
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycol-functionalized ionic liquids for high-temperature enzymatic ring-opening polymerization†
Hua Zhao *a,
Lennox O. Afriyiea,
Nathaniel E. Larmb and
Gary A. Bakerb
*a,
Lennox O. Afriyiea,
Nathaniel E. Larmb and
Gary A. Bakerb
aDepartment of Chemistry and Biochemistry, University of Northern Colorado, Greeley, CO 80639, USA. E-mail: hua.zhao@unco.edu; huazhao98@gmail.com
bDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA
First published on 23rd October 2018
Abstract
Enzymatic ring-opening polymerization (ROP) is a benign method for preparing polyesters, such as polylactides and other polylactones. These reactions are typically carried out at relatively high temperatures (60–130 °C), however, there is a deficiency of enzyme-compatible solvents for such thermally-demanding biocatalytic processes. In this study, we have prepared a series of short-chained glycol-grafted ionic liquids (ILs) based on a phosphonium, imidazolium, pyridinium, ammonium, or piperidinium cationic headgroup. Most of these glycol-grafted ILs exhibit relatively low dynamic viscosities (33–123 mPa s at 30 °C), coupled with excellent short-term thermal stabilities with decomposition temperatures (Tdcp) in the 318–403 °C range. Significantly, the long-term thermal stability under conditions matching those for enzymatic ROP synthesis (130 °C for 7 days) is excellent for several of these task-specific ILs. Using Novozym 435-catalyzed ROP, these ILs are demonstrated to be viable solvents for the enzymatic production of reasonable yields (30–48%) of high molecular mass (Mw ∼20 kDa) poly(L-lactide) and poly(ε-caprolactone) compared to solventless conditions (12–14 kDa).
Introduction
Enzymatic ring-opening polymerization (ROP) is a benign approach compared with metal-catalyzed methods for synthesizing biodegradable polyesters such as polylactides (PLAs) and polylactones. Among various reaction systems, ionic liquids (ILs) have been identified as novel reaction media alternatives to conventional organic solvents (e.g., toluene, heptane) in enzymatic ROP reactions. Common ILs being evaluated for enzymatic ROP include those based on imidazolium and pyridinium cations (such as 1-butyl-3-methylimidazolium, BMIM+) paired to anions such as tetrafluoroborate (BF4−), hexafluorophosphate (PF6−), or bis(trifluoromethylsulfonyl)imide (Tf2N−).1–4 However, inconsistent results have been reported in terms of molecular mass and yields for polylactides and polylactones produced by enzymatic ROP in these ILs.1 As indicated by our group previously, one important reason is the lack of fine control over reaction conditions such as the water content in ILs and enzymes.2 Another reason is tied to the requirement for long-term thermal stability of ILs due to the fact that the reaction is conducted at fairly high reaction temperatures (up to 130–150 °C) over a period of as long as 7–10 days. Although conventional ILs are typically reported to be thermally stable at these temperatures, they can slowly decompose when subjected to long-term thermal stresses. Thermal decomposition of alkylammonium salts follows a Hoffmann-type β-elimination in the presence of base to yield an amine and alkene while imidazolium ionic liquids degrade to alkylimidazole and imidazole, or even smaller fragments.5 The degraded ions can negatively impact the enzymatic polymerization reaction. Therefore, it is crucial to design thermally-stable ILs compatible with such prolonged, high-temperature enzymatic applications.Ionic liquids are frequently stated to possess high thermal stabilities; the onset decomposition temperatures (Tonset)‡ for many ILs reportedly range from 200–400 °C.6,7 In general, imidazolium salts are more thermally stable than alkylammonium, piperidinium, or pyridinium-based ILs. Meanwhile, ILs containing anions like Tf2N−, PF6−, and BF4− are generally more stable than those comprising dicyanamide, nitrate, halide, amino acid, or carboxylate anions.7 Phosphonium-based ILs typically have very high thermal stability (300–400 °C), however, alkyl-substituted phosphonium salts typically exhibit high melting points (i.e., solid or viscous/waxy liquid at room temperature).8,9 Ether-functionalized phosphoniums can, however, be prepared as room-temperature liquids possessing low viscosity (35–85 mPa s) coupled with high decomposition temperatures of ∼400 °C for a 10% mass loss.10 Toward further improving the thermostability of phosphonium solvents, the Wasserscheid group11 established that tetraphenylphosphonium bis(trifluoromethylsulfonyl)imide ([PPh4][Tf2N]) has a Tonset value of 420 °C and a long-term thermal stability (1% mass loss due to thermal decomposition per year at 301 °C). However, this salt displays a high melting point of 134 °C, although a eutectic composition of [PPh4][Tf2N]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cs[Tf2N] = 1:2.13 (i.e., 32 mol% [PPh4][Tf2N]) yields a slightly lower melting point of 98.6 °C. The Davis group12 evaluated the long-term thermal stability of triphenyl(P,P,P-triphenylphosphine imidato-κN)-phosphorus paired with various anions (mp > 120 °C) at 200 °C, 250 °C, and 300 °C for 96 h in air, and identified the most stable anions as C6H5SO3−, (C6H5SO2)2N−, benzene(disulfonyl)amidate (BDSA−), bis(perfluoroethanesulfonyl)imide (beti−), Tf2N−, and TfO−. Therefore, although alkyl-substituted phosphonium ILs are promising in terms of thermal stability, they can be associated with relatively high melting points or viscosities and may not otherwise represent ideal solvents for enzymatic ROP reactions.
:Cs[Tf2N] = 1:2.13 (i.e., 32 mol% [PPh4][Tf2N]) yields a slightly lower melting point of 98.6 °C. The Davis group12 evaluated the long-term thermal stability of triphenyl(P,P,P-triphenylphosphine imidato-κN)-phosphorus paired with various anions (mp > 120 °C) at 200 °C, 250 °C, and 300 °C for 96 h in air, and identified the most stable anions as C6H5SO3−, (C6H5SO2)2N−, benzene(disulfonyl)amidate (BDSA−), bis(perfluoroethanesulfonyl)imide (beti−), Tf2N−, and TfO−. Therefore, although alkyl-substituted phosphonium ILs are promising in terms of thermal stability, they can be associated with relatively high melting points or viscosities and may not otherwise represent ideal solvents for enzymatic ROP reactions.
