
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst synthesis of novel 2,4-bis((E)-styryl)quinoline-3-carboxylate derivatives and their antitumor activity†
Wentao Gao,
Zhiyuan Li,
Qiqi Xu and
Yang Li *
*
Institute of Superfine Chemicals, Bohai University, Jinzhou 121000, P. R. China. E-mail: bhuzh@163.com
First published on 19th November 2018
Abstract
A simple and flexible synthesis of a new series of 2,4-bis((E)-styryl)quinoline-3-carboxylates (3a–t) has been achieved for the first time in good yields via successive Arbuzov/Horner–Wadsworth–Emmons (HWE) reaction in one-pot using the newly-synthesized ethyl 4-(bromomethyl)-2-(chloromethyl)quinoline-3-carboxylate as the substrate. Our synthetic protocol is as attractive and powerful as it is simple, tolerates a wide range of substituents, and does not involve the use of expensive reagents or catalysts. These title compounds belong to a new class of quinoline derivatives and their antitumor activity was assessed on human cancer cell lines (A549, HT29 and T24). The MTT assay showed compounds 3h, 3k and 3t had significant inhibitory activity with IC50 values of 1.53, 1.38 and 2.36 μM against A549 and 1.50, 0.77 and 0.97 μM against HT29, respectively, much better than the reference cisplatin.
Introduction
Among various classes of quinoline derivatives, 2-styrylquinoline (SQL) derivatives represent a particularly fascinating class of pharmacologically active molecules,1 and members of this family are claimed to exhibit a broad spectrum of biological activities such as anti-HIV-1,2 antimicrobial,3 antimalarial4 and anti-Alzheimer activities.5 Especially, recent evaluation of their anti-proliferative effect on tumor cell lines have validated their importance as potential anti-tumor agents.6 For example, Chang et al.6b synthesized a range of styrylquinolines, which were proved to be useful as antitumor agents against the growth of MCF-7 (breast), NCI-H460 (lung), and SF-268 (CNS). In this respect, Mrozek-Wilczkiewicz et al.6c presented a similar series of compounds in a search for new anticancer agents against drug resistant lines and found that these compounds exhibited highly anti-proliferative activity against the human colon carcinoma cell lines. Recently, El-Sayed et al.6d reported a new series of 4,6-disubstituted 2-(4-(dimethylamino)styryl)quinolines, which exhibited significant anti-tumour activities against HepG2 and HCT116 cell lines.Due to their striking biological activities and in order to have structurally diversified molecules for bio-screening, considerable synthetic efforts have been devoted surrounding the 2-styrylquinoline molecular template for further modification and functionalization by both organic and medicinal chemists with the aim of enhancing the potency of this privileged class of compounds.7 For another, quinoline carboxylates are ubiquitous heterocyclic units found extensively in many natural products and pharmaceuticals.8 Despite the existence of extensive reports for their synthesis and functionalization,9 the literature related to synthesis of styrylquinoline carboxylates found to be scarce so far. In this regard, Dabiri et al.10a reported a facile one-pot synthesis of (E)-ethyl 4-phenyl-2-styrylquinoline-3-carboxylate derivatives through tandem Friedländer annulation and Knoevenagel condensation reaction using 1-methylimidazolium trifluoroacetate ([Hmim] TFA) as a Brønsted acidic ionic liquid as shown in Scheme 1a. Similarly, Kumar et al.10b recently described such synthesis with good yields by using In(OTf)3 as the catalyst (Scheme 1b).
 | ||
| Scheme 1 Synthetic route designed for (styryl)quinoline-3-carboxylates. | ||
Promoted by the above reports and in view of structural diversity playing a prominent role towards new drug discovery,11 we felt that it would be a worthwhile endeavor to extend this work to look into the synthesis of 2,4-bis(styryl)-substituted derivatives for current medicinal chemistry needs. As far as we know, the routes available to specifically synthesize the bis(styryl)quinolines has remained virtually unexplored hitherto, probably due to the lack of efficient methods for their synthesis. As such, in connection with our continuing interest in the synthesis of highly valuable quinoline systems,12 herein we would like to report a facile one-pot synthesis and preliminary antitumor activity evaluation of a range of structurally novel and intriguing 2,4-bis((E)-styryl)quinoline-3-carboxylate derivatives through one-pot successive Arbuzov/HWE reaction sequence (Scheme 1c).
