
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxylation and self-assembly of colloidal hydrogenated nanodiamonds by aqueous oxygen radicals from atmospheric pressure plasma jet†
Vít
Jirásek
 *ab,
Štěpán
Stehlík
a,
Pavla
Štenclová
a,
Anna
Artemenko
a,
Bohuslav
Rezek
ac and
Alexander
Kromka
a
*ab,
Štěpán
Stehlík
a,
Pavla
Štenclová
a,
Anna
Artemenko
a,
Bohuslav
Rezek
ac and
Alexander
Kromka
a
aInstitute of Physics, Czech Academy of Sciences, Cukrovarnická 10, 160 21 Prague 6, Czech Republic. E-mail: jirasek@fzu.cz; Tel: +420-266053113
bInstitute of Plasma Physics, Czech Academy of Sciences, Za Slovankou 1782/3, 182 00 Prague 8, Czech Republic
cFaculty of Electrical Engineering, Czech Technical University, Technická 2, 166 27 Prague 6, Czech Republic
First published on 9th November 2018
Abstract
Plasma chemical surface modification of nanoparticles in gas–liquid type reactors enables a controllable, specific, low-cost, and environmentally friendly alternative to wet chemistry methods or thermal and dry plasma treatments. Here the atmospheric pressure radio-frequency microplasma jet (µ-APPJ) operating with 0.6% O2 in He is used to deliver aqueous oxygen radicals (AOR) to the surface of ∼3 nm hydrogenated detonation nanodiamonds (H-DNDs) suspended in water. The AOR-treated H-DND samples are characterized by FTIR and XPS spectroscopies and by AFM and SEM imaging. The main chemical reaction mechanism is identified as the abstraction of surface hydrogen atoms by O or OH radicals and a consequent attachment of the OH group, thereby increasing concentration of alcohols, carboxyls, and aldehydes on the DND's surface. FTIR spectra reveal also a structural re-arrangement of the surface water on the AOR-treated H-DNDs. Yet zeta-potential of AOR-treated H-DNDs still remains positive (decreases from +45 mV to +30 mV). The chemical modification gives rise to formation of nanoscale chain-like aggregates when AOR-treated H-DNDs are deposited on Si substrate.
1. Introduction
Diamond nanoparticles, or nanodiamonds (NDs), recently established themselves in a family of nanomaterials extensively studied in both basic and applied research. The uniqueness of NDs is given by the high rigidity of the diamond core and variable surface chemistry provided by the character of carbon as a building element of organic compounds.1–3 Engineering of NDs' surface in terms of chemistry and structure (e.g. sp3/sp2 carbon ratio, termination of surface carbons) is an essential step in developing suitable nanomaterial especially for bioapplications.4,5Grafting of numerous functional groups on ND's surface is being used to establish a linking interface to other substances, like biomolecules or drugs. Another aim in modifying the surface of NDs' is to tune their physico-chemical properties, like the surface potential or hydrophilicity/hydrophobicity. Such a tuning might, for instance, control the NDs' aggregation,6 electrostatically-driven attachment to various substrates7–9 or efficiency of filling the composites.10,11
A plasma chemical approach as a surface chemistry modification tool brings several benefits over other more conventional methods represented by wet chemistry or thermal treatments. Firstly, it usually does not require the use of toxic or hazardous chemicals. Secondly, production of specific reactive species, namely free radicals, can be optimized by adjusting discharge parameters, gas mixtures, and experimental arrangement. This may promote specific modifications of the nanoparticles' surface which are not easily achieved by the conventional methods.
Various attempts were examined to modify the surface chemistry of a dry nanodiamond powder in established types of plasmatic reactors, including the hydrogenation in microwave CVD reactor,12 oxidation in RF O2 plasma,13 or hydrogenation14 and amination15 in dielectric coplanar surface barrier discharge. However, these “dry” plasma chemical methods commonly suffer from inhomogeneous and inefficient treatment of the whole particles' surface, especially if the particles are in an aggregated state such as conventional detonation nanodiamond (DND).
Modern non-equilibrium atmospheric pressure plasma sources enable to ignite a discharge in or above liquid supplying highly reactive environment without any thermal load on the sample.16 Therefore, approaches working with the NDs in a liquid suspension are considered promising. Prior experiments in that way17–19 resulted predominantly in moderate, specific oxidations of the ND's surface. The underwater pulsed streamer corona discharge treatment of DNDs in saline solution17 induced a moderate oxidation with epoxides as a specific moiety created by plasma oxidation. The treatment, however, also caused a pronounced change of nanodiamonds' physical properties. Their zeta potential (ZP) changed from +40 mV to −20 mV, leading subsequently to their agglomeration and formation of long filaments. Nanodiamonds in water suspension were also treated by RF (450 MHz) quartz capillary jet.18 This treatment increased the oxygen content and decreased ZP from +58 mV to +31 mV as well. Milder approach used the afterglow of DC corona or transient-spark discharge in the air above the suspension.19 The observed minor chemical changes were interpreted as a rearrangement of oxygen-containing surface functional groups rather than direct oxidation of the NDs.
Recently, significant progress in understanding the atmospheric pressure plasma jets (APPJ) physics20,21 and chemistry21 was achieved. This gave rise to an increasing number of proposed APPJ applications, namely in medicine22,23 and material processing.24,25 Low temperature (LT) APPJs operating with noble gas with O2 or H2O admixtures were found to generate high concentrations of strongly oxidizing O or OH radicals in water solutions.26,27 By measuring degradation of phenol in water, it was recently shown27 that for He/O2 plasma, oxidation was driven mainly by O atoms, and, by contrast, in He/H2O and He/O2/H2O plasmas, oxidation appeared to be driven by OH radicals. Several other studies26,28–30 have stated that introducing humidity into working gas dramatically decreases O atoms concentration. Briefly, it can be said that introducing water vapour into the plasma jet may switch from O-rich to OH-rich conditions.
The oxidation of carbon nanomaterials with oxygen radicals provided by LT APPJ was reported only rarely. The Ar/O2 (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) plasma jet fully immersed in the liquid was used for the treatment of carbon nanowalls.31 As a consequence of the plasma treatment, the chemical composition at surface evolved towards the incorporation of oxygen functionalities. The treatment also favored the attachment of smaller particles to larger particles. As another example, the APPJ with 1% O2 in He was successfully used to remove non-diamond carbon from raw DND powder treated in the form of pellets.32 In addition, the process was applied to a direct writing onto an ND/polymer composite layer to create an arbitrary pattern with high spatial resolution and high purity of ND. The atomic oxygen was mentioned as a probable oxidizing specie in this case.
:1) plasma jet fully immersed in the liquid was used for the treatment of carbon nanowalls.31 As a consequence of the plasma treatment, the chemical composition at surface evolved towards the incorporation of oxygen functionalities. The treatment also favored the attachment of smaller particles to larger particles. As another example, the APPJ with 1% O2 in He was successfully used to remove non-diamond carbon from raw DND powder treated in the form of pellets.32 In addition, the process was applied to a direct writing onto an ND/polymer composite layer to create an arbitrary pattern with high spatial resolution and high purity of ND. The atomic oxygen was mentioned as a probable oxidizing specie in this case.
In this paper we characterize activity of the APPJ supplied reactive oxygen species (ROS) towards the surface modification of individual hydrogenated NDs in a colloidal solution. We identify and describe specific oxidation mechanisms via aqueous oxygen-containing radicals (AOR) like O or OH species. We also show impact of the modification on self-assembly of nanodiamonds on substrates.
2. Experimental
2.1 Plasma treatment
The apparatus consists of the µ-APPJ plasma source connected to a small chamber with the working solution (see Fig. 1). The plasma source is a clone of the so-called COST Reference jet33 and the experimental arrangement is similar to that described in Hefny et al. 27 Briefly, plasma is ignited within the small, 1 × 1 × 30 mm volume by the 13.56 MHz sinusoidal voltage of 235 V root-mean-square (the absorbed power < 2 W). | ||
| Fig. 1 Experimental setup. Closed-up views of the discharge chamber (a), arrangement of the jet with the liquid suspension (b and c) and the whole reactor (d). | ||
As a feed gas, helium with the flow of 1.4 standard liters per minute (slm) was used, with the admixture of 0.6% (8.4 sccm) of oxygen. The outlet of jet nozzle was placed 4 mm above the liquid surface so that the visible plasma channel was not in direct contact with the liquid. The chamber consisted of a small, 6 ml glass cylinder filled by 3 ml of the working colloidal solution. The whole system was closed from the surrounding atmosphere by an aluminum cover with an integrated gas exhaust line sealed by the flat rubber ring. Before each experiment, the whole gas inlet tubing was purged by 0.2 slm He + 8.4 sccm O2 for at least 30 min in order to remove ambient air. The sealed chamber with the working solution was then yet purged for 2 min by the final working gas mixture to obtain saturated steady-state conditions. Optimizing this purge time lead to the absence of detectable (>1 × 10−6 M) amount of NO2 − and very low concentration of H2O2. The described conditions were found to lead to the production of O atoms as the major reactive oxygen species (ROS).34 Nevertheless, since also the OH radical takes part in the process, the described plasma treatment is referenced in the text as the “aqueous oxygen-containing radical” treatment or the AOR treatment.
