
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal free oxidation of vinamidine derivatives: a simple synthesis of α-keto-β-diimine ligands†
Monika Tripathi,
Vianney Regnier,
Zakaria Ziani,
Marc Devillard,
Christian Philouze and
David Martin *
*
Univ. Grenoble Alpes, CNRS, DCM, 38000 Grenoble, France. E-mail: david.martin@univ-grenoble-alpes.fr
First published on 14th November 2018
Abstract
Oxidation of vinamidinium salts with meta-chloroperbenzoic acid is the key synthetic step towards new persistent 1,3-di(amino)oxyallyl radical cations. When applied to parent vinamidines, this protocol allows for a simple straightforward synthesis of α-keto-β-diimine ligands, for which no convenient synthesis was previously available.
β-Diketiminates, so-called NacNac ligands (Scheme 1), have been a focus in coordination chemistry for decades.1 Structural modifications include a large variety of N-substituents, as well as bulky,2 electron-withdrawing,3 or electron donating4 R groups. Substitution at the central carbon atom (R′ ≠ H) has also been explored as a strategy to tame this reactive position and enhance the chemical stability of the complex.5 The α-keto-β-diimines are among rare representatives with a more significant modification at the central carbon. These electron-deficient ligands have found applications in the design of highly active nickel(II) initiators for the synthesis of high molecular weight polyethylenes and poly-α-olefins.6 Interestingly, low-disperse semi-crystalline polymers could be obtained under living conditions and remarkable enantiomorphic site control could be achieved.7
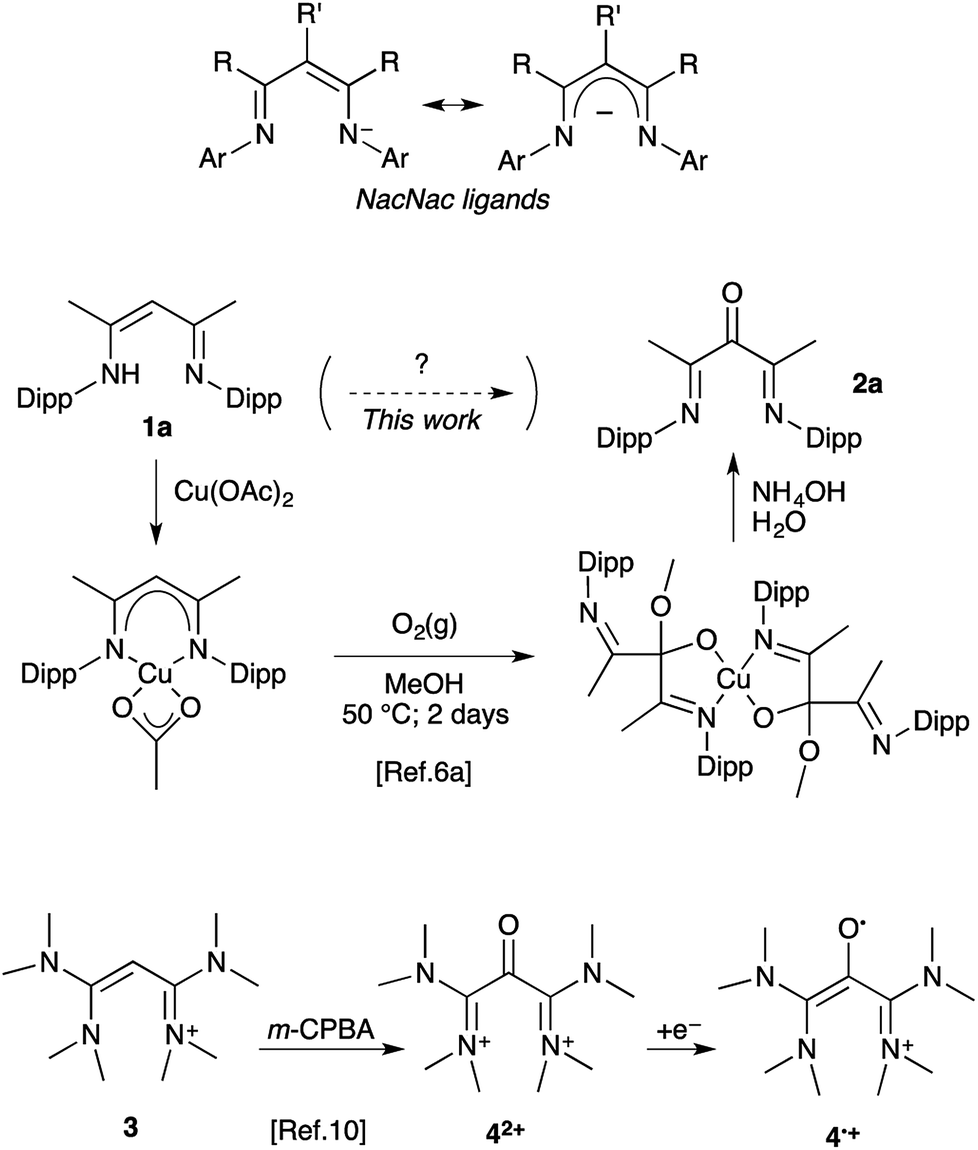 | ||
| Scheme 1 Previously reported synthesis of α-keto-β-diimines and synthesis of air-persistent radical 4˙+ from vinamidinium 3. | ||
The low availability of α-keto-β-diimines has clearly hampered further development. Their metal complexes have been known for long, but only as occasional by-products from the air-decomposition of unprotected NacNac complexes (R′ = H).8 To date only a rare selective oxygen-degradation of copper(II) complexes allows for the synthesis of a handful of 2,4-di(arylimino)pentan-3-ones 2 from the corresponding vinamidines 1.6a,d,9 The procedure requires (i) the synthesis of the NacNac–Cu(II) complex, (ii) oxidation at the ligand with dioxygen in a methanol/dichloromethane mixture, (iii) the decomplexation and hydrolysis of the resulting hemiacetal ligand (Scheme 1). In turn, we had to synthesize 2,4-bis((2,6-diisopropylphenyl)imino)pentan-3-one 2a and experienced firsthand the length and limitations of this methodology. Among the three steps, the oxidation of the NacNac–copper complex is especially inconvenient and wasteful, as it consists of a continuous bubbling of pure dioxygen in a warm solution for two days.6a Recently, we released a parented, though in principle far simpler, oxidation of tetrakis(dimethylamino) vinamidinium 3 into di(amidinium)ketone 42+ with meta-chloroperbenzoic acid (m-CPBA) as oxidant. Our initial focus was on the corresponding radical 4˙+, which was found remarkably air-persistent, despite minimal steric hindrance.10 Herein we report how further assessment of such 1,3-(diamino)oxyallyl radical cations ultimately led to a straightforward protocole for the synthesis of α-keto-β-diimines from NacNac precursors.
