellectual and commercial importance.1,2 In particular, assembled materials comprising either small molecules or polymer-based systems have applications in the fields of supramolecular device fabrication, electronics and biomedicine.3,4 Indeed, numerous natural processes involve the use of the robust molecular aggregation phenomenon to perform certain tasks in response to the environment.5 In the past two decades, extensive effort has been devoted towards the evolution of molecular self-assembly utilizing H-bonding, π–π stacking, van der Waals forces, anion or cation-π interactions and charge transfer interactions, resulting in the generation of a vast number of functional materials.6–8 In this context, molecular gels are a class of assembled materials gaining remarkable interest owing to their spontaneous formation of fibers, sheets, ribbons, helices, tubules, micelles, vesicles, etc.9–11 The intrinsic molecular features of low molecular-weight gelators (LMWGs) trigger the molecular level hierarchical supramolecular arrangement via non-covalent crosslinking and physical entanglement. Consequently, such materials display a 3D network structure where solvent molecules are entrapped, resulting in good viscoelastic gel materials.12–15 It is important to note that a large library of LMWGs derived from natural and synthetic compounds for various applications have been reported in the literature.16–20 However, new soft materials could be developed by intelligent design of small molecules and proper understanding of complex self-assembly mechanism. Thus, the present investigation unveils the synthesis and molecular self-assembly of functional glycolipids and their utility in wound closure. Carbohydrates are fundamental and important constituents in biological processes;21 examples include ribose and deoxyribose in RNA and DNA, glycogen in animals and cellulose in plants. For example, discoside, a natural glycolipid composed of acylated sugar unit, was isolated from a marine sponge Discodermia dissoluta.22,23 The pool of chiral carbons and multiple hydroxyl groups present in the sugar or its derivatives are some of the interesting features that have been extensively utilized for the generation of complex structured materials.24,25 Furthermore, these complex structures frequently offer diverse functionalities, including cell–cell communication, immune response, embryogenesis and blood coagulation.26 On account of renewability, biocompatibility, biodegradability, non-toxicity, economic viability and structural diversity, several research groups have established synthesis strategies for glycolipid production and studied their self-assembly.27,28 Recently, Bhattacharya et al. have reviewed the molecular self-assembly and diverse application of glycolipids.29 Nevertheless, the use of harsh reaction conditions, metal catalysts, formation of undesired products and intense purification steps limit the commercial value of glycolipids.30–32 The present study details a facile method for the generation of glycolipids from inexpensive commercially available raw materials derived from renewable resources using an enzyme as a catalyst. Generally, enzyme-triggered reactions are desirable for the precise synthesis of glycolipids under mild reaction conditions, with control in stereo- and regioselectivities, structure, substrate and functional groups.33–36 Furthermore, enzymatic reactions display high catalytic efficiency and simple workup process either by filtration or centrifugation.37–39 For the synthesis of acylated sugar, immobilized form of Candida antarctica lipase B (CALB),40 Novozyme 435® is the best option because of its potential catalytic activity towards polyol ester of fatty acid synthesis, reusability, thermal stability and stable catalytic performance under various conditions.41–43 Recently, John et al. have briefly discussed the biocatalytic synthesis of amphiphilic systems, which can create a variety of self-assembled soft materials with value added properties.44 Ulrich et al. highlighted the significance of Novozyme 435-catalyzed synthesis of glycolipids in deep eutetic solvents, and in view of green principles and technology, this method is promising for developing functional materials.35 For synthesising anesthetic glycolipids for wound healing studies, we have selected α-chloralose, a stable long lasting anaesthetic used for non-surgical purpose.45 Moreover, it produces minimal cardiovascular and respiratory system depression.46 In this study, we present a green protocol for the synthesis of anesthetic glycolipids with versatile hydrophilic and hydrophobic character using transesterification. Furthermore, molecular self-assembly of these glycolipids resulted in the formation of gels, in which natural hydrophobic drug, curcumin,47 has been encapsulated and used for anestethic wound healing studies.Results and discussion
To develop a chloralose-based anesthetic supramolecular material, a complementary hydrophobic unit has to be included, so that the supramolecule can attain amphiphilic character responsible for self-assembly.48 In this regard, α-chloralose, also referred to as monoglucochloralose or chloralosane,45,46 was selected as the functional hydrophilic core, while vinyl esters of fatty acids was chosen as the complementary hydrophobic part. Enzyme catalyzed transesterification of chloralose with various vinyl esters of fatty acids resulted in the formation of glycolipids 3a–e with good yields (Scheme 1). The as-synthesized glycolipids were characterized by NMR and mass spectral analysis. The gelation ability of these glycolipids was tested in water, different organic and binary solvents, and vegetable and mineral oils, and the critical gelation concentration is shown in Table S1.†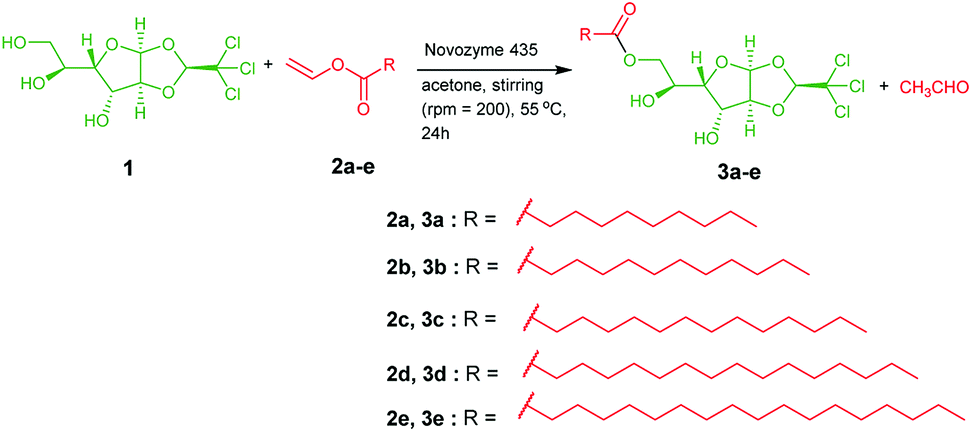 | ||
| Scheme 1 Synthesis of anesthetic glycolipids 3a–e. | ||
These glycolipids were unable to form gels in most of the organic solvents tested. However, glycolipids 3d and 3e were able to form gels in eucalyptus oil, DMSO + water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) and paraffin oil (Fig. S1 and S2†). In addition, glycolipids 3a–e did not form gels in acidic, basic and neutral pH solutions, and displayed stability even at elevated temperature. To our delight, glycolipid 3d formed a gel in paraffin oil with CGC of 1% (w/v), which was referred to as oleogel, via molecular self-assembly guided by intermolecular non-covalent interactions (Fig. S3 and S4†).