Results and discussion
2-Styrylquinoline derivatives are commonly prepared by the condensation reaction of 2-methylquinolines with aromatic aldehydes.3,6b,13 Accordingly, in initial experimental we attempted to synthesize the desired 2,4-bis(styryl)quinoline-3-carboxylate (3a) by using ethyl 2,4-dimethylquinoline-3-carboxylate (I) obtained from literature14 as the substrate for the analogous transformations (Route 1 of Scheme 2). Although the route looked straightforward and attractive for the adaptation in our synthesis, the purported approach was ineffective in our hands, giving poor yields of highly impure products. Moreover, the protocol described for the reaction was plagued by constraints like harsh reaction conditions and the use of a large excess of aldehydes. Attempts to use other reaction conditions such as NBS/TBHP,7c NaOAc/H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOH (1:1),15 and Bmim[BF4]16 also failed.
:AcOH (1:1),15 and Bmim[BF4]16 also failed.
 | ||
| Scheme 2 Synthetic route designed for 2,4-di(styryl)quinoline-3-carboxylate (3a). | ||
As well known, arylmethyl halides are important synthetic intermediates for various transformations in organic synthesis.17 For example, they can proceed to Arbuzov reaction with phosphites to furnish phosphonates.18 On the other hand, the Horner–Wadsworth–Emmons (HWE) olefination reaction of phosphonates with aldehydes has served as the most powerful method for the construction of double bonds.19 Thus, by combination of the two name reactions some versatile methodologies for the synthesis of arylvinyl-substituted compounds have been developed.20 On the basis of these observations, we envisioned that quinoline-3-carboxylate upon bearing two active halomethyl functional groups at its 2- and 4-positions might be a feasible building block to undergo the Arbuzov reaction and subsequent HWE olefination reaction process to access structurally novel and intriguing 2,4-bis((E)-styryl)quinoline-3-carboxylate as shown in Route 2 of Scheme 2.
With this synthetic plan in mind, the first stage involved the preparation of the requisite ethyl 2,4-bis(halomethyl)quinoline-3-carboxylate. In fact, prior to the current investigation we were well aware that the preparation of bis(halomethyl)quinoline would be very interesting and attractive because such quinoline scaffold would have important potential for the flexible construction of a large range of novel and complex quinoline-based derivatives. To our knowledge, no related reports are available concerning its synthesis. To this end, we first conducted the radical bromination reaction of ethyl 2,4-dimethylquinoline-3-carboxylate (I) with N-bromosuccinimide (NBS) (2.2 equiv.) for the construction of 2,4-bis(bromomethyl)quinoline-3-carboxylate (II) as shown in Scheme 3(a). Unfortunately, the reaction was fraught with difficulties associated with excess byproducts, from which the desired bis(bromomethyl)quinoline II could not be easily separated by recrystallization or column chromatography due to their very close polarities. Further varying the amount of NBS used resulted in no improvement as well. Recently, Aitken et al.21 reported the application of benzotrifluoride as the solvent in the NBS bromination reaction of 2,3-dimethylbenzene for the efficient preparation of 2,3-bis(bromomethyl)benzene. Disappointly, by employing this method the reaction always proceeded heterogeneously and no desired product could be obtained.
 | ||
| Scheme 3 Synthesis of ethyl 2,4-bis(halomethyl)quinoline-3-carboxylate (1). | ||
By a literature survey we found that in previous work Ryabukhin et al.22a and Degtyarenko et al.22b reported independently on the synthesis of ethyl 2-chloromethyl-4-methylquinoline-3-carboxylate (IV) through chlorotrimethyl silane (TMSCl)-mediated Friedländer reaction between o-aminoacetophenone (III) and 4-chloroacetoacetic ester. On this basis, it occurred to us that the synthetic route to the required 2,4-bis(halomethyl)quinoline might be achieved by the further radical bromination reaction of IV at its 4-methyl moiety as shown in Scheme 3(b). Thus, we first conducted the condensation reaction of III with ethyl 4-chloro-3-oxobutanoate, closely followed the literature process, in which the corresponding 2-chloromethylquinoline IV was readily obtained in 76% yield. Subsequently, the resulting IV was further subjected to the radical bromination reaction conditions with 1.1 equiv. of NBS. To our delight, in this case the radical bromination reaction proceeded well, giving the expected ethyl 4-(bromomethyl)-2-(chloromethyl)quinoline-3-carboxylate (1) in a good yield of 81% yield with a trace amount of other byproducts as observed by TLC.