To overcome these obstacles, we aimed to formulate so-called “task-specific ionic liquids” designed to be thermally stable, highly fluid, and biocompatible. Task-specific ionic liquids are a subset of ILs functionalized to display properties or reactivities suitably targeted for particular applications.13,14 It is known that symmetrical alkyl chains or a long-chain alkyl group on the cation can dramatically increase the melting point and viscosity of the resulting IL. On the other hand, appending glycols or their derivatives are known to be beneficial for achieving low melting point salts with reduced viscosities. Therefore, polyglycols have been incorporated into cationic or anionic units to produce the liquid state of ion-conductive polymers.15,16 In particular, various ILs have been synthesized by grafting alkyloxy substituents (ether or alcohol groups) onto an imidazolium17–28 or pyridinium ring.29,30 The inclusion of alkyloxy or alkyloxyalkyl groups can adequately lower the melting points of the resulting organic salts, yielding room-temperature ILs in many cases. The low viscosity arising from the incorporation of alkoxy chains has been justified by molecular dynamics (MD) simulations as resulting from the less effective assembly between flexible alkoxy chains as compared to their more rigid alkyl chain counterparts.31 In addition, MD simulations also suggest a reduction of intermolecular correlation (particularly tail–tail segregation) and cation–anion specific interactions due to the incorporation of the ether chain, which is responsible for the faster dynamics in ether-functionalized imidazolium ILs compared to alkyl-substituted ones.32 Following this rationale, our strategy toward enzymatic ROP compatible ILs involved the synthesis of a series of ILs carrying polyether-substituted cations paired with anions such as Tf2N−, as ILs containing this anion are frequently enzyme-compatible.33–35 With the targeted glycol-functionalized IL series in hand, we further evaluated how the thermal stability of the resulting IL contributed to the performance of high-temperature enzymatic ring-opening polymerization.
Experimental
Materials
The following enzymes and chemicals were purchased from Sigma-Aldrich (St. Louis, MO): L-(−)-lactide (Catalog #367044), Candida antarctica lipase B (CALB) immobilized on acrylic resin (Novozym 435; Catalog #L4777, Batch #SLBP0766V, purchased in March 2016), and polymer-bound triphenylphosphine (100–200 mesh, extent of labeling: ∼3.0 mmol g−1 loading, polystyrene with 2% cross-linked with divinylbenzene) (Catalog #366455). Both 2-bromoethyl methyl ether produced by BeanTown Chemical (Hudson, NH), and lithium bis(trifluoromethylsulfonyl)imide (Li[Tf2N]) produced by Matrix Scientific (Columbia, SC) were supplied by VWR (Radnor, PA). Diethylene glycol monomethyl ether, triethylene glycol monomethyl ether, lithium bis(pentafluoroethanesulfonyl)imide (Li[beti]), and ε-caprolactone were purchased from TCI America (Portland, OR). 1-Butyl-3-methylimidazolium hexafluorophosphate ([BMIM][PF6], high purity) and 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([BMIM][Tf2N], synthesis grade) were obtained through VWR and were products of Merck KGaA (EMD Millipore Corporation, Billerica, MA).Preparation of glycol-functionalized ILs
We modified literature methods36,37 to synthesize our glycol-functionalized ILs. Our preparation of phosphonium-type ILs is described next as a general procedure. The first step was the bromination of glycol, which followed the Appel reaction (see ESI of ref. 38) except for 2-bromoethyl methyl ether which was acquired from a commercial source. Glycol monomethyl ether (∼20 g, 1.0 molar equiv.) was dissolved in 100 mL of acetonitrile, followed by the addition of tetrabromomethane (1.2 molar equiv.). The mixture was stirred at room temperature until complete dissolution of CBr4. Polystyrene supported-triphenylphosphine (PS-Ph3P, 1.2 molar equiv.) was added in portions and the mixture was stirred at room temperature for 24 h. The solid was removed by filtration. Activated carbon was added into the light-brownish solution and stirred for 1 h to remove color. After filtering off the solid carbon, the solvent was evaporated under vacuum, producing bromoglycol monomethyl ether.Bromoglycol monomethyl ether (∼20 g) was mixed with 1.1 molar equiv. of tributylphosphine in anhydrous acetonitrile and refluxed for 24 h to produce the bromide salt. The silver nitrate test confirmed the presence of bromide anion. Following the reaction, solvent was evaporated under vacuum. The crude product was extracted three times by n-hexane (or n-heptane) to remove excess reactant (tributylphosphine) and any byproduct (such as tributylphosphine oxide).
An aqueous solution of lithium bis(trifluoromethylsulfonyl)imide (Li[Tf2N], 1.05 molar equiv.) was added dropwise to an equivalent of bromide salt predissolved in water. A turbid mixture formed immediately. The reaction mixture was stirred at room temperature for 20 min, and then allowed to sit for at least 2 h to allow the formation of two discrete layers. After adequate phase separation, the bottom IL layer was separated, taken up in CH2Cl2, and thoroughly washed with distilled water three times. Following this treatment, a silver nitrate test of the aqueous layer previously contacting the IL was negative for the presence of halides. After drying the organic solvent with Na2SO4 followed by filtration, the IL was obtained after removal of CH2Cl2 under vacuum. The resulting IL was collected, rinsed by n-hexane twice (for non-phosphonium salts, diethyl ether was used), and dried in a vacuum oven (25 mmHg) at 80 °C for three days. 1H, 13C, and 31P NMR data confirmed the structure and purity of the ILs.