:4) and paraffin oil (Fig. S1 and S2†). In addition, glycolipids 3a–e did not form gels in acidic, basic and neutral pH solutions, and displayed stability even at elevated temperature. To our delight, glycolipid 3d formed a gel in paraffin oil with CGC of 1% (w/v), which was referred to as oleogel, via molecular self-assembly guided by intermolecular non-covalent interactions (Fig. S3 and S4†).
In 1917, Gray reported the use of liquid paraffin in closure of war wounds.49 With the increase in applications of liquid paraffin in pharmaceutical, textile, cosmetics and food and beverage industry,50,51 research into the development of paraffin-based advanced healthcare materials has increased. The primary objective of this study is to investigate the use of oleogel in wound closures. In an oleogel matrix, any of the hydrophobic drugs can be encapsulated and used for developing healthcare materials. A recent report by M. Heng, clinical Professor of Dermatology, emphasized the effectiveness of topical curcumin gel in treating skin problems, such as burns and scalds.52 Based on literature reports, in the present investigation, in addition to oleogel, we have developed a composite gel by encapsulating curcumin (a natural drug) into the oleogel matrix. Before studying the wound closure behavior of the oleogel and the composite gel, we investigated the morphology and rheology of these gels. The morphology of the oleogel was investigated using optical microscopy and HRTEM. The existence of entangled fibrillar network of 200–500 nm thickness has been observed in the oleogel. However, the incorporation of curcumin influenced the morphology of the composite gel, resulting in the formation of fibers with the width of 200–400 nm (Fig. 1 and S5†).
 | ||
| Fig. 1 Optical microscopy images of (a and b) oleogel and (c and d) composite gel. HRTEM images of (e and f) oleogel and (g and h) composite gel. | ||
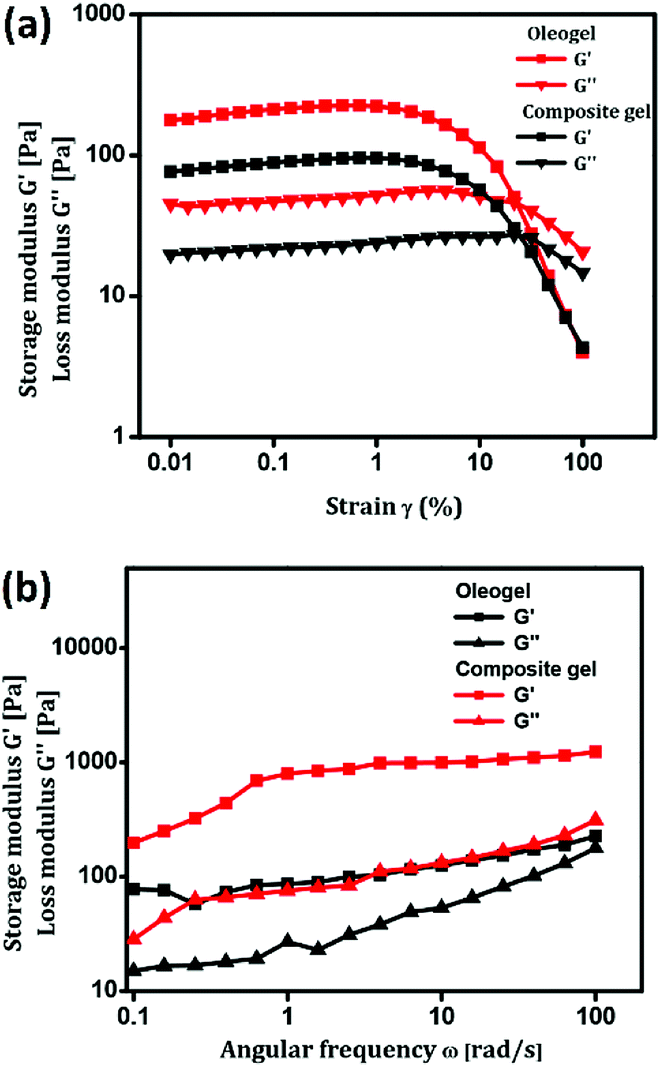
Mechanical and self-healing behaviours of the oleogel and composite gel were confirmed by rheological measurements. Generally, mechanical characteristics of a soft material are dependent on the hierarchical order of self-assembled structures.53,54 Rigidity and flow characteristics of both gels were identified using oscillatory rheology by measuring dynamic shear storage modulus (G′), which refers to the ability of a deformed soft-material to restore back to its original state, and loss modulus (G′′), which represents the flow behaviour under applied stress.55 For highly ordered soft materials, such as gels, G′ should be greater than G′′, and for deformed gels, G′ should be less than G′′.56 Dynamic frequency experiments of oleogel and composite gel at a constant strain 0.1% displayed G′ and G′′ values of 15 and 80 (oleogel), and 30 and 230 Pa (composite gel), respectively. In a frequency sweep across the entire range of analysis, G′ is greater than G′′, and the average difference is found to be 15 Pa for oleogel and 200 Pa for composite gel. This suggested that the incorporation of curcumin in oleogel increased the viscoelastic character of the gel by increasing the energy of dissipation (Fig. 2). In dynamic strain amplitude sweep experiment, upon the steady increase in strain on the oleogel and composite gel, G′ and G′′ remained constant up to a critical strain level ((γc) = 23%), following which a linear declining trend was observed (Fig. 2). Gels displaying injectable behaviour and healing from mechanical damage are often used for constructing advanced bio- and electronic materials. Mostly, macromolecules displaying viscoelastic behaviour undergo irreversible gel-to-sol transition via the collapse of the assembled structure. However, self-assembled gel systems derived from small molecules undergo partial change in physical interactions under the influence of the surroundings to the extent that the deformed system recovers back to its original form following an exact reversible path; this is often called as the thixotropic property. With the proper selection of gelator and solvent, it might be possible to fine-tune the physiochemical behaviour of gels. Discrete experiments were performed to identify the mechanical and thermoreversible behaviours of the oleogel and composite gel. An oscillatory temperature ramp was conducted between 23 and 45 °C at a constant strain of 0.1% to identify the critical gelation temperature and thermoprocessability.
 | ||
| Fig. 2 (a & b) Strain amplitude and angular frequency dependence of G′ and G′′ of injectable oleogel and composite gel. | ||
As shown in Fig. 3, with the increase in temperature from 23 °C to 45 °C with respect to time, G′ was found to be greater than G′′ throughout the entire time period without any decline in value, suggesting the thermal stability of oleogel. In addition, the oleogel did not display any change in viscoelastic behavior throughout the entire range of temperature under investigation (Fig. 3a). However, the composite gel displayed a steady decline in G′ and G′′ without crossing the trend lines, revealing the thermo-reversibility and processability of the gel under physiological conditions (Fig. 3b). Injectable nature of the oleogel and composite gel could be easily identified from continuous step strain measurements.57–59 Fig. 3c and d represent the strain sweep of the oleogel and composite gel, respectively. To access the thixotropic behavior of the gels, a strain sweep from 0.01% to 100% was performed for more than 3 cycles. An interesting progression in viscoelastic behavior was observed with respect to the strain sweep. On applying a higher strain of 100%, the gels behaved like fluids, whereas a release in strain resulted in the reorganization of molecular arrangement to form the gels (Fig. 3c and d). Rheological investigation revealed the suitability of the oleogel and composite gel for topical applications, particularly for the treatment of wounds, burn and scalds.