With the newly-synthesized bis(halomethyl)quinoline 1 in hand, our attention was turned to its Arbuzov reaction with triethyl phosphite. Initially, ZnBr2 was used as a catalyst in dichloromethane media at room temperature in accordance with the reaction conditions of the literature.18 Although the methodology is elegant and impressive, our attempt to extend this approach to our synthesis was unfruitful, and the reaction did not proceed satisfactorily, giving a poor yield of the corresponding product (27%). Interestingly, we found that by only refluxing 1 in an excess of triethyl phosphite (8 mL) without added catalyst, the Arbuzov reaction could be performed very smoothly,12c nearly quantitative conversion to the corresponding (quinolinylmethyl)phosphonate A within 3 h as monitored by TLC (Rf = 0.64, 10% ethyl acetate/petroleum ether in a 3 time run) and LC-MS (Scheme 4). As the reaction did not involve the use of additional organic solvents or catalysts and the intermediate A was obtained in nearly quantitative yield, we speculated that purification at this stage was unnecessary and the subsequent HWE olefination reaction would proceed successively in a one-pot manner as shown in Scheme 4.
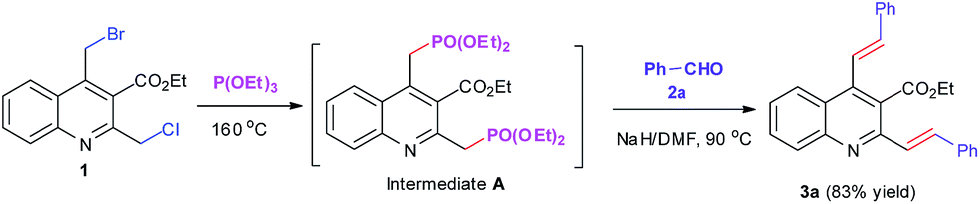 | ||
| Scheme 4 One-pot synthesis of 2,4-bis((E)-styryl)quinoline-3-carboxylate (3a). | ||
Accordingly, we simply evaporated the excess triethyl phosphite to dryness under reduced pressure and conducted in situ the HWE olefination reaction by adding directly a solution of 2.2 equiv. of benzaldehyde (2a) in DMF with the presence of NaOMe as the base to the residue according to a similar literature report for the preparation of bis-styrylbenzenes.23 However, our attempt to follow the reported protocol was frustrated by a poor yield with a number of side-products being evident. We further investigated the reaction using ButOK as the base in THF solution according to the protocol of literature.20a However, once again we obtained a complex mixture of the products, from which the expected 2,4-bis((E)-styryl)quinoline-3-carboxylate (3a) was isolated only in 14% yield. Attempt to use phase-transfer catalysis system, which was reported to yield good results in this type of reaction,24 was also to no avail without the formation of 3a even in trace amounts. After these unsuccessful trials, we were delight to find that the use of NaH as the base in DMF media was very suitable to promote the reaction, and 2.2 equiv. of NaH at 90 °C reaction temperature was found to be optimal to complete the reaction, giving 3a in a good yield of 83%. To show the effect of the one-pot successive synthesis, we also attempted to a step-wise procedure by isolating the intermediate A and then subjecting it to the HWE reaction. Compared with the one-pot sequential procedure, the step-wise reaction proceeded remarkably slower and the yield of 3a obtained was much lower (61%).