Enzymatic ROP of lactide
The water contents of all solvents, monomers, and enzymes were assayed by Karl Fischer (KF) titration (Mettler Toledo C20X compact coulometric titrator; detection limit ∼1 ppm water) at 20 °C. Hydranal® Coulomat AG was used as the analyte for the titration. A typical enzymatic ROP reaction was conducted as follows: the substrate (0.5 g L-lactide) was mixed with 0.25 mL of solvent (or noted otherwise), followed by the addition of 100 mg of Novozym 435. The reaction vial was tightly sealed and then immersed in an oil bath (130 °C) after initiating stirring (210 rpm). At the completion of reaction, the reaction mixture was cooled to room temperature and then 2.0 mL of deuterochloroform (CDCl3) was added to dissolve the polyester under vigorous agitation. For 1H NMR analysis, a 50 μL reaction aliquot was withdrawn and diluted with an additional 1.0 mL of CDCl3, followed by centrifugation (2000×g, 2 min). For gel permeation chromatography (GPC) analysis—a type of size exclusion chromatography, SEC—a 50 μL reaction aliquot was withdrawn and diluted into 1.0 mL of THF. This mixture was centrifuged before GPC injection (the GPC experimental conditions are described in the Polymer characterization section below). To obtain the final solid product, after evaporating chloroform, the polymer was precipitated by adding ice-cold methanol and was then separated by centrifugation or vacuum filtration. The polyester was air-dried for 24 h.Enzymatic ROP of ε-caprolactone
The substrate (0.5 g of ε-caprolactone; density: 1.03 g mL−1) was dissolved in 0.25 mL of solvent, followed by the addition of 100 mg of Novozym 435. The reaction mixture was sealed tightly and stirred (210 rpm) at 70 °C in an oil bath. At the completion of reaction, the reaction mixture was cooled to room temperature and 2.0 mL of CDCl3 was added to dissolve the polyester under vigorous agitation. For 1H NMR analysis, a 50 μL aliquot was withdrawn and diluted with 1.0 mL of CDCl3, followed by centrifugation. For GPC analysis, 50 μL aliquot was withdrawn and diluted with 1.0 mL THF, and the mixture was centrifuged prior to GPC injection. To obtain the final solid product, after evaporating chloroform, the polymer was precipitated by adding ice-cold methanol and was then separated by centrifugation or vacuum filtration. The recovered polyester was air-dried for 24 h.Polymer characterization
The L-lactide conversion was monitored via 1H NMR analysis (Bruker Avance II 400 MHz NMR) by comparing the peak area of the methine group (CH) adjacent to the carbonyl in the monomer (5.04 ppm) and in the polymer (5.16 ppm).3 Similarly, monomer ε-caprolactone conversion was determined from 1H NMR experiments by comparing the integrated signal areas for the methylene groups next to the carbonyl group within the monomer ε-caprolactone (CL, 4.23 ppm) and the poly(ε-caprolactone) (PCL, 4.07 ppm).39 The mass-average molecular mass (Mw) and polydispersity index (PDI = Mw/Mn) of the polyester were determined by GPC equipped with a LC-20AD Shimadzu HPLC, SPD-20A UV-visible dual-wavelength detector at 210 nm, and two PLgel MIXED-B 10 μm, 300 × 7.5 mm columns (Agilent) eluted with 1.0 mL min−1 THF at 30 °C.40 Calibration was achieved by using polystyrene standards from 570 to 62500 (Mw).41
Results and discussion
Synthesis of glycol-functionalized ILs
As illustrated in Scheme 1, the preparation of glycol-grafted ILs comprises three primary steps: bromination of the glycol, quaternization of a phosphine (or imidazole, pyridine, alkylamine, or alkylpiperidine), and ion metathesis. The first reaction (i.e., bromination) is the most challenging step. Dibromination of glycol (HO–PEGn–OH) with PBr3 has been shown to be effective.36,37 The direct monobromination of glycol monomethyl ether (MeO–PEGn–OH) using PBr3 has been reported to prepare MeO–PEGn–Br,42–45 however, this reaction can also generate di-brominated by-product (Br–PEGn–Br) which can prove difficult to separate from the main product. To improve this reaction, Et3N was added as a base during bromination to neutralize hydrobromic acid produced in the presence of trace water (i.e., hydrobromic acid is known to induce the dibromination reaction46). However, this process typically involves highly exothermic neutralization between Et3N and HBr/H3PO3, resulting in a large quantity of ammonium salts, making the reaction difficult to control. On the other hand, the Appel reaction38 can be conducted under mild conditions using triphenylphosphine and tetrabromomethane. However, removal of triphenylphosphine (Ph3P) and triphenylphosphine oxide (Ph3PO) at the completion of reaction still poses a separations challenge. Traditionally, the removal is accomplished by tedious column chromatography using large volumes of solvents. The Lipshutz group47 reported that mixing the product with Merrifield resin and NaI in acetone led to an effective removal of trace Ph3P/Ph3PO. However, this process is highly sensitive to moisture and oxygen because iodide can be easily oxidized to I2, contaminating the product and coloring it brown. A recent study suggested that Ph3PO could be complexed by ZnCl2 in ethanol to form a precipitate whereas Ph3P can be oxidized to its oxide form by 10% H2O2 solution before performing the complexing step.48 However, this method reportedly fails to precipitate trace amounts of Ph3PO, with <1% still remaining in solution.48 On the other hand, polymer-bound triphenylphosphine (particularly, polystyrene-bound Ph3P with 2% cross-linking with divinylbenzene, designated PS-Ph3P) has shown the advantage of convenient workup via simple filtration.49–51 Therefore, after examining all of the above options, we adopted the PS-Ph3P method to carry out the bromination of glycol monomethyl ether, as detailed in the Experimental. | ||
| Scheme 1 Synthesis of glycol-functionalized phosphonium ionic liquids. | ||
A common side reaction occurring during alkylation of electron-rich phosphines (such as tributylphosphine, Bu3P) is oxidation to yield the corresponding phosphine oxide.52 Therefore, after completion of the second step in glycol-functionalized IL synthesis (i.e., the phosphine alkylation step), the byproduct oxide (e.g., Bu3PO) and excess starting phosphine are removed by n-hexane extraction. This extraction step was repeated again on the final Tf2N− IL obtained by anion metathesis, before subjecting the fluid to prolonged drying in a vacuum oven. It should be noted that common Tf2N− ILs are frequently rinsed with diethyl ether or ethyl acetate, however, both of these solvents are miscible with the glycol-appended phosphonium ILs prepared in this study.