 | ||
| Fig. 3 (a & b) Temperature dependence of G′ and G′′ of oleogel and composite gel. (c & d) Thixotropy-loop test via continuous step-strain measurements of oleogel and composite gel. | ||
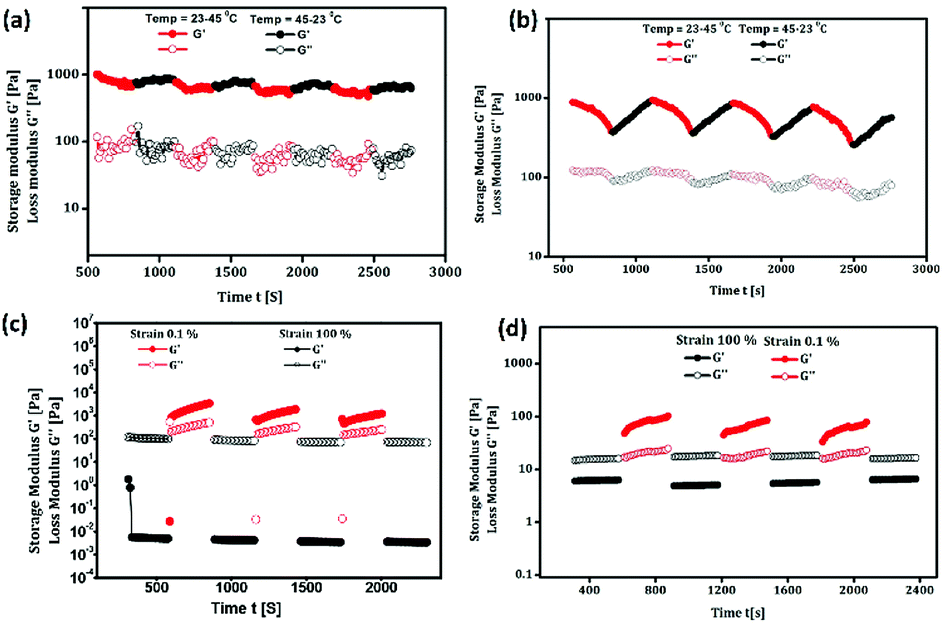
In particular, wound healing therapy remains challenging in the medical field because of its dynamic, complex and interactive process involving overlapping phases of inflammation, cell proliferation and extracellular matrix remodelling.60–62 The antecedent to optimal wound healing is to minimize tissue damage and provide sufficient tissue perfusion to restore the anatomical continuity.63,64 Owing to the inflation in lifestyle diseases, there is a great demand for the development of a multifunctional therapeutic agent to improve the healing of chronic impaired wounds.65,66 However, using gels as scaffold biomaterials for wound healing is considered to be a promising tool.67 Peppas et al. have discussed the use of hydrogels in the pharmaceutical and other domains and pointed out the potential of oleogels in biomedical fields.68 Recently, oleogels were identified as excellent candidates for biomedical applications because of their good physical, chemical and mechanical stability.69–71 Keeping all these aspects in mind, we intended to further explore the use of oleogel and composite gel in tissue regeneration, exclusively in wound exudates, fibroblast proliferation, secretion of ground substance, keratinocyte migration and neoangiogenesis, which are necessary for complete epithelialization and healing of the wound. The wound is protected from microbial infection on covering with the oleogels and also, bioactive molecules can be transported to the wound site.72 The enhanced wound healing potential of sunflower oil-based oleogel was reported earlier.73,74 Tropical gels prepared by using natural oils, such as black cumin and flax seed oil, displayed significant wound healing properties with enhanced reepithelization, which are comparable to those of standard silver sulfadiazine.75,76 European Medicines Agency (EMA) approved an oleogel-containing birch bark extract for the treatment of mid-dermal wounds.77 By considering the various reports discussed above, in the present study, the activity of the oleogel and composite gel in wound healing was evaluated in a full thickness (2 cm2) wound by applying 0.1 mL of paraffin oil as the control and 0.1 mL of oleogel 1 (1% w/v), oleogel 2 (2% w/v) and composite gel (1% w/v) as test samples. The percentage of wound closure in 6, 12, 18 and 21 days on treatment with 0.1 mL of oleogels and the composite gel is shown in Table 1 (Fig. S6†). Since the untreated group did not show progressive wound closure, in the present discussion, the paraffin oil-treated group is considered as the control. As shown in Fig. 4 and S6,† oleogels and the composite gel-treated rats displayed an increase in percentage of wound contraction to 96% and 95% on the 21st day, respectively. Oleogel-treated rats showed dose dependent wound contraction from day 6 onwards, indicating progressive wound closure. Furthermore, on day 12, more than 80% of the tissue structures were restored in the oleogel- and composite gel-treated rats (Table 1).
| Vehicles | Percentage of wound healing (mean + SEM) | |||
|---|---|---|---|---|
| Day 6 | Day 12 | Day 18 | Day 21 | |
| a Percentage value of mean ± SEM of each group. Significance was at P < 0.05 (*), P < 0.01 (**), or P < 0.001(***). Comparison of treated groups with the control group. The results were analysed statistically using one-way analysis of variance (ANOVA) followed by Dunnett's test for multiple comparisons. | ||||
| Control (paraffin oil) | 12.29 ± 2.06 | 58.82 ± 2.95 | 76.37 ± 166 | 82.83 ± 1.4 |
| Oleogel 1 | 18.07 ± 2.55 | 74.31 ± 4.7* | 90.34 ± 1.24* | 93.50 ± 0.99* |
| Oleogel 2 | 30.77 ± 3.67** | 83.71 ± 1.21*** | 94.27 ± 0.75** | 96.00 ± 0.68** |
| Composite gel | 24.69 ± 2.51* | 80.98 ± 1.41*** | 92.81 ± 0.61** | 95.33 ± 0.61* |
 | ||
| Fig. 4 The gross appearance of wound healing on day 18 (a–d) and 21 (e–h). (a and e) Wounds treated with 0.1 mL of paraffin oil (control); (b and f) wounds treated with 0.1 mL of oleogel (1% w/v); (c and g) 0.1 mL oleogel (2% w/v)-treated rats showing almost complete wound closure, and (d and h) 0.1 mL composite gel (1% w/v)-treated showing complete wound closure. Progress of wound healing with exact scale bar is given in Fig. S7a–d.† | ||
Visual observations of the wound healing process with exact scale bar are given in Fig. S7.† Interestingly, curcumin present in the composite gel promotes wound closure more than the oleogel 1 by forming a well-organized self-assembled structure, as identified by morphological and rheological studies. Furthermore, this self-assembly-facilitated wound closure has been studied in the aspects of collagen expression and stabilization, proliferation of fibroblasts and lipid peroxidation.