Theoretically, the structure of 3a should exist as (E)- and/or (Z)-geometry due to the presence of the exocyclic vinyl double bond. The most diagnostic evidence for the geometry of vinyl moiety was based on the analysis of its 1H NMR spectrum. As shown in Fig. 1, the 1H NMR spectrum clearly demonstrated four arising vinylic protons CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH appearing as four doublets at 6.99, 7.29, 7.81, and 8.03 ppm, respectively, with large coupling constants J ∼ 16.0 Hz, which are typical value for the E-stereoisomer of the 2,4-bis(styryl) moiety.
CH appearing as four doublets at 6.99, 7.29, 7.81, and 8.03 ppm, respectively, with large coupling constants J ∼ 16.0 Hz, which are typical value for the E-stereoisomer of the 2,4-bis(styryl) moiety.
 | ||
| Fig. 1 1H NMR spectrum of compound 3a. | ||
Due to the simplicity of the one-pot reaction procedure along with the satisfactory yield obtained, no further optimization in reaction conditions was necessary, and the above reaction condition was chosen for the following work. Thereafter, to investigate the scope and limitation of the synthetic method a variety of aromatic aldehydes 2b–n containing mono, di or tri substituents with differing electronic properties were subjected to the one-pot sequence in a similar fashion. As listed in Table 1, all these different substituted aromatic aldehydes readily engaged in the one-pot reaction procedure, delivering a series of 2,4-bis((E)-styryl)quinoline-3-carboxylate derivatives (3b–n) with good yields ranging from 70% to 85% (entries 2–14). It appeared that the nature and site of substituent present in the aromatic aldehydes had little impact on the success of the transformation, neither in product yield nor in reaction rate. For example, the compound 3c with electron-donating methyl group and 3n bearing slightly electron-withdrawing bromo group were obtained in similar yields of 85% and 84%, respectively, showing little distinction (entries 3 and 14). Moreover, the steric effect was not observed. When di-ortho substituted benzaldehyde (2i), as a consequence of its greater steric demand, was used, no significant difference in the reaction outcome was observed, giving product 3i in a comparable yield of 80% (entry 9). However, upon aromatic aldehydes bearing strong electron-withdrawing groups such as NO2 and CN, the reaction produced intractable mixtures as observed on TLC, from which we could not obtain any of the desired products in appreciable yields. Further, we extended its scope to naphthaldehydes (2o and 2p) and heteroaromatic aldehydes such as thiophene-2-carbaldehyde (2q), furan-2-carbaldehyde (2r) and picolinaldehyde (2s). As expected, these species were also viable substrates for this one-pot transformation, invariably furnishing the expected products 3o–s in satisfactory yields of 72–78% (entries 15–19). Encouraged by these results and with the aim of further diversifying our work on the synthesis of this class of 2,4-di((E)-styryl)quinoline derivatives accessible by this method, we became very interested in seeing whether ferrocenylalhehyde (2t) would exhibit a similar reactivity. Gratifyingly, ferrocenylalhehyde was equally amenable to the reaction process without any experimental difficulties, giving the corresponding 2,4-di(ferrocenylvinyl)-substituted analogue 3t in a good yield of 71% (entry 20, Table 1). The synthesized 3t would be very attractive as this class of ferrocenylvinyl heterocycles usually exhibit potent biological activities.25