Physical properties of glycol-functionalized ILs
Initially, we determined the dynamic (absolute) and kinematic viscosities, as well as the densities of the synthesized ILs at 30 °C (Table 1). All ILs were intensively dried in a vacuum oven to a final water content below 200 ppm. The studied ILs have densities within the range of 1.2–1.5 g cm−3 at 30 °C. In general, glycol-functionalized ILs carrying the Tf2N− anion show relatively low viscosities (η = 33–123 mPa s at 30 °C). As benchmarks, comparison is made with the prototypical, well-studied ILs [BMIM][Tf2N] (11) (η = 41.4 mPa s at 30 °C) and [BMIM][PF6] (12) (η = 205.8 mPa s). Different cation cores have a major influence on the dynamic viscosities, as illustrated by the observed general trend of decreasing viscosity in the order piperidinium (10) > ammonium (9) > pyridinium (8) > phosphonium (1) > imidazolium (7). Glycol-functionalization also leads to a reduced viscosity, as shown by the lower viscosity of 33.1 mPa s for [MeOCH2CH2-Et-Im][Tf2N] (7) versus 41.4 mPa s for [BMIM][Tf2N] (11). Likewise, a longer glycol chain generally reduces the viscosity, as exemplified by the [RPBu3][Tf2N] type ILs, where R is the glycol group Me(OCH2CH2)n (the number of ethylene oxide (EO) units, n = 1, 2, 3 and 4; see entries 2 and 4–6 in Table 1). A less symmetrical cation tends to have a lower viscosity as evidenced by a viscosity of 36.0 mPa s for [MeOCH2CH2-PEt3][Tf2N] (1) compared with 122.5 mPa s for [MeOCH2CH2-PBu3][Tf2N] (2). It is important to note that our physical characterization data are consistent with known literature values. For example, our measured dynamic viscosity for [MeOCH2CH2-PEt3][Tf2N] (1) of 36.0 mPa s at 30 °C accords with the literature value of 44 mPa s at 25 °C.10 Similarly, a dynamic viscosity of 61.4 mPa s at 30 °C for [MeOCH2CH2-Et3N][Tf2N] (9) falls in line with the reported value of 85 mPa s at 25 °C.10 The anions have some impact on the viscosity as well: the Tf2N− anion tends to lead to a lower viscosity than beti− and PF6− (2 vs. 3, 11 vs. 12 in Table 1).| IL | Dynamic viscositya (mPa s) | Kinematic viscositya (mm2 s−1) | Densitya (g cm−3) | Tderb (°C) | Tdcpc (°C) | Transition shape d | Residual char e | |
|---|---|---|---|---|---|---|---|---|
| a The viscosity and density data were acquired using an Anton Paar SVM 3000 viscometer; measurements were made at 30 °C. Thermogravimetric analysis (TGA) scans were measured on a TA Instruments TGA Q50 under a nitrogen atmosphere (60 mL min−1) using Pt pans with a heating rate of 10 °C min−1.b Tder is determined from the maximum in the first-derivative profile of the TGA scan.c Tdcp is the decomposition temperature measured as the onset of decomposition, using the common criterion of 10% total mass loss. Uncertainties in the temperatures are estimated to be on the order of ±2–3 °C.d The TGA mass loss behavior is qualitatively characterized on the basis of whether it occurs essentially in a single, discrete step (S) or exhibits multiple step (M) thermal decomposition. The latter is generally associated with an early mass loss step which occurs at a temperature 50–100 °C below the primary event within the vicinity of Tder. It should be noted that, for this reason, profiles that display some multi-step thermal decomposition character generally have lower effective Tdcp values.e The amount of carbon char residue is determined from the relative mass remaining at 600 °C; a residue amount on the order of ±1–2% should be considered within the error of the measurement baseline. | ||||||||
| 1 | [MeOCH2CH2-PEt3][Tf2N] | 36.0 | 26.3 | 1.368 | 468 | 362 | M | 2.6% |
| 2 | [MeOCH2CH2-PBu3][Tf2N] | 122.5 | 99.0 | 1.237 | 449 | 354 | M | 1.0% |
| 3 | [MeOCH2CH2-PBu3][beti] | 163.4 | 127.1 | 1.286 | 425 | 318 | M | 0.9% |
| 4 | [Me(OCH2CH2)2-PBu3][Tf2N] | 95.5 | 78.6 | 1.215 | 445 | 319 | M | 1.4% |
| 5 | [Me(OCH2CH2)3-PBu3][Tf2N] | 79.8 | 66.8 | 1.195 | 448 | 318 | M | 1.7% |
| 6 | [Me(OCH2CH2)4-PBu3][Tf2N] | 90.2 | 75.0 | 1.203 | 450 | 335 | M | 2.2% |
| 7 | [MeOCH2CH2-Et-Im][Tf2N] | 33.1 | 22.7 | 1.458 | 468 | 401 | S | 3.4% |
| 8 | [MeOCH2CH2-Py][Tf2N] | 44.5 | 29.5 | 1.508 | 456 | 403 | M | 2.8% |
| 9 | [MeOCH2CH2-Et3N][Tf2N] | 61.4 | 44.4 | 1.383 | 427 | 380 | S | 0.0% |
| 10 | [MeOCH2CH2-Me-Pip][Tf2N] | 84.2 | 59.0 | 1.426 | 453 | 399 | S | 0.3% |
| 11 | [BMIM][Tf2N] | 41.4 | 28.9 | 1.430 | 464 | 406 | S | 1.5% |
| 12 | [BMIM][PF6] | 205.8 | 151.1 | 1.362 | 472 | 424 | S | 0.5% |
The short-term thermal stabilities of the prepared ILs were evaluated on the basis of two parameters obtained from scanning thermogravimetric analysis (TGA): the derivative temperature Tder, determined from the maximum of the first-derivative profile of the TGA thermogram, and the decomposition temperature Tdcp, defined as the decomposition temperature measured using the prevalent criterion of a 10% total mass loss (Fig. S1–S12 in ESI† and Table 1). In terms of Tder values, glycol-functionalized ILs generally have stabilities (Tder ≈ 420–470 °C) similar to conventional saturated hydrocarbon side chain ILs such as 11 and 12, with a thermal stability that decreases to some extent in the order phosphonium (1) ≈ imidazolium (7) > pyridinium (8) > piperidinium (10) > ammonium (9). Short-chain glycol functionalization has no outward effect on thermal stability (7 vs. 11). Similarly, for a given cationic moiety, the glycol chain length has minimal impact on Tder (compare 4–6 with 2). In terms of Tdcp values, again short-chain glycol functionalization reveals no significant effect on apparent thermal stability (7 vs. 11), however, this parameter reveals that lengthening of the glycol chain (compare 2, 4, 5, and 6) lowers the thermal stability by some 19–36 °C. Based on Tdcp values, the cation core exerts a greater influence over the thermal stability, according to the trend of decreasing thermostability: pyridinium (8) ≈ imidazolium (7) ≈ piperidinium (10) > ammonium (9) > phosphonium (1). A comparison between 2 and 3 reveals that the Tf2N− anion is marginally more thermally stable than the beti− anion, in terms of Tder. Similarly, the difference in the long-term thermal stability observed for the Tf2N− and beti− salts of [MeOCH2CH2–PBu3]+ (2 and 3 in Table 2) is consistent with the observation that ΔHvap values for beti−-containing 1-alkyl-3-methylimidazolium ILs are ∼5% lower than their Tf2N− counterparts of the same alkyl chain length.53