Generally, wound contracture involves migration and proliferation of endothelial cells.78 Even though several binding proteins are responsible for the biological activities of the skin tissue, collagen is considered as the main extracellular protein in the granulation tissue and plays an important role in haemostasis and epithelisation at the later phase of wound healing.79 The use of oleogels 1 and 2 and the composite gel on the wound surface of rats led to an increase in hydroxyproline content at 6.8 ± 0.7, 8.1 ± 1.8 and 12.3 ± 2.9 mg/100 mg, respectively, when compared with that in control rats (5.3 ± 0.2 mg/100 mg). The resultant enhancement in the rates of wound contraction in oleogel- and composite gel-treated groups is due to the rapid formation of granulation tissues. Moreover, increased synthesis of protein around the wounded area with the assistance of the assembled fibrillar architecture provides strength and integrity to the tissue matrix (Table 2, Fig. S8†). The self-assembled architecture displayed by oleogels and the composite gel increases the content of hydroxyproline in the granulation tissue, which directly induces higher collagen content, and its high turnover resulted in rapid wound healing with greater tissue strength. The presence of curcumin in oleogel, i.e. the composite gel, and its corresponding self-assembled architecture rapidly increases the concentration of hydroxyproline in the granulation tissue. However, hexosamine, a component of glycosaminoglycan, strengthens the collagen fibres via assembly through molecular interactions.78,79 In addition to hydroxyproline, the increased levels of hexosamine in oleogel (1.19 ± 0.22)- and composite gel (1.27 ± 0.05)-treated groups stabilized collagen via the assembly process and facilitated the production of new extracellular matrix (Table 2, Fig. S8†). As evidenced from histopathological data, the enhanced production of hydroxyproline and hexosamine in oleogel- and composite gel-treated rats induces rapid wound healing and renders strength to the repaired tissue.
| Hydroxy proline (mg/100 mg) | Hexosamine (mg/100 mg) | Lipid peroxide (mg/100 mg) | Vitamin C (mg/100 mL) | |
|---|---|---|---|---|
| a Values noted as mean ± SE of each group. P < 0.05 (*), P < 0.01 (**), P < 0.001(***). Comparison of treated groups with control group. Results were analysed statistically using one-way analysis of variance (ANOVA) followed by Dunnett's test for multiple comparisons. | ||||
| Control (paraffin oil) | 5.3 ± 0.2 | 0.39 ± 0.22 | 9.7 ± 1.3 | 5.9 ± 0.7 |
| Oleogel (1% w/v) | 6.8 ± 0.7 | 0.72 ± 0.23 | 6.8 ± 1.7 | 7.9 ± 0.6* |
| Oleogel (2% w/v) | 8.1 ± 1.8** | 1.19 ± 0.22*** | 4.2 ± 1.9 ** | 8.4 ± 1.0** |
| Composite gel (1% w/v) | 12.3 ± 2.9*** | 1.27 ± 0.05*** | 4.7 ± 2 ** | 7.5 ± 0.1** |
Ascorbic acid, another necessary component in wound healing, facilitates the activation of dermal fibroblasts in tissue regeneration process. In addition, the antioxidant property displayed by ascorbic acid is responsible for diminution of macrophages, increased proliferation of fibroblasts, blood vessel repair and immunological defence.72–79 In the present study, elevated content of ascorbic acid in the oleogel 2-treated rats was observed when compared with that of oleogel 1-treated, composite gel-treated, control and untreated rats. It is important to mention that the presence of curcumin in the composite gel has less effect on ascorbic acid production in the wound area. The enhanced level of ascorbic acid by oleogel facilitates collagen deposition, increases tensile strength of the tissue and prevents cell damage caused by free radicals during the wound closure process. Free-radical oxidation of polyunsaturated fatty acids occurs in the biological system, and is referred to as “lipid peroxidation”.80 Hydroxyl radicals produced in the wound area stimulate lipid peroxidation, which affects the functions of endothelial cells, collagen and fibroblast metabolism and induces cell damage.80 Generally, superoxides generated on the wound surface are converted to hydrogen peroxide (H2O2) by superoxide dismutase (SOD), which is further converted to water by glutathione peroxidase (GPx) or lysosomes present in mitochondria.81 Topical application of oleogel 2 and the composite gel enhanced the activities of SOD, lysosomes and GPx, thus precluding the harmful consequences of free radicals generated in rats at the wound site. As indicated in Table 2 and Fig. S8,† decreased levels of lipid peroxide in oleogel 2- and composite gel-treated rats at 4.2 ± 1.9 and 4.7 ± 2, respectively, revealed the increased viability of collagen fibrils and activities of enzymes preventing cell damage. Inhibition of lipid peroxidation by the self-assembled structures of oleogel and composite gel minimizes the defect in Vascular Endothelial Growth Factor (VEGF) expression82 and favours the diabetes-induced tissue closure.
Furthermore, histopathological examination82 of haematoxylin and eosin stained sections of various treatment groups displayed different wound healing rates (Fig. 5). Histological analysis of oleogels and composite gel treated wounds displayed a high degree of collagen synthesis, fibroblast proliferation and neovascularization. The use of oleogel 2 and composite gel led to effective tissue regeneration when compared with other groups without any edema and congestion.