|
|||
|---|---|---|---|
| Entry | R | Product | Yielda (%) |
| a Isolated yield. | |||
| 1 | Ph | 3a | 83 |
| 2 | 2-MeC6H4 | 3b | 79 |
| 3 | 4-MeC6H4 | 3c | 85 |
| 4 | 2-MeOC6H4 | 3d | 76 |
| 5 | 4-MeOC6H4 | 3e | 80 |
| 6 | 4-EtOC6H4 | 3f | 71 |
| 7 | 2,4-diMeC6H3 | 3g | 84 |
| 8 | 3,4-diMeOC6H3 | 3h | 79 |
| 9 | 2,5-diMeOC6H3 | 3i | 80 |
| 10 | 5-Piperonyl | 3j | 75 |
| 11 | 3,4,5-TriMeOC6H2 | 3k | 77 |
| 12 | 2,4,5-TriMeOC6H2 | 3l | 70 |
| 13 | 2,6-DiClC6H4 | 3m | 78 |
| 14 | 4-BrC6H4 | 3n | 84 |
| 15 | 1-Naphthyl | 3o | 75 |
| 16 | 2-Naphthyl | 3p | 78 |
| 17 | 2-Furyl | 3q | 76 |
| 18 | 2-Thienyl | 3r | 72 |
| 19 | 2-Pyridyl | 3s | 73 |
| 20 | Ferrocenyl | 3t | 71 |

Having a series of the targeted compounds 3a–t in hand, a preliminary investigation for their in vitro anti-tumor activities against human cancer cell lines such as A549, HT29 and T24 was carried out, in which the potency was expressed as inhibition rate with cisplatin as a reference. Cell viability was assessed 72 h after treatment by conducting an MTT assay. The preliminary screening results were listed in Table 2. We observed that all of the tested compounds at 2 μg ml−1 concentration showed poor inhibition of cancer cell lines growth on the three studied cell lines, whereas at the concentration of 20 μg ml−1 the methoxyl-substituted compounds 3d, 3e and 3h–l were observed to exert relatively good anticancer activity against A549 and HT29 compared to other substituents. Further, the presence of the substituent in the position of ortho-, meta-, and/or para- of phenyl ring allowed us for the structure–activity-relationship (SAR) study. We observed that compounds 3h, 3j and 3k having methoxyl groups at both meta and para-position of the benzene ring showed better inhibitory activity (>90%). The results suggested that position of methoxyl group on meta and para-position of the styryl moiety played an important role for inhibitory activity of the compounds. The naphthyl and hetaryl-substituted compounds 3o–s displayed low activities for these types of cancer cell lines. The SAR demonstrated that the modification of phenyl ring of styryl moiety by naphthyl or heteroaromatic ring could not significantly affect the inhibitory activity of the compounds. In contrast, ferrocenyl-substituted compound 3t was found to exhibit significant antitumor activity with the inhibitory ratios of 86.17% against A549 and 104.17% against HT29, much better than the reference cisplatin. This result indicated that introduction of a ferrocenyl group could also contribute to improve the antitumor properties significantly.
| Compd | A549 | HT-29 | T-24 | |||
|---|---|---|---|---|---|---|
| 2 μg ml−1 | 20 μg ml−1 | 2 μg ml−1 | 20 μg ml−1 | 2 μg ml−1 | 20 μg ml−1 | |
| 3a | 12.77 | 38.41 | 20.12 | 48.30 | 15.08 | 37.79 |
| 3b | 5.50 | 62.54 | 29.96 | 52.71 | 12.92 | 28.20 |
| 3c | 26.36 | 41.17 | 29.34 | 55.83 | 13.55 | 39.71 |
| 3d | 21.89 | 66.83 | 27.53 | 64.75 | 14.23 | 46.84 |
| 3e | 33.48 | 72.86 | 35.75 | 75.56 | 11.07 | 28.35 |
| 3f | 22.05 | 65.51 | 9.16 | 48.74 | 6.94 | 31.08 |
| 3g | 12.00 | 26.10 | 7.32 | 26.46 | 15.27 | 46.23 |
| 3h | 28.69 | 92.65 | 51.63 | 94.76 | 8.22 | 25.72 |
| 3i | 24.93 | 78.92 | 36.02 | 74.13 | 16.86 | 40.58 |
| 3j | 37.30 | 94.45 | 61.46 | 92.85 | 30.19 | 53.46 |
| 3k | 18.83 | 97.78 | 69.54 | 98.81 | 29.18 | 40.78 |
| 3l | 49.40 | 86.66 | 43.20 | 85.51 | 33.84 | 51.70 |