2.2 DNDs preparations and physical characterization
As nanodiamonds we used a commercial DND powder from New Metals and Chemicals, Japan, with a declared nominal size of 5 nm. In order to remove possible residual sp2 carbon and to oxidize the DND surface 400 mg of the as-received DNDs were annealed in air at 450 °C for 30 minutes. About 200 mg of such treated DND powder was then subjected to annealing in pure hydrogen gas performed in a quartz reactor at 700 °C for 6 hours at an atmospheric pressure of hydrogen. 20 mg of the obtained H-DNDs were then dispersed in DI water by means of ultrasonication using a sonotrode (Hielscher 200) lasting for one hour. The 2 ml dispersion was centrifuged (Eppendorf Mini plus) at 14000 × g for 1 h and 1 ml of clear, brownish (approx. H-DND concentration was 2–3 mg ml−1) supernatant was then carefully separated by a micropipette. Dynamic light scattering (DLS) and zeta potentials were measured on Malvern Zetasizer Nano ZS equipped with a helium–neon laser (633 nm) at 25 °C; the scattering angle was 173°. A disposable folded capillary cells were used to eliminate sample cross contamination. DLS measurements were carried out at 20 °C, refractive index of bulk diamond (2.4) and viscosity of pure water (1002 Pa s) at 20 °C was used. The DLS data were plotted as volume/size distributions. The H-DND as well as µ-APPJ plasma treated H-DNDs were deposited on clean p-type Si wafers by immersion of the Si substrates into the ND colloids while 10 min sonication in an ultrasonic bath. After the deposition, the Si wafers were rinsed by DI water and blown by nitrogen. Atomic force microscopy (AFM, Ntegra Prima, NT-MDT) images were acquired in a non-contact regime with approximate cantilever (Nanosensors PPP 150 kHz) oscillation amplitude of 5 nm. The scan range was 200 to 500 nm at the resolution of 512 × 512 points. SEM (MAIA 3, Tescan) images of the deposited DNDs on Si wafers were obtained at acceleration voltage of 15 kV and 300k magnification in the regime of secondary electrons.
2.3 FTIR spectroscopy
Surface chemistry of DND particles before and after the AOR treatment was characterized by grazing angle reflectance Fourier transform infrared (GAR-FTIR) spectroscopy. IR absorbance spectra were measured using an N2 purged Thermo Nicolet 870 spectrometer equipped with the KBr beam splitter and MCT-D detector cooled by liquid nitrogen. A 100 µl of the DND aqueous colloidal solution was applied on the Au mirror by a drop casting just before the GAR-FTIR measurement. Solutions on Au mirrors were heated at 100 °C for 2 min to evaporate water, these samples are denoted as “short-dried”. All samples were additionally dried at 10−7 mbar while heating to 100 °C for 4 days. These samples are denoted as “in vacuo dried”. Optical absorbance was calculated in the standard absorbance units as A = −log(R/R 0), where R is the spectrum measured with NDs and R 0 is the reference (background) spectrum recorded using clean Au mirror prior to the NDs application. In all cases, the spectra represent an average of 128 scans recorded with a resolution of 4 cm−1.The interpretation of the taken FTIR spectra was based on general tables and books35,36 as well as on other works.37–41 For reference, the assignments of the peak wavenumbers to individual vibrational modes are summarized in Table 1.
| ω, cm−1 | Vibrational modea | ω, cm−1 | Vibrational mode |
|---|---|---|---|
| a ν: stretch, δ: deformation, HB – hydrogen bonds. | |||
| 3695 | νOH, free (non HB) | 1515–1670 | δN–H |
| 2800–3650 | νOH, HB: alcohols (3310), carboxyls (3200) | 1645, 1653 | δH2O, (HB H2O⋯H2O), “bulk” |
| 1615, 1635 | δH2O, (HB H2O⋯H2O) | ||
| 2943–47 | νCH3, asymmetric | 1589 | δH2O, non HB, close to C–OH |
| 2952–57 | νC–H, unassigned | 1560–72 | δH2O, non HB, close to C–H |
| 2925–29 | νCH2, asymmetric | 1466 | δCH2, scissoring |
| 2878–84 | νCH3, symmetric | 1385 | δCH3 |
| 2850–55 | νCH2, symmetric | 1331 | νC–C, phonon, N-induced |
| 2742–60 | δC–H, aldehyde (1st overtone) | 1379 | δC–H, aldehyde |
| 2660–70 | νC–H, aldehyde | 1270–1358 | δC–O–H, alcohol, carboxyl, νC–O–C, ether, ester (asymmetric) |
| 1780–1820 |
νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, anhydride, lactone O, anhydride, lactone |
1000–1220 | νC–O, alcohol, carboxyl, νC–O–C, ether, ester (symmetric) |
| 1720–40 |
νCO, carboxyl |
1195 | δCH2 (wagging, twisting)? |
| 1690–1720 |
νCO, aldehyde, ketone |
836 | δCH2 (rocking)? |
2.4 XPS spectroscopy
The X-ray photoelectron spectrometer (Kratos, AXIS Supra) equipped with a hemispherical analyser and a monochromatic Al Kα X-ray source (1486.6 eV) was used for estimation of the chemical composition of the NDs. The XPS spectra were acquired at a constant 90° take-off angle from the analysed area about 700 × 300 mm2. The survey XPS spectra were recorded with the pass energy of 80 eV, whereas the high-resolution spectrum scans were recorded with the pass energy of 20 eV. The measured spectra were processed in Casa XPS software with Shirley background correction procedure and Gaussian line shapes of variable widths. The samples for XPS analysis were prepared in the same way as for the IR analysis, which means by drop casting the colloidal suspension on Au substrate and heating on a hot plate at 100 °C until dry.3. Results and discussion
3.1 Chemical analysis by FTIR
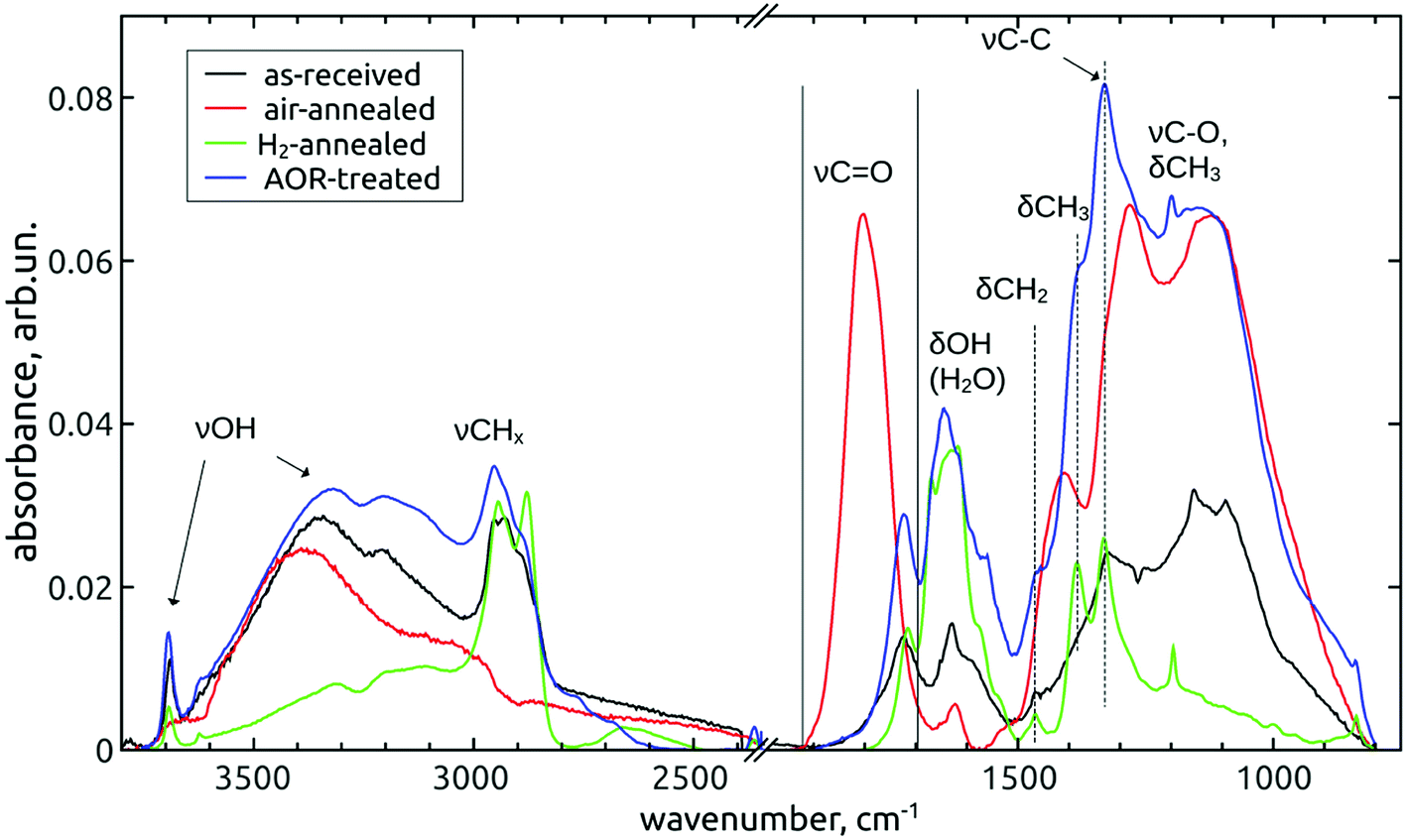 | ||
| Fig. 2 FTIR spectra of as-received and subsequently air-annealed, H2-annealed and AOR-treated NDs. All samples were short-dried. | ||
The as-received DND's are decorated with various oxygen-containing groups, i.e. hydroxyls (3800–2800 cm−1), carbonyls (1718 cm−1), ethers and epoxides (mixed with C–O–H bending and various CH x deformation modes in the broad “fingerprint” region 1500–800 cm−1). There is also a significant contribution of hydrogen-saturated carbon as manifested by the characteristic region of C–H stretching vibrations (3050–2800 cm−1).