| IL | % Mass loss and color change for 130 °C treatment for 7 days | % Mass loss and color change for 200 °C treatment for 24 h | |
|---|---|---|---|
| a The mass loss was determined after open air heating in an oven at a fixed temperature. | |||
| 1 | [MeOCH2CH2-PEt3][Tf2N] | No change | 0.60% (light brown) |
| 2 | [MeOCH2CH2-PBu3][Tf2N] | No change | 1.5% (dark brown) |
| 3 | [MeOCH2CH2-PBu3][beti] | 3.3% (no color change) | 5.0% (light brown) |
| 4 | [Me(OCH2CH2)2-PBu3][Tf2N] | 2.5% (no color change) | 7.5% (v. dark) |
| 5 | [Me(OCH2CH2)3-PBu3][Tf2N] | 4.3% (no color change) | 10.4% (v. dark) |
| 6 | [Me(OCH2CH2)4-PBu3][Tf2N] | 14.6% (no color change) | 13.0% (v. dark) |
| 7 | [MeOCH2CH2-Et-Im][Tf2N] | No change | 0.27% (light brown) |
| 8 | [MeOCH2CH2-Py][Tf2N] | No change | 0.27% (dark brown) |
| 9 | [MeOCH2CH2-Et3N][Tf2N] | No change | 0.58% (light brown) |
| 10 | [MeOCH2CH2-Me-Pip][Tf2N] | No change | 0.30% (dark brown) |
| 11 | [BMIM][Tf2N] | No change | 0.28% (v. light brown) |
| 12 | [BMIM][PF6] | 14.9% (white precipitate) | 8.5% (dark brown) |
Longer-term thermal stability was examined by incubating the ILs at 130 °C for 7 days (reaction conditions which match those for PLA synthesis) or at 200 °C for 24 h. As summarized in Table 2, many ILs investigated exhibit excellent long-term stability at 130 °C, with the notable exceptions of ILs 3, 4, 5, 6, and 12. It also appears that longer glycol-chains lead to lower long-term thermal stability. This can be seen by comparison of ILs 2, 4, 5, and 6 (one, two, three, and four EO units, respectively) which display mass losses of 0.0%, 2.5%, 4.3%, and 14.6%, respectively, after being held at 130 °C for 7 days. The reduced long-term stability (i.e., 14.9% mass loss after 7 days at 130 °C) of [BMIM][PF6] (12) suggests the inherent instability of the PF6− anion since [BMIM][Tf2N] (11) is comparably far more stable under these conditions. In the course of a 24 h exposure to 200 °C, many ILs show relatively minor mass losses <1.0% (Table 2), although visual changes are universally apparent (i.e., development of light to deep brown coloration). It is already well-established that trivial levels of IL decomposition undetectable by standard TGA or NMR analysis can be observed by changes in spectroscopic features (i.e., colored and/or fluorescent impurities).54 Notably, the ILs 3, 4, 5, 6, and 12 showed significant mass losses after 24 h at 200 °C (5.0%, 7.5%, 10.4%, 13.0%, and 8.5% mass loss, respectively). Based on these outcomes for long-term thermal exposure relevant to ROP conditions, we conclude that, with the exception of ILs 3, 4, 5, 6, and 12, the remaining ILs appear promising as thermally-suitable media for enzymatic ROP at 130 °C.
Enzymatic ROP of L-lactide and ε-caprolactone
Firstly, we examined the enzymatic ROP of L-lactide catalyzed by Novozym 435 at 130 °C under various solvent conditions (Table 3). As pointed out in our earlier study, the enzymatic polymerization can be greatly affected by the batch conditions and water content of the enzyme.2 When no solvent was employed, poly(L-lactide) (PLLA) was produced with a molecular mass (Mw) of 12400 Da in 36% yield and with a PDI of 1.14 (trial 1 in Table 3). This solventless result serves as a reference point for comparison. When an organic solvent (i.e. dimethylacetamide, DMA), or a conventional IL (i.e., [BMIM][PF6] or [BMIM][Tf2N]) was employed as solvent (trials 2–4), similar molecular masses in the range of 11500–13000 Da were obtained, although there appears to be wide variation in the observed yield (6–46%) and PDI (1.48–2.15). Using the same batch of lipase (Batch #SLBP0766V, purchased in March 2016) in [BMIM][PF6], this study synthesized PLLA with Mw = 11900 Da, PDI 2.15 and yield 24% (trial 3 in Table 3) while our earlier study (see trial 6 in Table 1 of ref. 2) obtained PLLA with a Mw of 11500 Da, PDI 1.41 and yield 12%; the molecular masses are comparable, however, the higher yield reported in this study is likely due to the lower water content (0.01 wt%) in [BMIM][PF6] (vs. 0.02 wt% in the earlier study2).
| Trial | Substrate | Solvent (water content) | T (°C) | Reaction time (days) | Conversion (%) | Yield (%) | Mwb (Da) | PDI |
|---|---|---|---|---|---|---|---|---|
| a General reaction conditions (unless otherwise noted): 0.5 g of L-lactide or ε-caprolactone, 0.25 mL of solvent, 100 mg of Novozym 435, gentle stirring (210 rpm) at 130 or 70 °C for 7 or 2 days. GPC-derived Mw values were based on results calibrated using polystyrene standards.b Based on GPC analysis. | ||||||||
| 1 | L-Lactide | No solvent | 130 | 7 | 85.3 | 36 | 12400 |
1.14 |
| 2 | L-Lactide | Dimethylacetamide (0.01 wt%) | 130 | 7 | 95.2 | 46 | 11500 |
1.54 |
| 3 | L-Lactide | [BMIM][PF6] (0.01 wt%) | 130 | 7 | 92.4 | 24 | 11900 |
2.15 |
| 4 | L-Lactide | [BMIM][Tf2N] (0.01 wt%) | 130 | 7 | 94.5 | 6 | 13000 |
1.48 |
| 5 | L-Lactide | [CH3OCH2CH2-PEt3][Tf2N] (0.01 wt%) | 130 | 7 | 84.9 | 12 | 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 100 100 |
1.89 |
| 6 | L-Lactide | [CH3OCH2CH2-PBu3][Tf2N] (0.01 wt%) | 130 | 7 | 86.3 | 34 | 17600 |
2.13 |
| 7 | L-Lactide | [CH3OCH2CH2-PBu3][Tf2N] (1.7 wt%) | 130 | 7 | 96.5 | 28 | 12600 |
2.38 |
| 8 | L-Lactide | [CH3OCH2CH2-PBu3][Tf2N] (0.01 wt%, 0.5 mL) | 130 | 7 | 75.5 | 10 | 12200 |