Prior to 4˙+, only two oxyallyl radical cations had been synthesized from the reaction of rare stable electrophilic carbenes with carbon monoxide.11–13 In principle, the oxidation of vinamidinium salts should provide for a more simple and general route, with no need of sophisticated N-substituents. To probe this assumption, we first considered the oxidative functionalization of the chloride salt of vinamidinium 5+, which was synthesized from N,N-dimethyl-benzamide and 1-dimethylamino-1-phenylethene.14 Addition of m-CPBA at room temperature yielded 2-chlorovinamidium 6 in 71% yield (Scheme 2). This result indicated the competitive formation of meta-chlorobenzoyl hypochlorite Cl(C6H4)CO2Cl, from the reaction of chloride anions with m-CPBA.15 Therefore we proceeded to an anion metathesis. The resulting tetrafluoroborate salt reacted with m-CPBA, but this time to afford 7·H+, which was fully characterized and isolated in 74% yield. Of note, the NMR spectra of vinamidiniums 6 and 7·H+ mostly differ in the 13C chemical shift of their central carbon: 94 and 127 ppm, respectively. They were unambiguously characterized by mass spectrometry analyses and X-ray diffraction studies (Fig. 1).
 | ||
| Scheme 2 Redox transformations of di(imidazole)methane and 1,3-di(phenyl) vinamidinium derivatives. | ||
 | ||
| Fig. 1 X-ray structures of 6, 7·H+ and 2c,d with thermal ellipsoids drawn at 50% probability level. Most hydrogen atoms, solvent molecules and counter-anions were omitted for clarity. | ||
Solutions of 7·H+ featured an EPR signal upon exposure to air. This slow reaction could be brought to fast completion in presence of potassium hydrogenocarbonate. The remarkable persistency under aerobic conditions of the resulting radical (in aerated solutions for several hours) was reminiscent of our previously reported stable oxyallyl radicals, some being similarly synthesized by auto-oxidation of parented enol-cations.11a Therefore, we hypothesized the formation of radical 7˙+. Although the radical ultimately decayed and couldn't be isolated, the excellent fit between the experimental EPR hyperfine coupling constants16 and the calculated values17 for 7˙+ strongly supported this reasonable assumption (Fig. 2a). The reaction of 7·H+ with excess meta-chloroperbenzoic acid or stronger oxidants, such dibromine or potassium ferricyanate, led to over oxidation and directly afforded EPR-silent mixtures of trione 8 and the corresponding hydrated gem-diol 8·H2O,18 likely through the formation and subsequent hydrolysis of electron-poor di(iminium)ketone 72+.