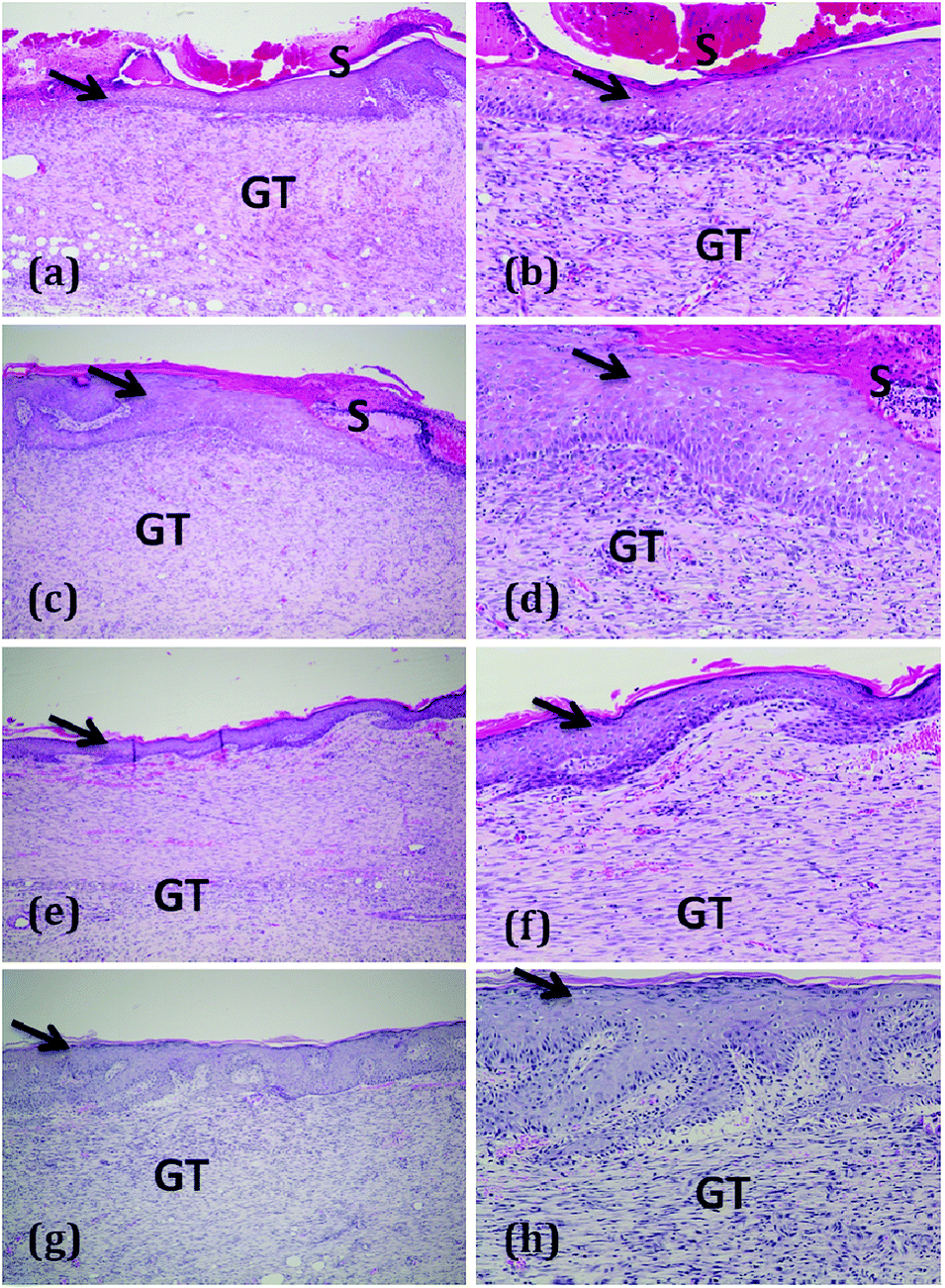 | ||
| Fig. 5 Haematoxylin and eosin (H & E) staining of sections of healed skin of experimental groups: (a and b) control group shows moderate epithelialization with early fibroblast phase; (c and d) skin section of oleogel 1 treated group shows granulation tissue with marked epithelialization and late fibroblast phase; (e and f) oleogel 2 treated group shows granulation tissue with complete epithelialization; and (g and h) composite gel group displays complete epithelialisation at 4× and 10× magnifications. Arrow – epidermis; D – dermis; S – scab; GT – granulation tissue. | ||
Conclusions
We synthesized a new class of glycolipids at good yields from α-chloralose and various fatty acid vinyl esters using Novozyme 435 as a catalyst. Supramolecular self-assembly of these glycolipids formed gels in highly hydrophilic solvents, DMSO–H2O mixture and hydrophobic oils, specifically in eucalyptus oil and paraffin oil, via H-bonding, π–π interaction and van der Waals forces. Interestingly, glycolipid 3d forms efficient oleogel in paraffin oil, which is routinely used as a chemical ingredient in medicines and cosmetics, including wound closure. A natural drug, curcumin, was successfully incorporated in the oleogel to generate a composite gel. The formation of supramolecular 3-D fibrous networks in oleogel and composite gel was identified by optical microscopy and HRTEM. Rheological studies demonstrated viscoelastic behaviour, thermoreversibility, thixotropicity and injectable nature of the oleogel and composite gel. As evidenced from the rheological studies, the incorporation of curcumin substantially meliorates the viscoelasticity of the oleogel by forming a more integrated architecture. We have successfully demonstrated the use of injectable oleogel and composite gel for wound closure by regulating the overlapping phases of inflammation, cell proliferation and extracellular matrix remodelling. Since α-chloralose alone or in combination with urethane displays long-lasting mild anesthesia with low cardiovascular and respiratory depression and intact autonomic reflexes, the oleogel and composite gel reported herein may establish a new tool to develop anesthetic wound healing gels in the future.Experimental section
Materials and methods
All essential chemicals, reagents and solvents used for the synthesis of glycolipids were purchased from Sigma Aldrich, Merck, TCI chemicals, Alfa aesar, SRL, and Avra chemicals. LR grade solvents were used for compound purification and AR grade solvents were used for the synthesis and gelation studies. Distilled solvents were used whenever necessary. Novozyme 435 was obtained from Novozymes A/S, Denmark as a gift sample for our research purpose. Progress of the reactions was monitored by thin-layer chromatography (TLC) on pre-coated silica gel plates purchased from Merck and visualized by UV detection using sulfuric acid spray or molecular iodine. Column chromatography was performed on silica gel (100–200 mesh) purchased from Avra chemicals, India.Characterization
1H and 13C NMR spectra were recorded on a Bruker Avance 300 MHz instrument in CDCl3, DMSO-d6 or a mixture of CDCl3 and DMSO-d6 at room temperature. Chemical shifts (δ) are reported in parts per million (ppm) with respect to internal standard TMS and coupling constants (J) are given in Hz. Proton multiplicity is assigned using the following abbreviations: singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m). High resolution MS analyses were performed on an Agilent 6520 Q-TOF instrument by dissolving the solid sample in either chloroform or methanol. SAXD measurements were performed on a BRUKER-binary V3 Diffractometer system.Synthesis of glycolipids 3a–e
To a solution of α-chloralose (1 mmol) in acetone (5 mL), vinyl ester (3 mmol) and Novozyme 435 (100 mg) were added. The reaction mixture was kept in an orbital shaker incubator at 55 °C and 300 rpm for 24 h. Progress of the reaction was monitored using TLC. After completion of the reaction, the reaction mixture was cooled to room temperature and filtered to remove Novozyme 435, which was washed well with acetone. The filtrate was concentrated using a rotary evaporator at reduced pressure, diluted with DCM and washed well with water. The organic layer was extracted and dried over Na2SO4 and concentrated to obtain a whitish crude solid. The crude residue was purified by silica gel column chromatography using hexane-ethyl acetate mixture as the eluent.Gelation studies
The gelation property of glycolipids was consistently studied in different solvents, such as water, toluene, xylene, DMF, DMSO, and cyclohexane and also with oils, including olive oil, eucalyptus oil, hazelnut oil, sesame oil, jojoba oil, linseed oil, paraffin oil, neem oil and castor oil. Gelation studies were performed by mixing a known quantity of gelator with a suitable amount of solvent in a sealed vial, and the entire content was heated until a homogeneous solution was formed. Through this procedure, the boiling point of the solvent becomes higher than that at standard atmospheric pressure. The hot solution was slowly allowed to cool to room temperature, and gelation was visually observed by the “test tube inversion” method. A gel sample was obtained that exhibited no gravitational flow in inverted tube and denoted as “G”. If the content remains as a solution at the end of the test, the sample is referred to as “S” (solution) and if the content is present as a precipitate, then the system is denoted as “P” (precipitation). If the gelator is not soluble even at the boiling point of the solvent, the system is called an insoluble system (I).Gel–sol melting temperature (Tg)
Gel melting temperature was determined by the test tube inversion method. Gels obtained via molecular self-assembly are thermally reversible. Upon heating the vial above the gelation temperature, the gel phase is transferred to solution phase and upon cooling, the solution could self-assemble to form a gel. A gel was prepared in a 5 mL glass vial following the standard protocol, and the vial was immersed in an oil-bath ‘upside down’ and slowly heated. The temperature at which the gel melted to solution was recorded as the gel melting temperature (Tg).Preparation of composite gel