1.84 |
| 9 | L-Lactide | [CH3OCH2CH2-PBu3][Tf2N] (0.01 wt%, 1.0 mL) | 130 | 7 | 79.2 | 6 | 6800 | 1.84 |
| 10 | L-Lactide | [CH3OCH2CH2-PBu3][beti] (0.02 wt%) | 130 | 7 | 69.2 | 32 | 14500 |
2.19 |
| 11 | L-Lactide | [CH3(OCH2CH2)2-PBu3][Tf2N] (0.02 wt%) | 130 | 7 | 74.5 | 12 | 15400 |
1.38 |
| 12 | L-Lactide | [CH3(OCH2CH2)3-PBu3][Tf2N] (0.02 wt%) | 130 | 7 | 87.2 | 14 | 13400 |
1.38 |
| 13 | L-Lactide | [CH3(OCH2CH2)4-PBu3][Tf2N] (0.01 wt%) | 130 | 7 | 87.5 | 4 | 14000 |
1.71 |
| 14 | L-Lactide | [CH3(OCH2CH2)4-PBu3][Tf2N] (0.04 wt%) | 130 | 7 | 80.0 | 20 | 13200 |
1.97 |
| 15 | L-Lactide | [MeOCH2CH2-Et-Im][Tf2N] (0.03 wt%) | 130 | 7 | 90.0 | 10 | 23000 |
1.55 |
| 16 | L-Lactide | [MeOCH2CH2-Py][Tf2N] (0.03 wt%) | 130 | 7 | 78.6 | 14 | 17300 |
1.29 |
| 17 | L-Lactide | [MeOCH2CH2-Et3N][Tf2N] (0.03 wt%) | 130 | 7 | 92.5 | 20 | 19000 |
1.58 |
| 18 | L-Lactide | [MeOCH2CH2-Me-Pip][Tf2N] (0.03 wt%) | 130 | 7 | 88.0 | 16 | 11300 |
1.76 |
| 19 | ε-Caprolactone | No solvent | 70 | 2 | 96.8 | 37 | 13800 |
1.71 |
| 20 | ε-Caprolactone | [BMIM][PF6] (0.01 wt%) | 70 | 2 | 87.8 | 55 | 18500 |
1.94 |
| 21 | ε-Caprolactone | [CH3OCH2CH2-PBu3][Tf2N] (0.02 wt%) | 70 | 2 | 96.9 | 48 | 18900 |
1.59 |
| 22 | ε-Caprolactone | [CH3OCH2CH2-PBu3][beti] (0.02 wt%) | 70 | 2 | 52.1 | 15 | 9100 | 1.19 |
| 23 | ε-Caprolactone | [CH3(OCH2CH2)2-PBu3][Tf2N] (0.02 wt%) | 70 | 2 | 70.1 | 30 | 14300 |
1.35 |
| 24 | ε-Caprolactone | [MeOCH2CH2-Et-Im][Tf2N] (0.03 wt%) | 70 | 2 | 95.7 | 42 | 12300 |
1.60 |
| 25 | ε-Caprolactone | [MeOCH2CH2-Py][Tf2N] (0.03 wt%) | 70 | 2 | 94.3 | 32 | 16200 |
1.34 |
| 26 | ε-Caprolactone | [MeOCH2CH2-Et3N][Tf2N] (0.03 wt%) | 70 | 2 | 96.4 | 11 | 17300 |
1.39 |
| 27 | ε-Caprolactone | [MeOCH2CH2-Me-Pip][Tf2N] (0.03 wt%) | 70 | 2 | 96.1 | 11 | 18100 |
1.69 |
On the other hand, the glycol-functionalized phosphonium ILs [CH3OCH2CH2-PEt3][Tf2N] (1) and [CH3OCH2CH2-PBu3][Tf2N] (2) afforded significantly improved Mw (21100 and 17600 Da, respectively) whilst the latter IL gave a higher yield (34%) (see trials 5 and 6 in Table 3). The water content within the studied ILs was generally controlled within the 0.01–0.03 wt% range (100–300 ppm). We observed lower molecular masses at higher water contents, illustrated by the ROP of L-lactide in [CH3OCH2CH2-PBu3][Tf2N] containing 0.01% (trial 6) versus 1.70% water (trial 7), leading to Mw values of 17600 and 12600 Da, respectively; a similar trend was observed for trials 13 and 14. Employing ILs with longer glycol chains (i.e., two to four EO units) attached to the phosphonium cation as ROP media led to lower molecular masses (15400, 13400 and 14000 Da for trials 11, 12 and 13, respectively) in comparison to trial 6 involving a phosphonium IL bearing a single EO unit glycol pendant chain. In addition, we observed that the choice of cationic core had a heavy influence over the ROP reaction (trials 15–18). Using the imidazolium-based IL [MeOCH2CH2-Et-Im][Tf2N] (trial 15) gave the highest molecular mass (23000 Da), despite a relatively low yield of 10%. The rank of IL cations in terms of the resulting PLLA Mw was observed to be: imidazolium (23000 Da) > ammonium (19000 Da) > phosphonium (17600 Da) ≈ pyridinium (17300 Da) > piperidinium (11300 Da). The ranking based on PLLA yield differs as the following: phosphonium (34%) > ammonium (20%) > piperidinium (16%) ≈ pyridinium (14%) > imidazolium (10%). Through comparing trials 10 and 6 in Table 3, it appears the beti−-based IL produces a slightly lower Mw than the Tf2N− analogue. The observed PLLA Mw, yield, and PDI results are illustrated by Fig. S13 in the ESI.†
We further examined the enzymatic ROP of ε-caprolactone at 70 °C in various glycol-functionalized ILs (Table 3, trials 20–27). When no solvent was used, poly(ε-caprolactone) (PCL) was produced with a Mw of 13800 Da in 37% yield with a 1.71 PDI (trial 19). In [BMIM][PF6] (trial 20 in Table 3), PCL was obtained with a Mw of 18500 Da, 55% yield and 1.94 PDI, which is comparable with our earlier results (a Mw 20700 Da, 46% yield and 1.86 PDI)2 although different batches of Novozym 435 were used. The PCL Mw is again influenced by the identity of the cationic headgroup of the IL: phosphonium (18900 Da) > piperidinium (18100 Da) ≈ ammonium (17300 Da) > pyridinium (16200 Da) > imidazolium (12300 Da). The PCL yield decreases in the order phosphonium (48%) > imidazolium (42%) > pyridinium (32%) > ammonium (11%), piperidinium (11%). A corresponding summary of Mw, yield, and PDI for ROP-produced PCL is provided in Fig. S14 in the ESI.† Overall, the phosphonium-based IL [CH3OCH2CH2-PBu3][Tf2N] (2) enabled the synthesis of a relatively high molecular mass PCL in high yield.
Based on the data compiled within Tables 1 and 3, it appears that neither Mw nor yields of ROP-derived polyesters exhibit any straightforward correlation with the short-term thermal stability of the studied ILs. Although ILs with poor long-term stability (ILs 3, 4, 5, 6, and 12 in Table 2) all led to relatively low Mw (trials 10, 11, 12, 13, and 3, respectively, in Table 3), some thermally-stable ILs also gave low Mw polymers (e.g., trials 4 and 18, Table 3). Therefore, there are clearly other important and confounding factors operative beyond the long-term thermal stability of ILs. The viscosity shows a more puzzling effect on Mw, as demonstrated in Fig. 1. For PLLA synthesis at 130 °C, a loose correlation suggests a higher viscosity leading to a lower Mw (Fig. 1a), however, for PCL synthesis at 70 °C (Fig. 1b), there is a slow increase in Mw with an increase in viscosity. These trends possibly arise due to mass transfer being the limiting factor for PLLA synthesis, making a reduced viscosity beneficial, whereas the rapid dissolution of ε-caprolactone (a liquid) into low-viscosity ILs becomes disadvantageous for PCL synthesis, resulting in more substrate stabilization.