The air-annealing resulted in the total removal of these C–H bonds and the introduction of new carbonyl functions (1800 cm−1: lactones, anhydrides, strongly coupled carboxyls13,42) and ethers13 (1275 cm−1, 1115 cm−1). The subsequent hydrogenation introduced again C–H bonds and removed oxygen in ethers and other C–O bonds. The chemical reduction of highly oxidized carbons (lactones, anhydrides, carboxyls) led to the shift of the residual carbonyl peak back to low wavenumber representing lower oxidation states such as aldehydes and ketones. The substantial decrease of C–O contributions in the fingerprint region by H2-annealing augmented the ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CH2 scissoring (1466 cm−1) and –CH3 bending (1385 cm−1) modes, as well as the peak at 1331 cm−1, attributed to the one-phonon diamond mode which was found to appear in FTIR spectra due to the nitrogen substitutions in the diamond lattice.39 The sharp peaks emerging at 1195 cm−1 and 836 cm−1 would also correspond to CH2 deformation modes, namely the twisting or wagging and rocking vibrations, respectively. Although these assignments are somewhat questionable, since still some residual C–O stretch vibration may appear here, the correlation with the hydrogenation process favours the CH
x
deformation vibrations. Note that the free O–H stretch (3695 cm−1) is missing in the spectra of air-annealed NDs.
CH2 scissoring (1466 cm−1) and –CH3 bending (1385 cm−1) modes, as well as the peak at 1331 cm−1, attributed to the one-phonon diamond mode which was found to appear in FTIR spectra due to the nitrogen substitutions in the diamond lattice.39 The sharp peaks emerging at 1195 cm−1 and 836 cm−1 would also correspond to CH2 deformation modes, namely the twisting or wagging and rocking vibrations, respectively. Although these assignments are somewhat questionable, since still some residual C–O stretch vibration may appear here, the correlation with the hydrogenation process favours the CH
x
deformation vibrations. Note that the free O–H stretch (3695 cm−1) is missing in the spectra of air-annealed NDs.
After the AOR treatment, a substantial increase of single-bonded oxygen groups (3800–2800 cm−1 and 1450–1000 cm−1) was found. However, in comparison with the air-annealing, the CH
x
signatures and the CO band were much less affected.
Fig. 3 shows the evolution of the vibrational signatures with increasing time of the AOR treatment.
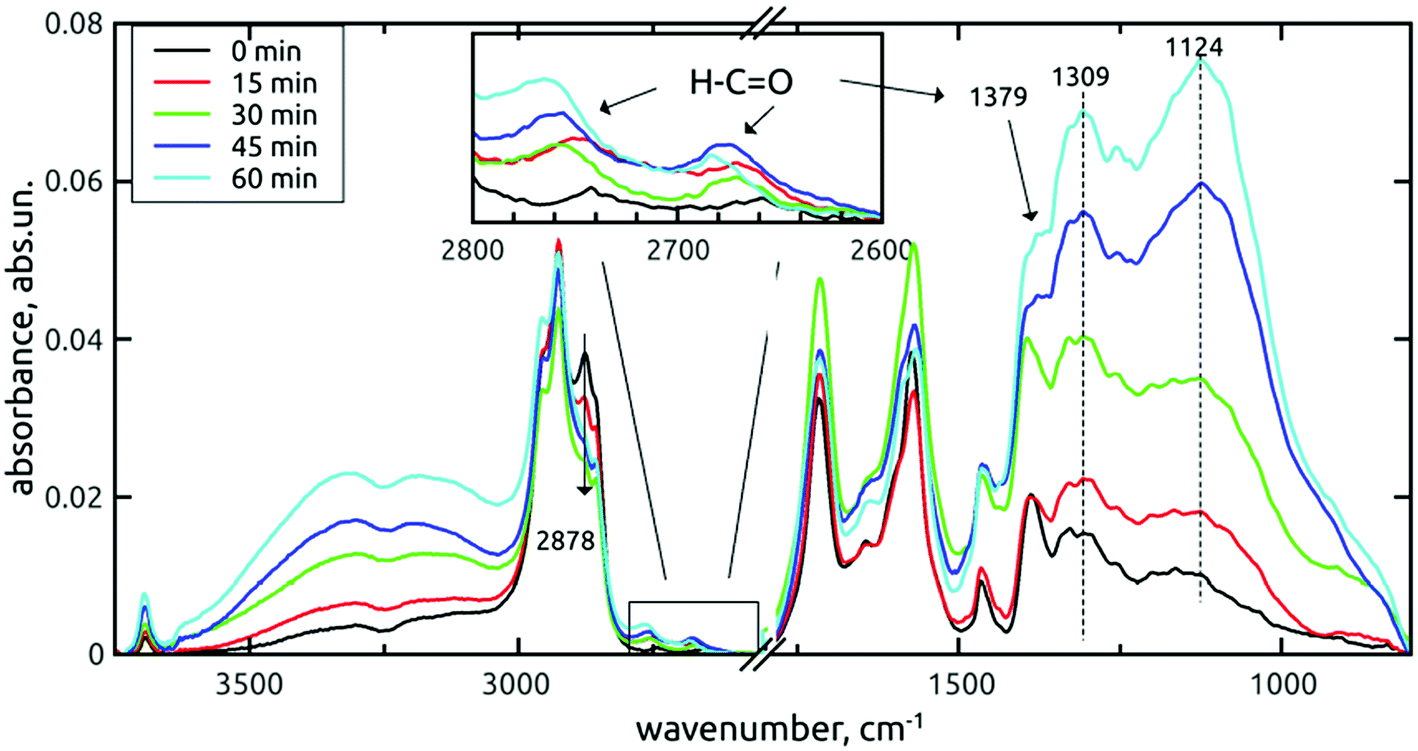 | ||
| Fig. 3 FTIR spectra of H-DNDs modified by the AOR treatment for 0–60 min. Samples were dried in vacuo. | ||
In these survey spectra, a subsequent increase of oxygen-containing surface groups, namely the O–H stretching (3800–2800 cm−1 and 3695 cm−1) and C–O single bond vibrations (1450–1000 cm−1) with the treatment time is clearly visible. The fine structure of the fingerprint band almost does not change up to 30 min, but at 45 and 60 min, increased contributions at 1124 cm−1 (C–O stretch) and 1309 cm−1 (C–O–H bend) are apparent. Due to the correlation with O–H stretches increase, these contributions can be assigned to increasing concentration of alcohols or carboxyls on the AOR treated H-DNDs.
The above mentioned changes were similar on both short-dried and in vacuo dried samples – see Fig. 4.
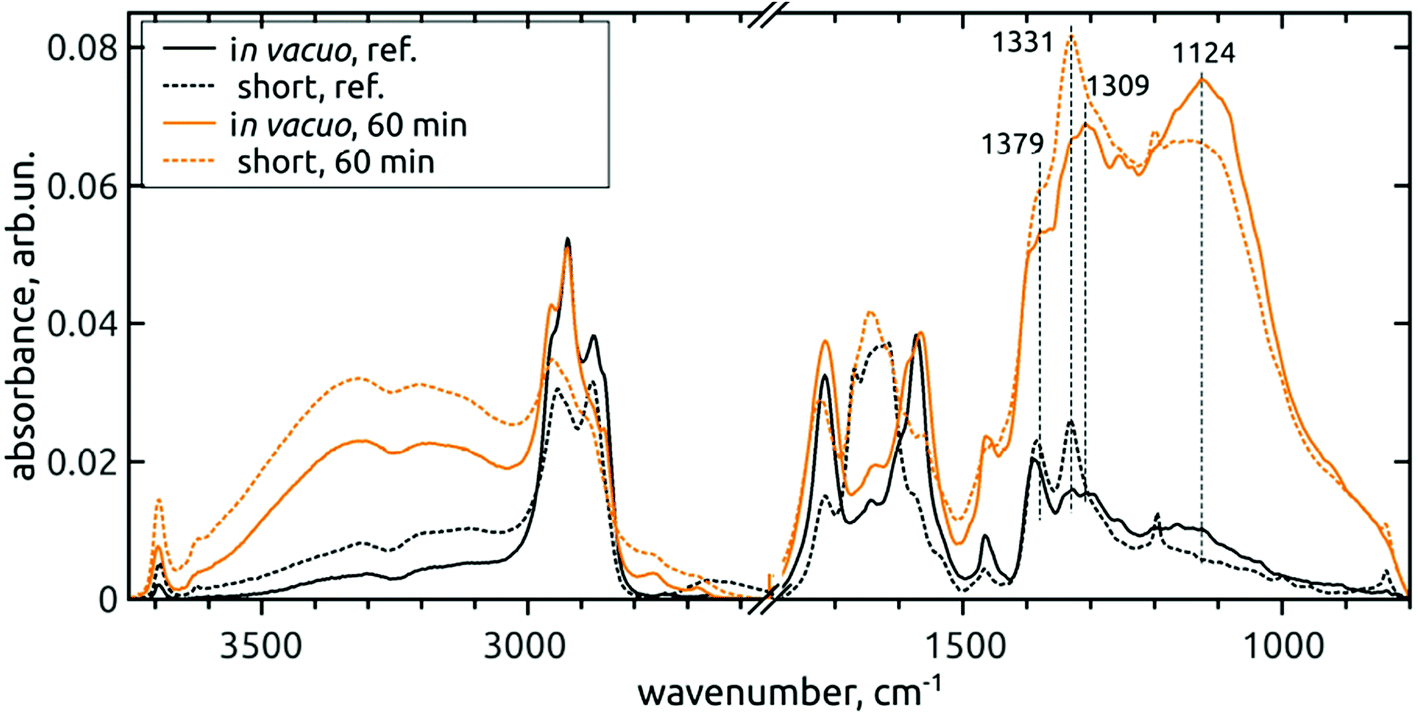 | ||
| Fig. 4 Comparison of FTIR spectra of in vacuo dried (full line) and short-dried (dashed line) reference DNDs and 60 min AOR-treated H-DNDs. | ||
However, local differences were found in the region of C–O vibrations. From the spectra of short-dried samples, we see that the increase of 1309 cm−1 peak is masked by the proximity of the one-phonon diamond peak (1331 cm−1). In addition, the whole 1400–800 cm−1 band, as it increases, its fine-structure is somewhat disappearing, although the two sharp peaks attributed to CH2 deformation vibrations remain well pronounced. On the other hand, the fine structure remains well resolved after 60 min AOR treatment in the case of in vacuo dried samples. This indicates a sensitivity of C–O stretch and C–O–H bend modes to the formation of hydrogen bonds (HB) with adsorbed water molecules. In the case of alcohol surface C–OH groups, high diversity exists as a result of primary, secondary and tertiary alcohols sitting on the different type of possible sites, and forming HB not only with the adsorbed water, but also between each other. This can lead to a high dispersion of vibrational frequencies and formation of a broad, continuous absorption band. We assume that this dispersion can explain the lower resolution of the band for the AOR-oxidized H-DND's, with the CH2 peaks remaining intact.