| 3m | −10.35 | −8.33 | −9.69 | −9.09 | 2.72 | 24.04 |
| 3n | 6.81 | 37.61 | 18.33 | 47.54 | 12.12 | 28.68 |
| 3o | 25.19 | 65.57 | 30.55 | 34.66 | 13.66 | 32.89 |
| 3p | −5.64 | 45.30 | 9.43 | 47.94 | 14.57 | 35.81 |
| 3q | 18.58 | 22.86 | 16.29 | 54.26 | 27.14 | 42.18 |
| 3r | 12.11 | 13.78 | 5.23 | 43.80 | 26.51 | 39.42 |
| 3s | −2.88 | 3.38 | 11.55 | 38.42 | 3.61 | 24.85 |
| 3t | −6.61 | 86.17 | 60.49 | 104.17 | 15.69 | 31.29 |
| Cisplatin | 70.04 | 82.25 | 11.21 | 65.92 | 42.26 | 56.81 |
Basing from the above results, we decided to study the concentration-response of the potent derivatives 3h–l and 3t by obtaining their IC50 (i.e., the concentration required for 50% cell viability) values in suppressing cell growth of A549 and HT-29 cells. As listed in Table 3, compounds 3h, 3k, and 3t exhibited more potent in vitro anti-cancer activity against A549 and HT29 with IC50 values of 1.53, 1.38, 2.36 μM and 1.50, 0.77, 0.97 μM (entries 1, 4 and 6), respectively, which were much superior to cisplatin (entry 7). The data further confirmed that the presence of methoxy substituents at both meta and para-positions of benzene ring or the introduction of ferrocene moiety played an important role in the antitumor activity of 2,4-di((E)-styryl)quinoline-3-carboxylate derivatives.
Conclusions
In conclusion, we have described an easy access to a series of structurally novel and intriguing 2,4-bis((E)-styryl)quinoline-3-carboxylate derivatives through one-pot Arbuzov/HWE reaction procedure. Experimental simplicity, inexpensive reagents and satisfactory yields would contribute to the usefulness of this method. The preliminary bioassay for their antitumor activity revealed that some of the title compounds such as 3h, 3k and 3t exerted excellent anti-cancer activity against A549 and HT29 with IC50 values much better than the reference cisplatin. These results might provide valuable information for designing anticancer lead compounds, and currently further work, mainly focusing on structural optimization, application and exploration of the enormous potential of these compounds, is underway in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (Grant No. 21476028 and 21402011) and the Natural Science Foundation of Liaoning Province (Grant No. 201602006).References
- (a) M. V. Rubtsov, G. N. Pershin, N. A. Yanbuktin, L. A. Pelenitsina, T. J. Gurevich, N. A. Novitskaya, S. N. Milovanova and S. A. Vichkanova, J. Med. Chem., 1959, 2, 113 CrossRef; (b) K. Mekouar, J. F. Mouscadet, D. Desmaële, F. Subra, H. Leh, D. Savouré, C. Auclair and J. d'Angelo, J. Med. Chem., 1998, 41, 2846 CrossRef CAS PubMed.
- (a) M. Ouali, C. Laboulais, H. Leh, D. Gill, D. Desmaële, K. Mekouar, F. Zouhiri, J. d'Angelo, C. Auclair, J.-F. Mouscadet and M. L. Bret, J. Med. Chem., 2000, 43, 1949 CrossRef CAS PubMed; (b) J. Polanski, F. Zouhiri, L. Jeanson, D. Desmaële, J. d'Angelo, J. F. Mouscadet, R. Gieleciak, J. Gasteiger and M. L. Bret, J. Med. Chem., 2002, 45, 4647 CrossRef CAS PubMed.
- W. Cieslik, R. Musiol, J. E. Nycz, J. Jampilek, M. Vejsova, M. Wolff, B. Machura and J. Polanski, Bioorg. Med. Chem., 2012, 20, 6960 CrossRef CAS PubMed.
- B. F. Roberts, Y. Zheng, J. Cleaveleand, S. Lee, E. Lee, L. Ayong, Y. Yuan and D. Chakrabarti, Int. J. Parasitol.: Drugs Drug Resist., 2017, 7, 120 Search PubMed.
- X. Q. Wang, C. L. Xia, S. B. Chen, J. H. Tan, T. M. Ou, S. L. Huang, D. Li, L. Q. Gu and Z. S. Huang, Eur. J. Med. Chem., 2015, 89, 349 CrossRef CAS PubMed.