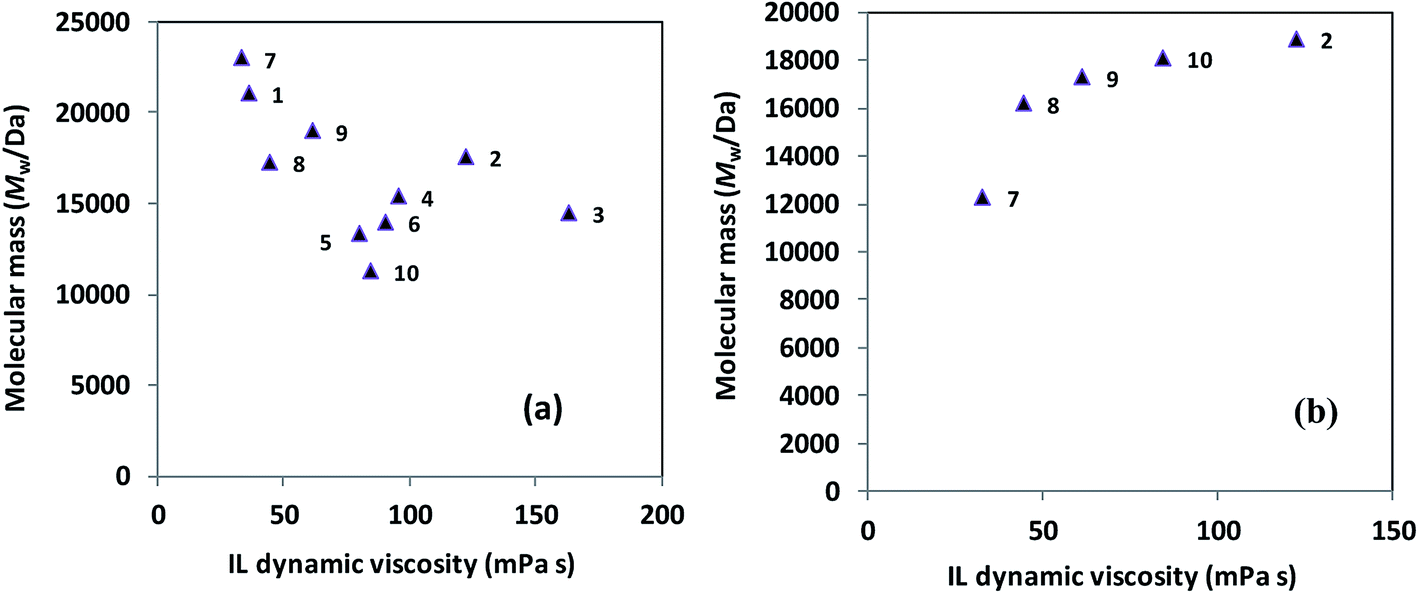 | ||
| Fig. 1 Correlation between the mass-average molecular mass (Mw) for Novozym 435-catalyzed ROP-produced polyester and the dynamic viscosity (30 °C) of the IL medium (the number next to each symbol indicates the IL shown in Table 1) for (a) poly(L-lactide) and (b) poly(ε-caprolactone) synthesis. | ||
Conclusions
We have synthesized a series of thermally-stable glycol-functionalized ILs possessing fairly low viscosities based on various cationic moieties, including phosphonium, imidazolium, pyridinium, ammonium, and piperidinium salts. We demonstrate that these new ionic solvents are suitable media for high-temperature enzymatic reactions, using Novozym 435-catalyzed ring-opening polymerization for illustration. Comparing with a solventless condition, these task-specific ILs led to higher molecular masses (Mw ≈ 20000 Da) of poly(L-lactide) and poly(ε-caprolactone) in reasonable yields. In addition to the importance of the long-term thermal stability of the ionic liquid, there appear to be other confounding factors responsible for achieving high yields and molecular masses, with the viscosity of the solvent also playing a non-trivial role.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. Z. acknowledges support from a Henry Dreyfus Teacher-Scholar Award (2012–2018) and startup funds from the University of Northern Colorado. G. A. B. acknowledges support for this research from the Research Corporation for Science Advancement.References
- H. Zhao, J. Chem. Technol. Biotechnol., 2018, 93, 9–19 CrossRef CAS.
- H. Zhao, G. A. Nathaniel and P. C. Merenini, RSC Adv., 2017, 7, 48639–48648 RSC.
- M. Yoshizawa-Fujita, C. Saito, Y. Takeoka and M. Rikukawa, Polym. Adv. Technol., 2008, 19, 1396–1400 CrossRef CAS.
- K. A. Barrera-Rivera, Á. Marcos-Fernández, R. Vera-Graziano and A. Martínez-Richa, J. Polym. Sci., Part A-1: Polym. Chem., 2009, 47, 5792–5805 CrossRef CAS.
- A. Efimova, L. Pfützner and P. Schmidt, Thermochim. Acta, 2015, 604, 129–136 CrossRef CAS.
- E. M. Siedlecka, M. Czerwicka, S. Stolte and P. Stepnowski, Curr. Org. Chem., 2011, 15, 1974–1991 CrossRef CAS.
- Y. Cao and T. Mu, Ind. Eng. Chem. Res., 2014, 53, 8651–8664 CrossRef CAS.
- K. J. Fraser and D. R. MacFarlane, Aust. J. Chem., 2009, 62, 309–321 CrossRef CAS.
- A. F. Ferreira, P. N. Simões and A. G. M. Ferreira, J. Chem. Thermodyn., 2012, 45, 16–27 CrossRef CAS.
- K. Tsunashima and M. Sugiya, Electrochem. Commun., 2007, 9, 2353–2358 CrossRef CAS.
- M. Scheuermeyer, M. Kusche, F. Agel, P. Schreiber, F. Maier, H.-P. Steinrück, J. H. J. Davis, F. Heym, A. Jess and P. Wasserscheid, New J. Chem., 2016, 40, 7157–7161 RSC.
- A. Benchea, B. Siu, M. Soltani, J. H. McCants, E. A. Salter, A. Wierzbicki, K. N. West and J. H. J. Davis, New J. Chem., 2017, 41, 7844–7848 RSC.
- J. H. J. Davis, Chem. Lett., 2004, 33, 1072–1077 CrossRef CAS.
- R. Giernoth, Angew. Chem., Int. Ed., 2010, 49, 2834–2839 CrossRef CAS PubMed.
- H. Ohno, Bull. Chem. Soc. Jpn., 2006, 79, 1665–1680 CrossRef CAS.