A very interesting result is the appearance of aldehydes with their typical doublet (2742 cm−1 and 2660 cm−1 for the reference H-DNDs, see the inset in Fig. 3), which are rather unique to nanodiamonds, taking into account the published literature. The 1379 cm−1 peak denoted in Fig. 3 and 4 was assigned to the aldehyde C–H bending mode, whose 1st overtone is in resonance with the C–H stretch and forms the second C–H mode at 2742 cm−1.35 The apparent, yet rather small concentration of aldehydes on the reference H-DNDs is probably a result of carboxyls reduction to aldehydes by H2-annealing and can be regarded as specific for the used sequence of modifications of the as-received DND powder. All three aldehyde peaks increase with the AOR treatment time. Furthermore, the 2742 cm−1 mode is shifted towards higher values due to dynamic dipole coupling,43 as the concentration of aldehydes increases. The aldehydes contribution to the carbonyl peak (1715 cm−1) is less apparent, probably due to their low concentration. However, certain changes on the carbonyl peak can be still seen under higher magnification. This band belongs to the spectral region (1800–1500 cm−1) the most affected by water adsorption, as demonstrated in Fig. 4 too. The corresponding spectral part will be discussed separately in Section 3.1.3.
The band representing the C–H stretches of alkane-type groups was modified, as indicated in Fig. 3, by decreasing the peak at 2878 cm−1 over the treatment time. This small, but specific oxidation of hydrogenated carbon is more corroborated and discussed in next section.
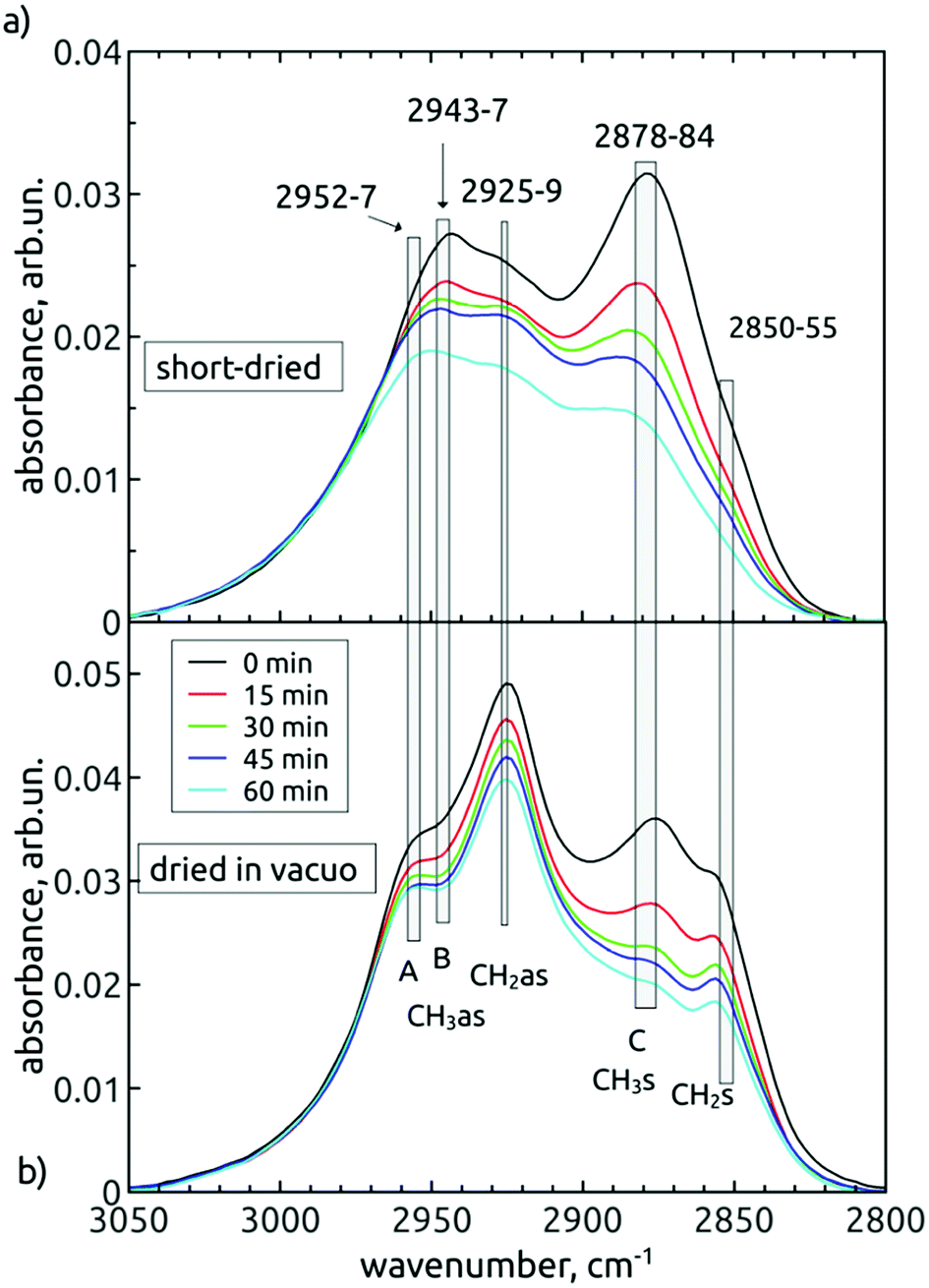 | ||
| Fig. 5 FTIR spectra of the alkane-type C–H stretch region of short-dried (a) and in vacuo dried (b) AOR-treated H-DNDs. The contributions with common temporal behaviour separated by more than 10 cm−1 are treated as individual vibrational modes. The span of their exact positions is denoted as the widths of the vertical grey bars. The spectra were baseline-corrected and rescaled for better clarity. | ||
In different FTIR studies of NDs, most frequently four peaks are resolved in this spectral band. According to literature,38,39,41 these peaks would have to be attributed in both our spectral sets to different wavenumbers. The “better” representation, i.e. the dried in vacuo measurement, leads us to attribute asymmetric (2925 cm−1) and symmetric (2857 cm−1) stretches of the CH2 group and asymmetric (2954 cm−1) and symmetric (2878 cm−1) stretches of the –CH3 group, respectively. However, in Fig. 5b, it follows that the peak “C” was steadily decreasing during the treatment duration relative to the strongest 2925 cm−1 mode. Since the decrease seems not to be accompanied by a substantial decrease of the 2954 cm−1 shoulder (“A”, the proposed asymmetric partner), this would indicate that the “C” peak could be a signature of a monohydride vibration. However, the monohydride vibrations are generally of weak intensity35 and are present rather as a termination of dangling bonds on the surface of truly diamond crystalline lattice of {111}, {110} or {100}-2 × 1 reconstructed surface. We suppose that area of such surfaces on our H-DND's will be much lower in comparison to more or less disordered sp3/sp2 carbon surface layer eventually decorated with small hydrocarbon residues.
The comparison of short-dried samples (Fig. 5a) with the in vacuo dried samples (Fig. 5b) becomes useful here. It is known that water adsorption influences relatively little the frequencies of collective symmetric and asymmetric CH
x
stretches.35 Consequently, we suggested five vibrational modes common to both spectral sets, little shifted by the water adsorption. The observed shifts (3–6 cm−1) are denoted by the width of the grey bars in Fig. 5, while the individual bars denote contributions of individual vibration modes, common to both spectral series. Thus, we distinguish two contributions at the higher-wavenumber edge, i.e. 2952–2957 cm−1 (contribution A) and 2943–2947 cm−1 (contribution B). While the contribution A does not change relatively to CH2as, the contribution B decreases in time, together with the peak C. The other common feature of “B” and “C” is their dominance in the spectra of short-dried samples over the CH2as peak. Interestingly, in the spectra of in vacuo dried samples, we found the reverse situation, i.e. the CH2as is dominant over the “B” and “C” peaks. The origin of this phenomenon is still unclear. Yet we can conclude that a common inspection of both short-dried and in vacuo dried time variations of spectral shapes enabled to reveal that there is a decrease in concentration of –CH3 groups (represented by CH3as – contribution B and CH3s – contribution C) during the AOR treatment.