- (a) B. Podeszwa, H. Niedbala, J. Polanski, R. Musiol, D. Tabak, J. Finster, K. Serafin, M. Milczarek, J. Wietrzyk, S. Boryczka, W. Mol, J. Jampilek, J. Dohnal, D. S. Kalinowskif and D. R. Richardsonf, Bioorg. Med. Chem. Lett., 2007, 17, 6138 CrossRef CAS PubMed; (b) F. S. Chang, W. Chen, C. Wang, C. C. Tzeng and Y. L. Chen, Bioorg. Med. Chem., 2010, 18, 124 CrossRef CAS PubMed; (c) A. Mrozek-Wilczkiewicz, E. Spaczynska, K. Malarz, W. Cieslik, M. Rams-Baron, Vl. Kryštof and R. Musiol, PLoS One, 2015, 10, e0142678 CrossRef PubMed; (d) M. A.-A. El-Sayed, W. M. El-Husseinya, N. I. Abdel-Aziz, A. S. El-Azab, H. A. Abuelizz and A. A.-M. Abdel-Aziz, J. Enzyme Inhib. Med. Chem., 2017, 33, 199 CrossRef PubMed.
- (a) R. Musiol, B. Podeszwa, J. Finster, H. Niedbala and J. Polanski, Monatsh. Chem., 2006, 137, 1211 CrossRef CAS; (b) F. Sliman, M. Blairvacq, E. Durieu, L. Meijer, J. Rodrigo and D. Desmaële, Bioorg. Med. Chem. Lett., 2010, 20, 2801 CrossRef CAS PubMed; (c) L. Gong, L. J. Xing, T. Xu, X. P. Zhu, W. Zhou, N. Kang and B. Wang, Org. Biomol. Chem., 2014, 12, 6557 RSC.
- (a) A. Baba, N. Kawamura, H. Makino, Y. Ohta, S. Taketomi and T. Sohda, J. Med. Chem., 1996, 39, 5176 CrossRef CAS PubMed; (b) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC; (c) Y. Liu, Y. Zhao, X. Zhai, X. Feng, J. Wang and P. Gong, Bioorg. Med. Chem., 2008, 16, 6522 CrossRef CAS PubMed; (d) V. Krishnakumar, F.-R. N. Khan, B. K. Mandal, S. Mitta, R. Dhasamandha and V. N. Govindan, Res. Chem. Intermed., 2012, 38, 1819 CrossRef CAS.
- (a) H. Huang, H. Jiang, K. Chen and H. Liu, J. Org. Chem., 2009, 74, 5476 CrossRef CAS PubMed; (b) H. Venkatesan, F. M. Hocutt, T. K. Jones and M. H. Rabinowitz, J. Org. Chem., 2010, 75, 3488 CrossRef CAS PubMed; (c) S. K. Gadakh, S. Dey and A. Sudalai, Org. Biomol. Chem., 2016, 14, 2969 RSC.
- (a) M. Dabiri, P. Salehi, M. Baghbanzadeh and M. S. Nikcheh, Tetrahedron Lett., 2008, 49, 5366 CrossRef CAS; (b) D. Kumar, A. Kumar, M. M. Qadri, M. I. Ansari, A. Gautam and A. K. Chakraborti, RSC Adv., 2015, 5, 2920 RSC.
- (a) A. M. M. Jørgensen and J. T. Pedersen, J. Chem. Inf. Comput. Sci., 2001, 41, 338 CrossRef; (b) R. E. Dolle and K. H. Nelson, J. Comb. Chem., 1999, 1, 235 CrossRef CAS PubMed.
- (a) W. T. Gao, J. Liu, Y. Jiang and Y. Li, Beilstein J. Org. Chem., 2011, 7, 210 CrossRef CAS PubMed; (b) W. T. Gao, G. H. Lin, Y. Li, X. Y. Tao, R. Liu and L. J. Sun, Beilstein J. Org. Chem., 2012, 8, 1849 CrossRef CAS PubMed; (c) W. T. Gao, X. D. Xing, Y. Li and S. Lan, Tetrahedron, 2014, 70, 2180 CrossRef CAS; (d) W. T. Gao, X. B. Fu, X. F. Zhang, Y. N. Zhao, D. F. Wang and Y. Li, Tetrahedron Lett., 2016, 57, 4145 CrossRef CAS.