- H. Ohno, Electrochemical Aspects of Ionic Liquids, John Wiley & Sons, Hoboken, NJ, 2005 Search PubMed.
- H. Ohno, Y. Nakai and K. Ito, Chem. Lett., 1998, 27, 15–16 CrossRef.
- M. Yoshizawa and H. Ohno, Chem. Lett., 1999, 28, 889–890 CrossRef.
- M. Yoshizawa and H. Ohno, Electrochim. Acta, 2001, 46, 1723–1728 CrossRef CAS.
- J. Pernak, A. Czepukowicz and R. Pozniak, Ind. Eng. Chem. Res., 2001, 40, 2379–2383 CrossRef CAS.
- L. C. Branco, J. N. Rosa, J. J. Moura Ramos and C. A. M. Afonso, Chem.–Eur. J., 2002, 8, 3671–3677 CrossRef CAS.
- J. Fraga-Dubreuil, M.-H. Famelart and J. P. Bazureau, Org. Process Res. Dev., 2002, 6, 374–378 CrossRef CAS.
- J. Pernak, A. Olszówka and R. Olszewski, Pol. J. Chem., 2003, 77, 179–187 CAS.
- U. Domanska and A. Marciniak, J. Chem. Thermodyn., 2005, 37, 577–585 CrossRef CAS.
- Q. Liu, M. H. A. Janssen, F. van Rantwijk and R. A. Sheldon, Green Chem., 2005, 7, 39–42 RSC.
- E. Kuhlmann, S. Himmler, H. Giebelhaus and P. Wasserscheid, Green Chem., 2007, 9, 233–242 RSC.
- M. Wang, X. Xiao, X. Zhou, X. Li and Y. Lin, Sol. Energy Mater. Sol. Cells, 2007, 91, 785–790 CrossRef CAS.
- G. Laus, G. Bentivoglio, H. Schottenberger, V. Kahlenberg, H. Kopacka, T. Röder and H. Sixta, Lenzinger Ber., 2005, 84, 71–85 CAS.
- A. M. Leone, S. C. Weatherly, M. E. Williams, H. H. Thorp and R. W. Murray, J. Am. Chem. Soc., 2001, 123, 218–222 CrossRef CAS PubMed.
- J. Pernak and M. Branicka, J. Surfactants Deterg., 2003, 6, 119–123 CrossRef CAS.
- L. J. A. Siqueira and M. C. C. Ribeiro, J. Phys. Chem. B, 2009, 113, 1074–1079 CrossRef CAS PubMed.
- G. D. Smith, O. Borodin, L. Li, H. Kim, Q. Liu, J. E. Bara, D. L. Gin and R. Nobel, Phys. Chem. Chem. Phys., 2008, 10, 6301–6312 RSC.
- F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757–2785 CrossRef CAS PubMed.
- H. Zhao, J. Chem. Technol. Biotechnol., 2016, 91, 25–50 CrossRef CAS PubMed.
- M. Moniruzzaman, K. Nakashima, N. Kamiya and M. Goto, Biochem. Eng. J., 2010, 48, 295–314 CrossRef CAS.
- Z. S. Breitbach and D. W. Armstrong, Anal. Bioanal. Chem., 2008, 390, 1605–1617 CrossRef CAS PubMed.
- C.-M. Jin, C. Ye, B. S. Phillips, J. S. Zabinski, X. Liu, W. Liu and J. M. Shreeve, J. Mater. Chem., 2006, 16, 1529–1535 RSC.
- R. F. Landis, A. Gupta, Y.-W. Lee, L.-S. Wang, B. Golba, B. Couillaud, R. Ridolfo, R. Das and V. M. Rotello, ACS Nano, 2017, 11, 946–952 CrossRef CAS PubMed.
- U. Piotrowska, M. Sobczak, E. Oledzka and C. Combes, J. Appl. Polym. Sci., 2016, 133, 43728 CrossRef.
- M. Mena, A. López-Luna, K. Shirai, A. Tecante, M. Gimeno and E. Bárzana, Bioprocess Biosyst. Eng., 2013, 36, 383–387 CrossRef CAS PubMed.
- D. Omay and Y. Guvenilir, Biocatal. Biotransform., 2013, 31, 132–140 CrossRef CAS.
- J. Olejniczak, J. Sankaranarayanan, M. L. Viger and A. Almutairi, ACS Macro Lett., 2013, 2, 683–687 CrossRef CAS PubMed.
- M. B. Harney, R. R. Pant, P. A. Fulmer and J. H. Wynne, ACS Appl. Mater. Interfaces, 2009, 1, 39–41 CrossRef CAS PubMed.
- M. Chen, H. Zhu, S. Zhou, W. Xu, S. Dong, H. Li and J. Hao, Langmuir, 2016, 32, 2338–2347 CrossRef CAS PubMed.
- A. K. Mandal, S. Sreejith, T. He, S. K. Maji, X.-J. Wang, S. L. Ong, J. Joseph, H. Sun and Y. Zhao, ACS Nano, 2015, 9, 4796–4805 CrossRef CAS PubMed.
- M. M. Cecchini, A. Bendjeriou, N. Mnasri, C. Charnay, F. De Angelis, F. Lamaty, J. Martinez and E. Colacino, New J. Chem., 2014, 38, 6133–6138 RSC.
- B. H. Lipshutz and P. A. Blomgren, Org. Lett., 2001, 3, 1869–1871 CrossRef CAS PubMed.
- D. C. Batesky, M. J. Goldfogel and D. J. Weix, J. Org. Chem., 2017, 82, 9931–9936 CrossRef CAS PubMed.
- P. Hodge and E. Khoshdel, J. Chem. Soc., Perkin Trans. 1, 1984, 195–198 RSC.
- H. M. Relles and R. W. Schluenz, J. Am. Chem. Soc., 1974, 96, 6469–6475 CrossRef CAS.
- G. Anilkumar, H. Nambu and Y. Kita, Org. Process Res. Dev., 2002, 6, 190–191 CrossRef CAS.
- P. A. Sibbald, J. Chem. Educ., 2015, 92, 567–570 CrossRef CAS.
- H. Luo, G. A. Baker and S. Dai, J. Phys. Chem. B, 2008, 112, 10077–10081 CrossRef CAS PubMed.
- R. E. Del Sesto, T. M. McCleskey, C. Macomber, K. C. Ott, A. T. Koppisch, G. A. Baker and A. K. Burrell, Thermochim. Acta, 2009, 491, 118–120 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07733a |
| ‡ Tonset is the intersection of the baseline mass and the tangent of the mass dependence on the temperature curve as decomposition occurs. |
| This journal is © The Royal Society of Chemistry 2018 |