Such an observation is fully consistent with the increased concentration of aldehydes, resulting from the radical oxidation of –CH3 groups in two reaction sequences. In the first reaction, CH3 group is oxidized to primary alcohol as
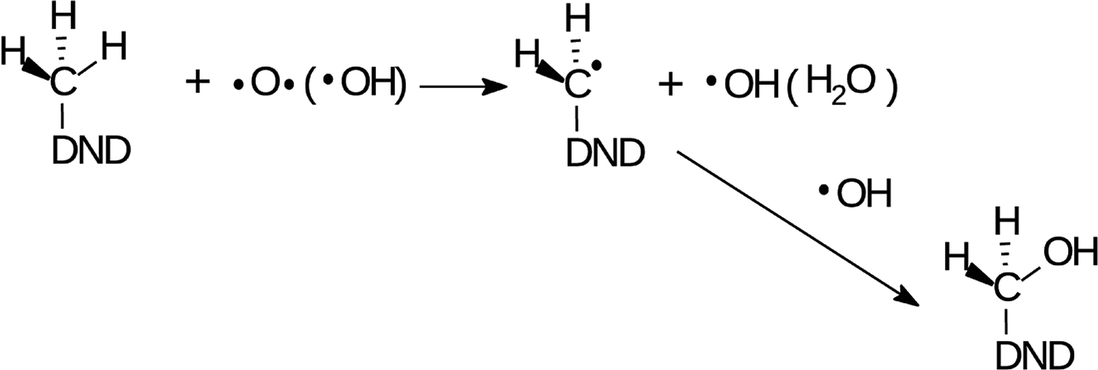
| (1) |
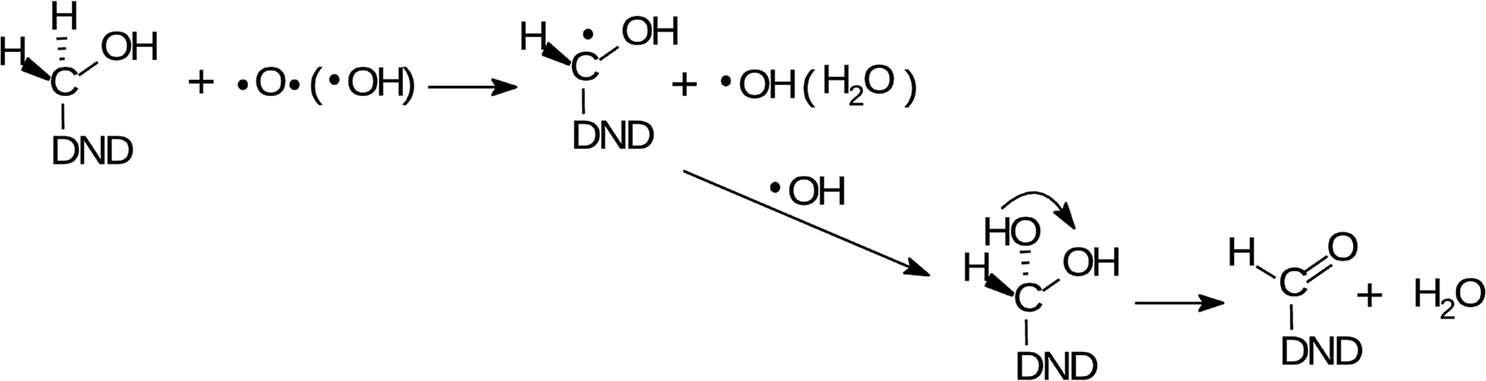
| (2) |
The O and OH radicals are denoted as the acting species in both reactions. It is known that the µ-APPJ jet working with the O2/He gas mixture provides a high flux of O atoms. At the O2/He ratio of 0.6% the two other reactive oxygen species, OH and O3, are an order of magnitude lower.34 Since the ozone is little soluble in water, we will discuss the O and OH radical species. Both species have similar effects on hydrocarbons – the abstraction of hydrogen from the C–H bond, forming the radical C˙ site (1st step in reactions (1) and (2)). This site will be saturated by single-bonding OH, resulting either from the 1st step, reaction of aqueous O atoms with H2O, or delivered from the jet. The efficient hydroxylation of phenol in the same reactor system was reported27,44 with the evidence that the majority of O atoms forming the –OH groups originates from the plasma jet itself and not from water. Nevertheless, the effect of either O or OH produced from the jet and interacting with hydrocarbon residues on the NDs surface seems to be very similar. This assumption was fully confirmed in our experiments when we used humidified He/O2 mixture (with 4000 ppm of water vapour). The FTIR spectra (short-dried) of H-DNDs treated under these “OH-rich” conditions (see Section 1) were very similar to those measured under “O-rich” conditions (see Fig. S1 in the ESI†). This supports the proposed reaction mechanisms (1) and (2) where the action of both radicals leads to the same products.
The two acting radicals are of course able to attack any CH x site on the H-DND surface. It is therefore very probable that alcohol groups of all three types were formed in reactions of the type as a reaction (1) and contribute substantially to already discussed O–H and C–O FTIR signatures.
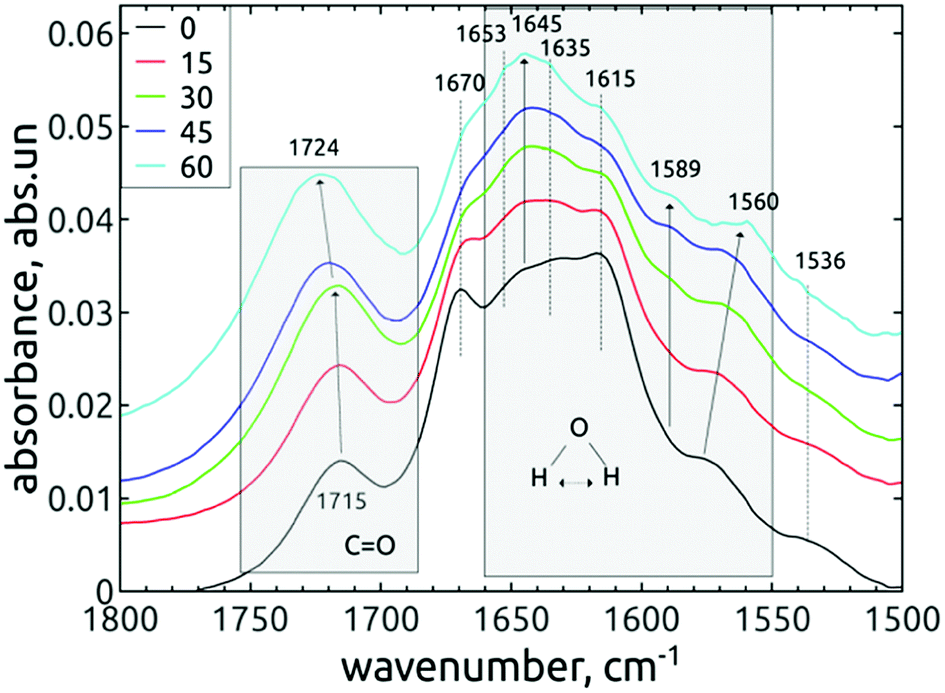 | ||
| Fig. 6 FTIR spectra of short-dried H-DNDs in dependence on the AOR treatment time. The individual spectral lines are stacked for better clarity. The modes which change their relative intensity or position are marked with arrows. | ||
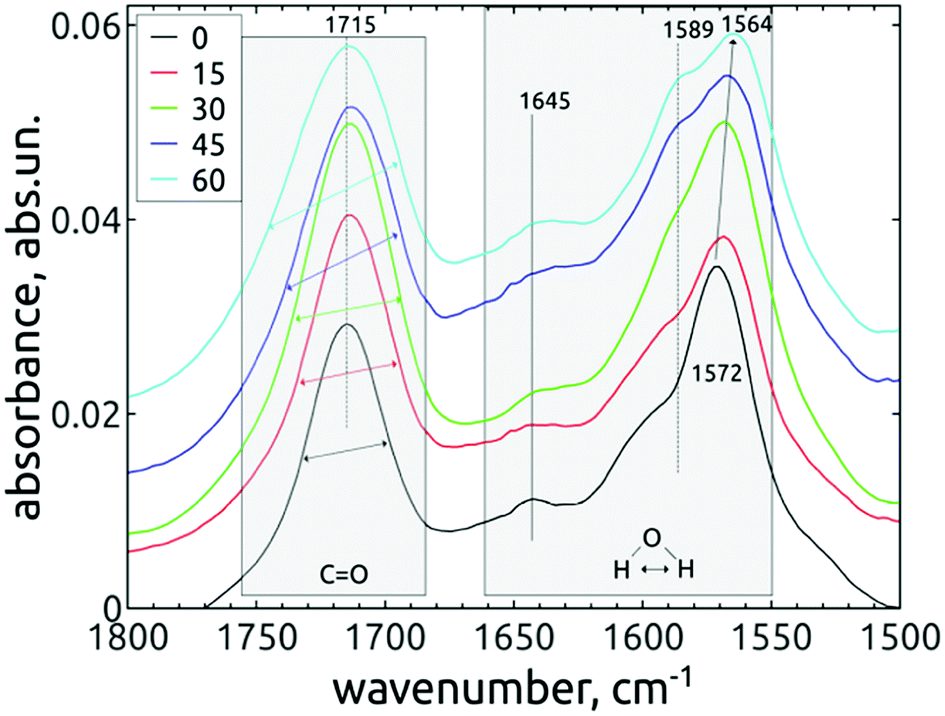 | ||
| Fig. 7 FTIR spectra of in vacuo dried H-DNDs in dependence on the AOR treatment time. The individual spectral lines are stacked for better clarity. The modes which change their relative intensity or position are marked with arrows. | ||
The difference observed between short-dried and in vacuo dried samples, together with conclusions from previous works37,45 indicates that almost all observed peaks in the range 1565–1660 cm−1 may be attributed to bending vibrations of the water molecules adsorbed on DND, although the contribution of other vibrations, e.g. the N–H bending modes, cannot be totally excluded.