- M. Normand-Bayle, C. Bénard, F. Zouhiri, J. F. Mouscadet, H. Leh, C. M. Thomas, G. Mbemba, D. Desmaële and J. d'Angelo, Bioorg. Med. Chem. Lett., 2005, 15, 4019 CrossRef CAS PubMed.
- R. Varala, R. Enugala and S. R. Adapa, Synthesis, 2006, 3825 CAS.
- A. Kamal, A. Rahim, S. Riyaz, Y. Poornachandra, M. Balakrishna, C. G. Kumar, S. M. A. Hussaini, B. Sridhar and P. K. Machiraju, Org. Biomol. Chem., 2015, 13, 1347 RSC.
- R. Sharma, M. Abdullaha and S. B. Bharate, J. Org. Chem., 2017, 82, 9786 CrossRef CAS PubMed.
- (a) E. Chorell, C. Bengtsson, T. S. L. Banchelin, P. Das, H. Uvell, A. K. Sinha, J. S. Pinkner, S. J. Hultgren and F. Almqvist, Eur. J. Med. Chem., 2011, 46, 1103 CrossRef CAS PubMed; (b) G. L. Lu, A. Ashoorzadeh, R. F. Anderson, A. V. Patterson and J. B. Smaill, Tetrahedron, 2013, 69, 9130 CrossRef CAS; (c) S. V. Amosova, I. A. Novokshonova, M. V. Penzik, A. S. Filippov, A. I. Albanov and V. A. Potapov, Tetrahedron Lett., 2017, 58, 4381 CrossRef CAS.
- G. G. Rajeshwaran, M. Nandakumar, R. Sureshbabu and A. K. Mohanakrishnan, Org. Lett., 2011, 13, 1270 CrossRef CAS PubMed.
- (a) J. T. Markiewicz, D. J. Schauer, J. Löfstedt, S. J. Corden, O. Wiest and P. Helquist, J. Org. Chem., 2010, 75, 2061 CrossRef CAS PubMed; (b) K. Kobayashi, K. Tanaka III and H. Kogen, Tetrahedron Lett., 2018, 59, 568 CrossRef CAS.
- (a) E. Diez-Barra, J. C. Garcia-Martinez and J. Rodríguez-López, Tetrahedron Lett., 1999, 40, 8181 CrossRef CAS; (b) L. Viau, O. Maury and H. L. Bozec, Tetrahedron Lett., 2004, 45, 125 CrossRef CAS; (c) M. Ordóñez, E. Hernández-Fernández, M. Montiel-Pérez, R. Bautista, P. Bustos, H. Rojas-Cabrera, M. Fernández-Zertuche and O. García-Barradas, Tetrahedron: Asymmetry, 2007, 18, 2427 CrossRef.
- R. A. Aitken, S. J. Jethwa, N. V. Richardson and A. M. Z. Slawin, Tetrahedron Lett., 2016, 57, 1563 CrossRef CAS.
- (a) S. V. Ryabukhin, D. M. Volochnyuk, A. S. Plaskon, V. S. Naumchik and A. A. Tolmachev, Synthesis, 2007, 1214 CAS; (b) A. S. Degtyarenko, A. A. Tolmachev, Y. M. Volovenko and A. V. Tverdokhlebov, Synthesis, 2007, 3891 CAS.
- D. P. Flaherty, S. M. Walsh, T. Kiyota, Y. Dong, T. Ikezu and J. L. Vennerstrom, J. Med. Chem., 2007, 50, 4986 CrossRef CAS PubMed.
- Q. Zhao, J. Sun, B. Liu and J. He, Dyes Pigm., 2013, 99, 339 CrossRef CAS.
- K. Kowalski, A. Koceva-Chyła, Ł. Szczupak, P. Hikisz, J. Bernasińska, A. Rajnisz, J. Solecka and B. Therrien, J. Organomet. Chem., 2013, 741–742, 153 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and NMR data of new compounds. See DOI: 10.1039/c8ra08023b |
| This journal is © The Royal Society of Chemistry 2018 |