To the thermally reversible oleogel (1% w/v) obtained via the self-assembly process, 1 mg of curcumin was added and the entire content was heated above Tg. The resulting solution was slowly allowed to cool to room temperature, and gelation was visually observed by inverting the vial. The addition of 2 mg of curcumin did not lead to the formation a composite gel; instead, the sample remained as a precipitate.Morphological analysis
Morphology of the oleogel and composite gel formed by the gelator was studied using Carl Zeiss AXIO Scope A1 fluorescent/phase contrast microscope. A small portion of the gel was mounted on a glass plate and its morphology was investigated under a Phase Contrast Microscope. Furthermore, both the oleogel and composite gel were dispersed in hexane using a sonicator, and the morphology of the gels was studied using a JEOL JEM 2100F field emission transmission electron microscope with the features of ultrahigh resolution and highest analytical performance.Rheological measurements
Flow behaviours of the oleogel and composite gel were identified using a stress-controlled rheometer (Anton Paar 302 rheometer) implemented with a steel-coated 25 mm diameter parallel-plate geometry. The gap between the two plates for rheological testing of the gel was fixed at 1 mm, and the experiments were performed at 23 °C. In order to investigate the viscoelastic behaviour of the gel, oscillation measurements were recorded at 23 °C. Initially, an amplitude sweep experiment was performed to identify the linear viscoelastic range. Then, the storage modulus G′ and the loss modulus G′′ were monitored as functions of frequency from 0.1 to 100 rad s−1. To identify the processability and stability of the gel, temperature ramp up (23–45 °C) and ramp down (45–23 °C) experiments were performed in cycles. Injectable nature of the gels and the recovery of their self-assembly under physiological conditions were identified using strain ramp experiments (100% strain and 0.1% strain were applied, alternatively).In vivo wound closure studies and ethical issues
Healthy Wistar Rattus norvegicus female rats (200–230 g), 8 to 9 weeks old, were procured from Central Animal Facility, SASTRA Deemed University, Thanjavur, Tamil Nadu, India. The rats were housed individually in standard laboratory environment for 7 days. During the course of the experiment, the rats were fed a standard pellet diet and water ad libitum. Experimental protocols used in this study were approved by the Institutional Animal Ethics Committee (IAEC), SASTRA Deemed University. Experiments were performed as per the guidelines of Committee for the Purpose of Control and Supervision of Experimental Animal (CPCSEA), New Delhi (Reg. no. 817/04/ac/CPCSEA), SASTRA Deemed University, Thanjavur, India, and its approval number is 336/SASTRA/IAEC/RPP.Experimentally inflicted wounds81
Healthy Wistar albino female rats were randomly segregated into four groups comprising six rats each and caged individually. The animals were anaesthetized with thiopentone sodium (60 mg kg−1, i.p.) prior to the wound creation. The rats were inflicted with excision wounds. Then, the dorsal fur of the rats was shaved and anticipated area of the wound created was marked. A full thickness excision wound was created by excising 200 mm2 areas of skin in length and 0.2 cm in depth from the dorsal region using a sharp surgical blade and pointed scissors.Topical application of gel
Wounds of Group 1 rats were treated with 0.1 mL paraffin oil, and this group served as the control. The wounds of Group 2, Group 3 and Group 4 rats were treated with 0.1 mL oleogel 1 (1% w/v), oleogel 2 (2% w/v) and composite gel (1% w/v), respectively. In this study, 0.1 mL of gel was applied on the wound surface once a day and the wound closure was monitored every day.Estimation of the rate of wound contraction
Progress of wound closure was monitored by tracking the wound area on a transparent graph sheet on alternate days for up to 21 days for all six animals in each group. A transparent graph sheet was laid over the wound and traced using a permanent marker. The area of wound size was studied using image j software. Its diameter was recorded and the percentage of reduction of wound size was calculated until the day of complete epithelialization. Standard and mean deviations were given in sq mm. Percentage of wound contraction was calculated using the initial area and the area under investigation as follows:| % of wound contraction = [(W1 − W0)/W1] × 100 |
On the 21st day post wounding, the rats were sacrificed, and blood and granulation tissue samples were collected for the estimation of free radicals, antioxidants, hexosamine and collagen tissue parameters.
Statistical analysis
All values were reported as mean ± SEM. Data obtained from the in vivo assay were assessed using Dunnett's Multiple Comparison Test and analysis of variance (ANOVA). A value of P < 0.05 was considered significant. Statistical analysis was performed using GraphPad Prism for Windows software.Histopathological examination
Wound tissue specimens from control and test groups were taken after complete healing of the excision wound. After usual processing, skin samples were cut with a thickness of 6 μm and stained with hematoxylin and eosin (H & E). Sections were qualitatively assessed under a light microscope and the results were recorded. Wound tissues collected were used for the estimation of hydroxy proline,83 hexosamine,84 and ascorbic acid as well as lipid peroxidation85 and antioxidant activity86 in the granulation tissue samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Department of Science and Technology (IFA11-CH-04 and #SB/FT/CS-024/2013), India and Board of Research in Nuclear Science (#37(1)/20/47/2014), Department of Atomic Energy, India. S. N. thanks National Institute of Technology, Warangal for RSM intramural research fund. We thank Novozymes A/S, Denmark for generous supply of Novozyme 435® for our research purpose. We also thank Dr C. Davidraj, Central Animal Facility, SASTRA Deemed University, Thanjavur for his help in analysing biochemical parameters.Notes and references
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- O. Ikkala and G. Ten Brinke, Chem. Commun., 2004, 2131–2137 RSC.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 104–116 CrossRef.