The free, non-HB bending mode of gaseous water is 1589 cm−1.37 The hydrogen bonding shifts the value to higher frequencies, in our case 1645 cm−1 for both short-dried and in vacuo dried H-DNDs and also 1615, 1635 and 1653 cm−1 for short-dried NDs. The highest mode, 1670 cm−1 (short-dried samples), is too far from the original free bending mode, but it is missing on in vacuo dried samples. It may be attributed to amide I band, since it is accompanied by 1536 cm−1 shoulder (possibly the amide II band)46 visible also only on the short-dried samples. The 1645 cm−1 mode was reported upon slight water adsorption on NDs with different surface functions, including hydrogenated NDs.37 This mode is probably the major “liquid” water component, since it greatly diminishes when the samples are dried in vacuo. Its increase in relation to other neighbouring modes by the AOR treatment (visible only on short-dried samples) indicates higher susceptibility to water adsorption on the AOR-treated NDs surfaces. This is different from DND's oxidized by air-annealing, which were found45 (by thermogravimetric analysis) to bound less water than H-DNDs. We assume that the main reason for this phenomenon is the different character of the oxidized surface after AOR treatment, preferring termination with OH groups, highly susceptible to water bonding.
In the work of Petit et al.,37 the 1589 cm−1 mode was attributed to non-HB water on ND's, as the frequency is identical with that of a water monomer in the gas phase. Fig. 6 and 7 show that this mode is missing on the reference H-DNDs and becomes more pronounced during the AOR treatment duration. Note that the peaks at 1589–1594 cm−1 can be attributed to CC bonds,41,45 but this can be excluded in our case since the formation of CC bonds is unlikely with the AOR treatment. The continuous enhancement of the 1589 cm−1 peak by the AOR treatment (apparent both on short-dried and in vacuo dried samples) is in correlation with the enhancement of the free O–H stretch at 3695 cm−1 (see Fig. 3). A presence of this free stretch mode has previously been reported for water molecules adsorbed on hydrophobic surfaces.47 However, newly formed carboxyl (COOH) or alcohol moieties may also contribute to this peak.
The lower-lying mode 1572 cm−1 shifts during the AOR treatment down to 1564 cm−1 (see Fig. 7). This value is close to the 1560 cm−1 mode reported for hydrogenated NDs,37 where it was hypothesized to originate from water molecules near to the hydrogenated diamond surface. It is expected that such a water molecule can accept an electron from the diamond hydrogenated surface layer with negative electron affinity. This extra electron would be accepted to the water's antibonding molecular orbital, weaken the dipole strength and decrease the vibrational frequency. 37
The changes of water signatures with the AOR treatment duration have to be related to the modification of the DND's surface chemistry. The surface regions decorated with polar groups, especially OH functions, increased in their area, attracting more water molecules. These water molecules were arranged on both hydrophilic and hydrophobic surface regions, resulting in the increase of contributions of weakly (1560–1589 cm−1) as well as strongly (1616–1653 cm−1) HB-bonded water. In the FTIR spectra in Fig. 3 we can see that the O–H bend region (1565–1645 cm−1) does not follow the highly increasing trend in the broad 3600–2800 cm−1 O–H stretch band with the AOR treatment time. Therefore, we assume that both water (less) and OH groups formed by radical treatment (more) contribute to this O–H stretch band. In Fig. 6 and 7, details of the CO stretch region (maxima at 1715–1724 cm−1) can also be resolved. The observed formation of aldehydes by radical oxidation means that also carboxyls can be formed by further oxidation of aldehydes:
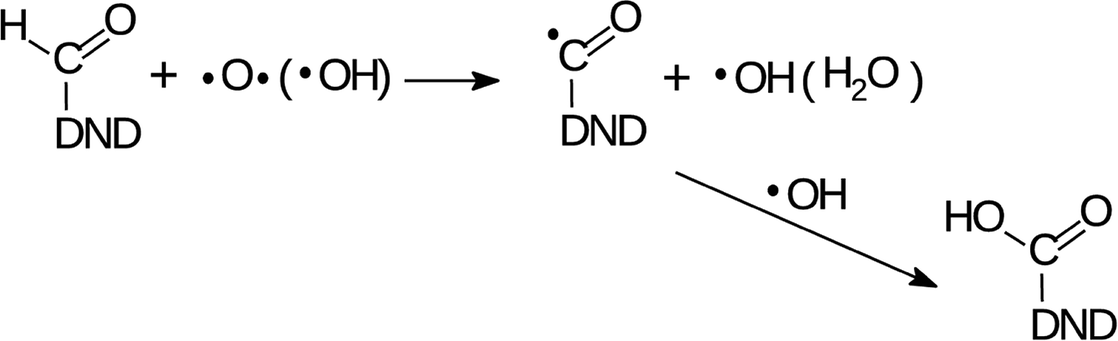
| (3) |
In the spectra of short-dried samples (Fig. 6), the shift of the maxima of the carbonyl peak from 1715 cm−1 to 1724 cm−1 indicates the increasing ratio of carboxyls to aldehydes or ketones. On the other hand, in the spectra of in vacuo dried samples (Fig. 7), the temporal change is observed as a peak broadening. We may explain this different observation between short-dried and in vacuo dried samples by the creation of HB between H2O molecules and polarized CO bond. Simultaneous formation of new aldehydes and conversion of existing aldehydes to carboxyls will broaden the carbonyl band to both sides, as it is shown on in vacuo dried samples. However, a higher amount of adsorbed water, as in the case of short-dried samples, will affect more COOH groups than aldehydes thanks to the O–H⋯O–H hydrogen bond and highlight the COOH contribution, as demonstrated in Fig. 6.
3.2 Morphology, size, and zeta potential of modified H-DNDs
Fig. 8 shows a dependence of dynamic light scattering (DLS) volumetric size and zeta potential (ZP) on the AOR treatment time.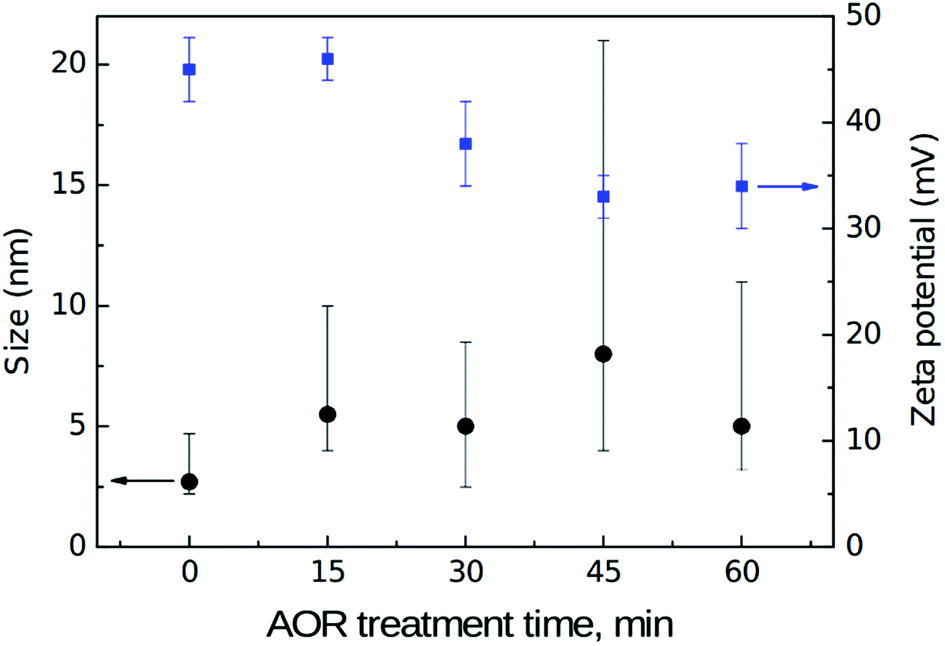 | ||
| Fig. 8 DLS volumetric size (black, left) and zeta potential (blue, right) values in dependence on the AOR treatment time. Note that the “error” bars for size represent FWHM of the asymmetrical size distribution. | ||
DLS volumetric mean size of the reference, initial H-DNDs is about 3 nm which is a significantly smaller size compared to the generally accepted mean size of single DND particles which is typically reported in the 4–6 nm range. The DLS mean size around 3 nm has been indeed reported several times for hydrogenated DNDs.6,7,48 Although the exact origin of this discrepancy remains unclear, several important factors can be still discussed. First, this is an effect of ND hydration shell which is involved in the measured DLS size. Different surface chemistry and related effects, such as the strength of hydrogen bonds between water molecules in the hydration shell, cause its different thickness on H-NDs and O-NDs. Specifically, due to weakening of the hydrogen bonds (HB) on the H-ND surface,37,45 the hydration shell of H-NDs is significantly thinner than of O-NDs resulting in DLS size discrepancy of particles with otherwise identical ND core.9 Second, hydrogen is known to be relatively efficient in disruption of the primary ND clusters (core agglutinates)48 thus helping to release truly individual DND particles. It is probably the hydrogen ability to penetrate into the ND inter-particle boundaries and react there with the ND surface,49 thus passivating the DND surface and efficiently breaking up the core agglutinates. From the results presented here and arguments, it seems that the mean size of truly de-agglutinated DND particles is around 3 nm. Indeed, in the recent work,50 ND mean size of 3.3 nm was reported in the latest generation of NanoAmando™ (NanoCarbon Research Institute, Japan) colloid, which is produced by zirconia microbeads milling of a DND powder. Complex size distribution characterization of the reference H-DNDs by means of atomic force microscopy (AFM), DLS and analytical ultracentrifuge9 can be found in ESI (Fig. S2 in the ESI).† With increasing time of the AOR treatment the DLS size values increases and the size distribution broadens which indicates an aggregation of the primary 3 nm H-DNDs. This is expressed by the (asymmetrical) error bars which represent FWHM values of an average volume distribution obtained from three subsequent DLS scans. The ZP of the reference H-DND colloid is 45 ± 3 mV. The origin of positive ZP of hydrogenated NDs remains unclear although it has been tentatively attributed to protonation of π electrons on sp2-phase carbon48 on the H-ND surface. After the AOR treatment the ZP drops down to approx. 32 mV. Possible aggregation may be awaited from the decrease of ZP although none of the samples were below a colloidal stability limit and the AOR treated colloidal solutions remained visually stable in all cases.
Fig. 9 shows AFM images of reference (a) and AOR-treated (b–e) H-DNDs deposited on Si/SiO x substrate.
 | ||
| Fig. 9 AFM 200 × 200 nm scans of the H-DNDs on Si/SiO x substrate of reference particles (a) and after 15 min (b), 30 min (c), 45 min (d), and 60 min (e) AOR treatment. | ||
Fig. 9a demonstrates very dense and even deposition of the reference H-DNDs, forming preferentially a monolayer of mostly isolated single ∼3 nm particles which is accompanied by very low RMS value of 1.1 nm. With increasing treatment time we observe a gradual coarsening of the deposited layer accompanied with an increase of the RMS values up to nearly 5 nm for the 60 min sample (Fig. 9e). The deposition of positively charged H-NDs to negatively charged Si/SiO x substrate is driven by electrostatic attraction.7,48 It has been shown that intentional decrease of positive ZP of H-NDs to around +30 mV, i.e. to the colloidal stability limit, may lead to very dense coverage and nucleation density.6,9 However we do not observe such an effect here. Although the AOR treatment leads to the ZP decrease to around +30 mV, the AOR-treated H-DNDs rather form aggregates than a denser coverage.
The aggregation tendency is further demonstrated by the SEM analysis of the samples before and after 60 min AOR treatment. The typical SEM morphologies of the pristine and AOR-treated H-DNDs deposited on silicon substrate are shown in Fig. 10.
 | ||
| Fig. 10 SEM morphologies of reference H-DNDs (a) and 60 min AOR-treated H-DNDs (b) deposited on Si substrate. The chain-like structures are highlighted by red colour. | ||
SEM morphologies of both reference (Fig. 10a) and 60 min AOR-treated (Fig. 10b) samples corresponds well to the AFM images (Fig. 9), although DND coverage/particle density looks seemingly higher in AFM than in SEM images. This is an artifact of the AFM measurement caused by convolution of the AFM tip with the DND particles.9 Thus, the SEM provides to some extent complementary data which are not obscured by the AFM tip convolution with the sample. The SEM analysis of the reference and 60 min AOR-treated samples reveals an obvious transition from individual particle-like to chain-like cluster (up to 50 nm) deposits. This behaviour can be explained by electrostatic attraction of facets of specific crystallographic orientations which should be induced by selective oxidation of these facets by AOR.
Lai and Barnard51 calculated the strength of attraction between different pairs of hydrogenated and hydroxylated ND facets of {100}, {110}, and {111} orientation. They predicted that two hydroxylated {110} facets exhibit a substantially higher aggregation probability than any other pair. As we propose that the alcohol groups are the major newly formed moieties by the AOR treatment, the specific hydroxylation of {110} facets seems to be a possible explanation of the observed DNDs' chaining behaviour, which is hardly correlated with any particular value of ZP. The preferential attraction of selected facets would support the tendency to form chain-like aggregates rather than spherical clusters. Note that in our previous work,17 the as-received DNDs treated in the underwater corona discharge formed long filaments after settlement. However, those filaments were 2–15 µm wide and were formed from discharge-treated 0.5–1 µm clusters. Here we obtained shorter chains (<50 nm) mostly made from single DND particles.
3.3 Correlation of qualitative and quantitative analyses
The qualitative changes found in FTIR spectra can be compared with the changes in atomic composition as found by XPS. Table 2 summarizes the dependence of atomic concentrations of chemical elements (O, C, N) on the AOR treatment time.| AOR treatment time, min | O, at% | C, at% | N, at% |
|---|---|---|---|
| 0 | 1.9 | 95.4 | 2.7 |
| 15 | 2.5 | 95.6 | 1.9 |
| 30 | 3.4 | 94 | 2.6 |
| 45 | 3.8 | 94.1 | 2.1 |
| 60 | 3.7 | 94.3 | 2 |
The nitrogen content had not any trend and its value 2–3% corresponds to the intrinsic N usually found in detonation NDs as the remnant of the detonation synthesis.5 On the other hand, the content of oxygen increased two times after the 60 min AOR treatment compared to the reference sample, with almost the entire increment achieved in the first 30 min. This saturation-like trend in the overall oxidation as found by XPS is not entirely consistent with the continuous increase of oxygen-containing groups as revealed in the FTIR spectra. In the first estimation, the saturation should indicate a certain exhausting of the reaction sites on the H-DNDs’ surface. In other words, that radicals prefer to recombine with each other rather than to react with H-DNDs. However, the saturation should be also attributed to the conversion of primary alcohols to aldehydes, which does not increase the overall O content (see reaction (2)). With view to rapidly increasing O–H stretch and C–O related bands, the O content increase in the first 30 min can be most probably related to the formation of alcohol moieties.
The trend in oxygen content is well consistent with the trend in ZP, decrease of which also slows down after 30 min of the AOR treatment (see Fig. 8). It should be emphasized that the ZP represents an average value over the distribution of particles size and their possibly various surface chemistry. Since facets of different crystallographic orientation prefer chemisorption of different functional groups,52 these different facets will be charged in a different manner and contribute with both positive and negative surface charge to the overall surface potential. We have identified the formation of alcohols, aldehydes, and carboxyls during the AOR treatment. While the alcohols and aldehydes are acido-basically neutral (they do not release nor accept H+) under pH ∼ 7, the carboxylic acids release a proton and can contribute to the negative ZP. We thus propose that the decrease in ZP is caused by the replacement of positively charged hydrogenated places with neutral alcohols and aldehydes (major contribution) and formation of negatively charged places with carboxyls (minor contribution).
4. Conclusions
We demonstrated a capability of µ-APPJ in delivering reactive oxygen species towards the surface of individual DNDs in colloidal solution, promoting mild surface chemistry modifications with high impact on self-assembly of nanodiamonds. The main reactive mechanism was proposed as an abstraction of a hydrogen atom by O or OH radical and its exchange with the OH group, resulting in the increased concentration of alcohols, aldehydes and carboxyls on the DND's surface. As a specific and rather unique process for DND's chemistry, the formation of aldehydes from the radical oxidation of –CH3 groups was identified. The cross-correlation of changes during the AOR treatment between short-dried and in vacuo dried DNDs was crucial for interpreting the changes in CH stretch (2800–3050 cm−1) and OH bend (1565–1660 cm−1) spectral regions. The structural arrangement of surface water molecules also changed after the AOR treatment. The surface water molecules re-arranged on both hydrophilic and hydrophobic surface regions, resulting in the increase of contributions of weakly (1560–1589 cm−1) as well as strongly (1616–1653 cm−1) HB-bonded water. Mild oxidation by AOR treatment was confirmed by the overall oxygen content increase from 1.9% to 3.7% after 30–60 min treatments assigned mostly to the OH alcohol moieties. This mild oxidation led to a decrease in ZP close to the colloid-stability limit of +30 mV without disrupting the colloid stability, but with a large impact on H-DND morphology and aggregation tendency. The observed formation of chain-like aggregates on the Si substrate was attributed to the attraction of specific nanodiamonds facets modified by the AOR treatment.The APPJ process can be thus an attractive alternative to more traditional methods allowing controllable, gradual, and specific surface chemistry modifications by means of radical reactions in the nanodiamond suspensions. At the same time, the AOR treatment seems as a suitable tool for tuning self-assembly of nanodiamonds on surfaces. This can be especially useful when producing ordered nanodiamond superstructures, like narrow nucleation rows for selective-area deposition of diamond in photonic devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Health of the Czech Republic grant No. NV15-33018A (V. J., A. A., A. K.) and by Operational Programme Research, Development and Education financed by European Structural and Investment Funds and the Czech Ministry of Education, Youth and Sports (Project No. SOLID21 - CZ.02.1.01/0.0/0.0/16_019/0000760). S. S. acknowledges the Czech Science Foundation (GACR) grant number 18-11711Y. We acknowledge O. Vanek for the AUC measurement.References
- D. Passeri, F. Rinaldi, C. Ingallina, M. Carafa, M. Rossi, M. L. Terranova and C. Marianecci, J. Nanosci. Nanotechnol., 2015, 15, 972–988 CrossRef CAS PubMed.