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671–679 CrossRef CAS PubMed.
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed., 1996, 35, 1154–1196 CrossRef.
- S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460–1494 CrossRef PubMed.
- L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382–2426 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- D. K. Smith, Molecular Gels – Nanostructured Soft Materials, in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, 2008, pp. 111–154 Search PubMed.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- P. Térech, in Molecular Gels: Materials with Self-Assembled Fibrillar Networks, 2006, pp. 275–324 Search PubMed.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- K. L. Caran, D. C. Lee and R. G. Weiss, in Soft Fibrillar Materials: Fabrication and Applications, 2013, pp. 1–75 Search PubMed.
- J. J. D. De Jong, B. L. Feringa and J. Van Esch, in Molecular Gels: Materials with Self-Assembled Fibrillar Networks, 2006, pp. 895–927 Search PubMed.
- J. de Jong, B. L. Feringa and J. van Esch, Responsive Molecular Gels, 2011, vol. 1 Search PubMed.
- A. Friggeri, K. J. C. Van Bommel and S. Shinkai, in Molecular Gels: Materials with Self-Assembled Fibrillar Networks, 2006, pp. 857–893 Search PubMed.
- J. Yan, B. S. Wong and L. Kang, in Soft Fibrillar Materials: Fabrication and Applications, 2013, pp. 129–162 Search PubMed.
- D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 279–293 RSC.
- K. Babooram and R. Narain, in Polymer Adhesion, Friction, and Lubrication, 2013, pp. 545–574 Search PubMed.
- L. Barbieri, V. Costantino, E. Fattorusso and A. Mangoni, J. Nat. Prod., 2005, 68, 1527–1530 CrossRef CAS PubMed.
- G. J. Florence, T. Aslam, G. J. Miller, G. D. S. Milne and S. J. Conway, Synlett, 2009, 3099–3102 CrossRef CAS.
- K. Lalitha, V. Sridharan, C. U. Maheswari, P. K. Vemula and S. Nagarajan, Chem. Commun., 2017, 53, 1538–1541 RSC.
- Y. S. Prasad, S. Miryala, K. Lalitha, K. Ranjitha, S. Barbhaiwala, V. Sridharan, C. U. Maheswari, C. S. Srinandan and S. Nagarajan, ACS Appl. Mater. Interfaces, 2017, 9, 40047–40058 CrossRef CAS PubMed.
- J. Luo and Z. K. Xu, in ACS Symposium Series, 2012, vol. 1120, pp. 253–275 Search PubMed.
- K. Lalitha, K. Muthusamy, Y. S. Prasad, P. K. Vemula and S. Nagarajan, Carbohydr. Res., 2015, 402, 158–171 CrossRef CAS PubMed.
- K. Muthusamy, V. Sridharan, C. U. Maheswari and S. Nagarajan, Green Chem., 2016, 18, 3722–3731 RSC.
- S. Bhattacharya and S. K. Samanta, Chem. Rev., 2016, 116, 11967–12028 CrossRef CAS PubMed.
- J. Xie and N. Bogliotti, Chem. Rev., 2014, 114, 7678–7739 CrossRef CAS PubMed.
- C. I. C. Crucho, P. Correia-da-Silva, K. T. Petrova and M. T. Barros, Carbohydr. Res., 2015, 402, 124–132 CrossRef CAS PubMed.
- L. Latxague, A. Gaubert and P. Barthélémy, Molecules, 2018, 23, 89 CrossRef PubMed.
- T. Fukuoka, T. Yanagihara, T. Imura, T. Morita, H. Sakai, M. Abe and D. Kitamoto, Carbohydr. Res., 2011, 346, 266–271 CrossRef CAS PubMed.
- M. Phnlein, C. Slomka, O. Kukharenko, T. Grtner, L. O. Wiemann, V. Sieber, C. Syldatk and R. Hausmann, Eur. J. Lipid Sci. Technol., 2014, 116, 423–428 CrossRef.
- M. Pöhnlein, J. Ulrich, F. Kirschhöfer, M. Nusser, C. Muhle-Goll, B. Kannengiesser, G. Brenner-Weiß, B. Luy, A. Liese, C. Syldatk and R. Hausmann, Eur. J. Lipid Sci. Technol., 2015, 117, 161–166 CrossRef.
- M. Pöhnlein, R. Hausmann, S. Lang and C. Syldatk, Eur. J. Lipid Sci. Technol., 2015, 117, 145–155 CrossRef.
- H. Yu, Y. Li, J. Zeng, V. Thon, D. M. Nguyen, T. Ly, H. Y. Kuang, A. Ngo and X. Chen, J. Org. Chem., 2016, 81, 10809–10824 CrossRef CAS PubMed.
- A. Sembayeva, B. Berhane and J. A. Carr, Tetrahedron, 2017, 73, 1873–1880 CrossRef CAS.
- F. L. Craven, J. Silva, M. D. Segarra-Maset, K. Huang, P. Both, J. E. Gough, S. L. Flitsch and S. J. Webb, Chem. Commun., 2018, 54, 1347–1350 RSC.
- A. Jahangiri, A. H. Møller, M. Danielsen, B. Madsen, B. Joernsgaard, S. Vaerbak, P. Adlercreutz and T. K. Dalsgaard, Food Chem., 2018, 268, 203–209 CrossRef CAS PubMed.
- C. Scheckermann, A. Schlotterbeck, M. Schmidt, V. Wray and S. Lang, Enzyme Microb. Technol., 1995, 17, 157–162 CrossRef CAS.
- R. T. Otto, H. Scheib, U. T. Bornscheuer, J. Pleiss, C. Syldatk and R. D. Schmid, J. Mol. Catal. B: Enzym., 2000, 8, 201–211 CrossRef CAS.
- J. Mutschler, T. Rausis, J. M. Bourgeois, C. Bastian, D. Zufferey, I. V. Mohrenz and F. Fischer, Green Chem., 2009, 11, 1793–1800 RSC.
- J. R. Silverman, M. Samateh and G. John, Eur. J. Lipid Sci. Technol., 2016, 118, 47–55 CrossRef CAS PubMed.
- D. J. Gaertner, T. M. Hallman, F. C. Hankenson and M. A. Batchelder, in Anesthesia and Analgesia in Laboratory Animals, 2008, pp. 239–297 Search PubMed.
- P. Flecknell, Laboratory Animal Anaesthesia, 4th edn, 2015 Search PubMed.