- J. C. Arnault, in Carbon Nanoparticles and Nanostructures, ed. N. Yang, X. Jiang and D.-W. Pang, Springer International Publishing, Cham, 2016, pp. 1–45 Search PubMed.
- J. C. Arnault and H. A. Girard, Curr. Opin. Solid State Mater. Sci., 2017, 21, 10–16 CrossRef CAS.
- J. Whitlow, S. Pacelli and A. Paul, J. Controlled Release, 2017, 261, 62–86 CrossRef CAS PubMed.
- O. A. Shenderova and G. E. McGuire, Biointerphases, 2015, 10, 30802 CrossRef PubMed.
- T. Yoshikawa, V. Zuerbig, F. Gao, R. Hoffmann, C. E. Nebel, O. Ambacher and V. Lebedev, Langmuir, 2015, 31, 5319–5325 CrossRef CAS PubMed.
- J. Hees, A. Kriele and O. A. Williams, Chem. Phys. Lett., 2011, 509, 12–15 CrossRef CAS.
- O. Shimoni, J. Cervenka, T. J. Karle, K. Fox, B. C. Gibson, S. Tomljenovic-Hanic, A. D. Greentree and S. Prawer, ACS Appl. Mater. Interfaces, 2014, 6, 8894–8902 CrossRef CAS PubMed.
- S. Stehlik, M. Varga, P. Stenclova, L. Ondic, M. Ledinsky, J. Pangrac, O. Vanek, J. Lipov, A. Kromka and B. Rezek, ACS Appl. Mater. Interfaces, 2017, 9, 38842–38853 CrossRef CAS PubMed.
- J. M. Rosenholm, I. I. Vlasov, S. A. Burikov, T. A. Dolenko and O. A. Shenderova, J. Nanosci. Nanotechnol., 2015, 15, 959–971 CrossRef CAS PubMed.
- V. N. Mochalin and Y. Gogotsi, Diamond Relat. Mater., 2015, 58, 161–171 CrossRef CAS.
- H. A. Girard, J. C. Arnault, S. Perruchas, S. Saada, T. Gacoin, J.-P. Boilot and P. Bergonzo, Diamond Relat. Mater., 2010, 19, 1117–1123 CrossRef CAS.
- H. Kozak, Z. Remes, J. Houdkova, S. Stehlik, A. Kromka and B. Rezek, J. Nanopart. Res., 15, 1568–1575 CrossRef.
- A. Kromka, J. Čech, H. Kozak, A. Artemenko, T. Ižák, J. Čermák, B. Rezek and M. Černák, Phys. Status Solidi B, 2015, 252, 2602–2607 CrossRef CAS.
- V. Jirásek, J. Čech, H. Kozak, A. Artemenko, M. Černák and A. Kromka, Phys. Status Solidi A, 2016, 213, 2680–2686 CrossRef.
- P. J. Bruggeman, M. J. Kushner and B. R. Locke, et al. , Plasma Sources Sci. Technol., 2016, 25, 53002 CrossRef.
- V. Jirásek, P. Lukeš, H. Kozak, A. Artemenko, M. Člupek, J. Čermák, B. Rezek and A. Kromka, RSC Adv., 2016, 6, 2352–2360 RSC.
- H. Kozak, A. Artemenko, J. Čermák, V. Švrček, A. Kromka and B. Rezek, Vib. Spectrosc., 2016, 83, 108–114 CrossRef CAS.
- P. Stenclova, V. Celedova, A. Artemenko, V. Jirasek, J. Jira, B. Rezek and A. Kromka, RSC Adv., 2017, 7, 38973–38980 RSC.
- X. Lu and K. Ostrikov, Appl. Phys. Rev., 2018, 5, 31102 Search PubMed.
- S. Reuter, T. von Woedtke and K.-D. Weltmann, J. Phys. D: Appl. Phys., 2018, 51, 233001 CrossRef.
- S. Wu, Y. Cao and X. Lu, IEEE Trans. Plasma Sci., 2016, 44, 134–151 CAS.
- K.-D. Weltmann and T. von Woedtke, Plasma Phys. Controlled Fusion, 2017, 59, 14031 CrossRef.
- K. Ostrikov, E. C. Neyts and M. Meyyappan, Adv. Phys., 2013, 62, 113–224 CrossRef CAS.
- O. V. Penkov, M. Khadem, W.-S. Lim and D.-E. Kim, J. Coat. Technol. Res., 2015, 12, 225–235 CrossRef CAS.
- Y. Gorbanev, D. O'Connell and V. Chechik, Chem.–Eur. J., 2016, 22, 3496–3505 CrossRef CAS PubMed.
- M. M. Hefny, C. Pattyn, P. Lukes and J. Benedikt, J. Phys. D: Appl. Phys., 2016, 49, 404002 CrossRef.
- S. Reuter, J. Winter, S. Iseni, A. Schmidt-Bleker, M. Dunnbier, K. Masur, K. Wende and K.-D. Weltmann, IEEE Trans. Plasma Sci., 2015, 43, 3185–3192 CAS.
- J. Benedikt, D. Schröder, S. Schneider, G. Willems, A. Pajdarová, J. Vlček and V. Schulz-von der Gathen, Plasma Sources Sci. Technol., 2016, 25, 45013 CrossRef.
- T. Murakami, K. Niemi, T. Gans, D. O'Connell and W. G. Graham, Plasma Sources Sci. Technol., 2013, 22, 45010 CrossRef.
- M. D. Ionita, S. Vizireanu and S. D. Stoica, et al. , Eur. Phys. J. D, 2016, 70, 31 CrossRef.
- S. P. Hong, T. H. Kim and S. W. Lee, Carbon, 2017, 116, 640–647 CrossRef CAS.
- J. Golda, J. Held, B. Redeker, M. Konkowski, P. Beijer, A. Sobota, G. Kroesen, N. S. J. Braithwaite, S. Reuter, M. M. Turner, T. Gans, D. O'Connell and V. Schulz-von der Gathen, J. Phys. D: Appl. Phys., 2016, 49, 84003 CrossRef.
- D. Ellerweg, J. Benedikt, A. von Keudell, N. Knake and V. Schulz-von der Gathen, New J. Phys., 2010, 12, 13021 CrossRef.
- R. M. Silverstein, F. X. Webster, D. J. Kiemle and D. L. Bryce, Spectrometric identification of organic compounds, Wiley, Hoboken, NJ, 8th edn, 2015 Search PubMed.
- G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, Wiley, Chichester, 3rd edn, as paperback, 2010 Search PubMed.
- T. Petit, L. Puskar and T. Dolenko, et al. , J. Phys. Chem. C, 2017, 121, 5185–5194 CrossRef CAS.
- T. Petit and L. Puskar, Diamond Relat. Mater., 2018, 89, 52–66 CrossRef CAS.
- T. Jiang and K. Xu, Carbon, 1995, 33, 1663–1671 CrossRef CAS.
- S. Ji, T. Jiang, K. Xu and S. Li, Appl. Surf. Sci., 1998, 133, 231–238 CrossRef CAS.
- O. Shenderova, A. M. Panich, S. Moseenkov, S. C. Hens, V. Kuznetsov and H.-M. Vieth, J. Phys. Chem. C, 2011, 115, 19005–19011 CrossRef CAS.
- V. Jirásek, H. Kozak and Z. Remeš, Adv. Sci., Eng. Med., 2015, 7, 275–278 CrossRef.
- T. A. Newton, J. A. Boiani and M. A. Hines, Surf. Sci., 1999, 430, 67–79 CrossRef CAS.
- J. Benedikt, M. Mokhtar Hefny, A. Shaw, B. R. Buckley, F. Iza, S. Schäkermann and J. E. Bandow, Phys. Chem. Chem. Phys., 2018, 20, 12037–12042 RSC.
- S. Stehlik, T. Glatzel, V. Pichot, R. Pawlak, E. Meyer, D. Spitzer and B. Rezek, Diamond Relat. Mater., 2016, 63, 97–102 CrossRef CAS.
- V. N. Mochalin, I. Neitzel, B. J. M. Etzold, A. Peterson, G. Palmese and Y. Gogotsi, ACS Nano, 2011, 5, 7494–7502 CrossRef CAS PubMed.
- L. F. Scatena, Science, 2001, 292, 908–912 CrossRef CAS PubMed.
- O. A. Williams, J. Hees, C. Dieker, W. Jäger, L. Kirste and C. E. Nebel, ACS Nano, 2010, 4, 4824–4830 CrossRef CAS PubMed.
- A.-I. Ahmed, S. Mandal, L. Gines, O. A. Williams and C.-L. Cheng, Carbon, 2016, 110, 438–442 CrossRef CAS.
- V. I. Korepanov, H. Hamaguchi, E. Osawa, V. Ermolenkov, I. K. Lednev, B. J. M. Etzold, O. Levinson, B. Zousman, C. P. Epperla and H.-C. Chang, Carbon, 2017, 121, 322–329 CrossRef CAS.
- L. Lai and A. S. Barnard, J. Phys. Chem. Lett., 2012, 3, 896–901 CrossRef CAS PubMed.
- A. S. Barnard, Nanoscale, 2018, 10, 8893–8910 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07873d |
| This journal is © The Royal Society of Chemistry 2018 |