- P. K. Vemula, J. Li and G. John, J. Am. Chem. Soc., 2006, 128, 8932–8938 CrossRef CAS PubMed.
- E. Parisini, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Soc. Rev., 2011, 40, 2267–2278 RSC.
- H. M. W. Gray, Br. Med. J., 1917, 2, 509 CrossRef CAS PubMed.
- S. Tamilvanan and S. Benita, Eur. J. Pharm. Biopharm., 2004, 58, 357–368 CrossRef CAS PubMed.
- D. P. Mehta, H. J. Rathod, D. P. Shah and C. N. Shah, Res. J. Pharm. Technol., 2015, 8, 118–126 CrossRef.
- M. Heng, Biodiscovery, 2017, 20, e11207 CrossRef.
- K. Lalitha, Y. S. Prasad, C. U. Maheswari, V. Sridharan, G. John and S. Nagarajan, J. Mater. Chem. B, 2015, 3, 5560–5568 RSC.
- K. Lalitha and S. Nagarajan, J. Mater. Chem. B, 2015, 3, 5690–5701 RSC.
- K. Lalitha, Y. S. Prasad, V. Sridharan, C. U. Maheswari, G. John and S. Nagarajan, RSC Adv., 2015, 5, 77589–77594 RSC.
- H. M. Wyss, in Fluids, Colloids and Soft Materials: An Introduction to Soft Matter Physics, 2018, pp. 149–164 Search PubMed.
- Y. S. Pek, A. C. A. Wan, A. Shekaran, L. Zhuo and J. Y. Ying, Nat. Nanotechnol., 2008, 3, 671–675 CrossRef CAS PubMed.
- C. H. Lee, V. Moturi and Y. Lee, J. Controlled Release, 2009, 136, 88–98 CrossRef CAS PubMed.
- J. Nanda, A. Biswas and A. Banerjee, Soft Matter, 2013, 9, 4198–4208 RSC.
- M. Calvin, Wounds, 1998, 10, 12–32 Search PubMed.
- M. A. Carlson and M. T. Longaker, Wound Repair Regen, 2004, 12, 134–147 CrossRef PubMed.
- C. K. Sen, Wound Repair Regen, 2009, 17, 1–18 CrossRef PubMed.
- V. Kant, A. Gopal, D. Kumer, S. Bag, N. P. Kurade, A. Kumar, S. K. Tandan and D. Kumar, Eur. J. Pharmacol., 2013, 715, 345–353 CrossRef CAS PubMed.
- B. Rybinski, J. Franco-Barraza and E. Cukierman, Physiol. Genomics, 2014, 46, 223–244 CrossRef CAS PubMed.
- H. Haga, C. Irahara, R. Kobayashi, T. Nakagaki and K. Kawabata, Biophys. J., 2005, 88, 2250–2256 CrossRef CAS PubMed.
- J. W. Choi, J. K. Park, J. W. Chang, D. Y. Kim, M. S. Kim, Y. S. Shin and C. H. Kim, Biomaterials, 2014, 35, 4911–4918 CrossRef CAS PubMed.
- D. R. Griffin, W. M. Weaver, P. O. Scumpia, D. Di Carlo and T. Segura, Nat. Mater., 2015, 14, 737–744 CrossRef CAS PubMed.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS PubMed.
- R. Balasubramanian, G. Damodar and A. Sughir, Asian J. Pharm., 2012, 6, 1–9 CrossRef.
- S. S. Sagiri, B. Behera, R. R. Rafanan, C. Bhattacharya, K. Pal, I. Banerjee and D. Rousseau, Soft Mater., 2014, 12, 47–72 CrossRef CAS.
- K. Rehman and M. H. Zulfakar, Drug Dev. Ind. Pharm., 2014, 40, 433–440 CrossRef CAS PubMed.
- V. K. Singh, S. Ramesh, K. Pal, A. Anis, D. K. Pradhan and K. Pramanik, J. Mater. Sci.: Mater. Med., 2014, 25, 703–721 CrossRef CAS PubMed.
- B. Behera, S. Dey, V. Sharma and K. Pal, Adv. Polym. Technol., 2015, 34, 2 CrossRef.
- B. Behera, V. K. Singh, S. Kulanthaivel, M. K. Bhattacharya, K. Paramanik, I. Banerjee and K. Pal, Eur. Polym. J., 2015, 64, 253–264 CrossRef CAS.
- T. K. Lim, Edible Medicinal and Non-Medicinal Plants, vol. 5, Fruits, 2013 Search PubMed.
- A. M. Bakry, S. Abbas, B. Ali, H. Majeed, M. Y. Abouelwafa, A. Mousa and L. Liang, Compr. Rev. Food Sci. Food Saf., 2016, 15, 143–182 CrossRef CAS.
- S. Ebeling, K. Naumann, S. Pollok, T. Wardecki, S. Vidal-y-Sy, J. M. Nascimento, M. Boerries, G. Schmidt, J. M. Brandner and I. Merfort, PLoS One, 2014, 9, e86147 CrossRef PubMed.
- G. Broughton, J. E. Janis and C. E. Attinger, Plast. Reconstr. Surg., 2006, 117, 1e-S–32e-S CrossRef PubMed.
- S. Sanon, D. A. Hart and E. E. Tredget, in Skin Tissue Engineering and Regenerative Medicine, 2016, pp. 19–47 Search PubMed.
- A. E. K. Loo, Y. T. Wong, R. Ho, M. Wasser, T. Du, W. T. Ng and B. Halliwell, PLoS One, 2012, 7, e49215 CrossRef CAS PubMed.
- R. M. El-Ferjani, M. Ahmad, S. M. Dhiyaaldeen, F. W. Harun, M. Y. Ibrahim, H. Adam, B. Mohd Yamin, M. M. J. Al-Obaidi and R. Al Batran, Sci. Rep., 2016, 6, 38748 CrossRef CAS PubMed.
- C. K. Sen, S. Khanna, B. M. Babior, T. K. Hunt, E. Christopher Ellison and S. Roy, J. Biol. Chem., 2002, 277, 33284–33290 CrossRef CAS PubMed.
- R. E. Neuman and M. A. Logan, J. Biol. Chem., 1950, 184, 299–306 CAS.
- L. A. Wagner, Anal. Bioanal. Chem., 1979, 94, 394–396 Search PubMed.
- S. T. Omaye, J. D. Turnbull and H. E. Sauberlich, Methods Enzymol., 1979, 62, 3–11 CAS.
- H. Ohkawa, N. Ohishi and K. Yagi, Anal. Biochem., 1979, 95, 351–358 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Gelation studies table, optical microscopy images, SAXD, FTIR, wound closure picture, graphical representation of biochemical profile, 1H NMR spectra, 13C NMR spectral, ESI-MS spectra. See DOI: 10.1039/c8ra07703g |
| This journal is © The Royal Society of Chemistry 2018 |