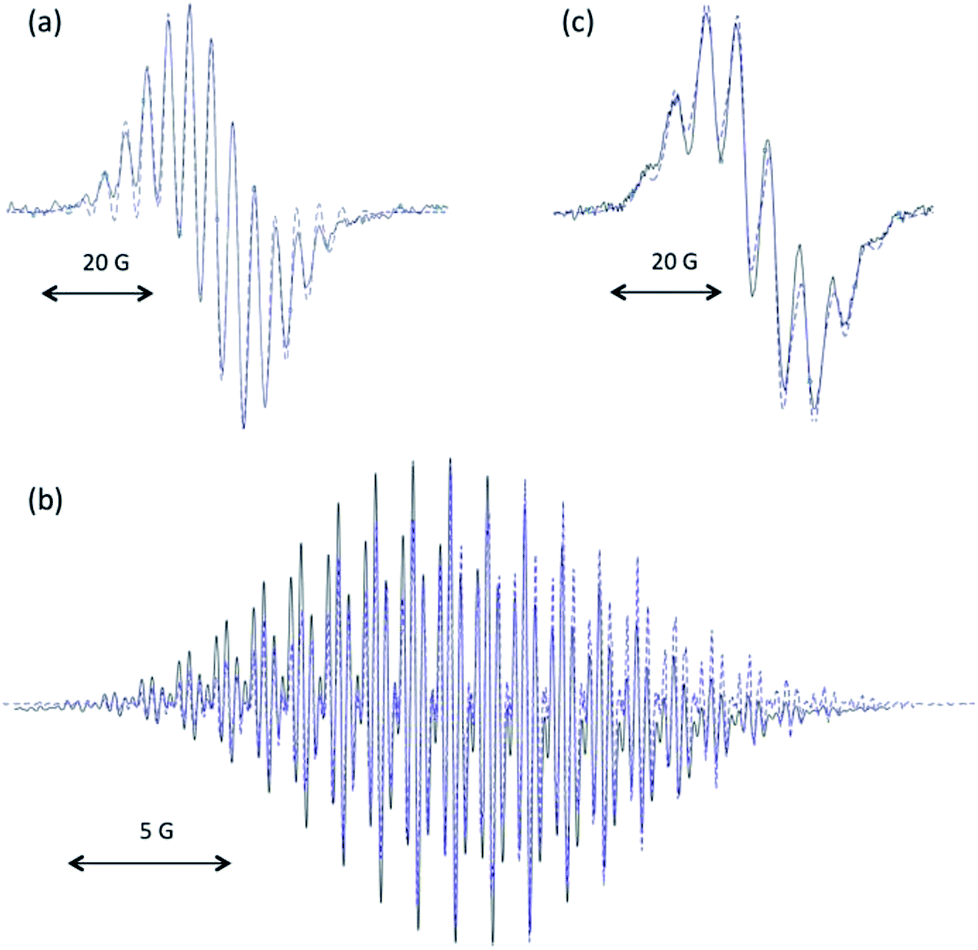 | ||
| Fig. 2 Experimental isotropic X-band EPR spectra in dichloromethane at room temperature (plain black line) of 7˙+ (a), 10˙+ (b) and a crude reaction mixture of m-CPBA and 1a (c). Simulated spectra (dashed blue line) were obtained with (a) a Lorentzian line-broadening parameter of 0.22 and the following set of hyperfine constants: a(14N) = 8.6 MHz (2 nuclei) and a(1H) = 12.0 MHz (12 nuclei); (b) with a Lorentzian line-broadening parameter of 0.013 and the following set of hyperfine constants: a(14N) = 3.1 MHz (4 nuclei), a(1H) = 6.5 MHz (12 nuclei) and a(1H) = 0.86 MHz (4 nuclei); (c) with a Lorentzian line-broadening parameter of 0.37 and the following set of hyperfine constants: a(14N) = 7.8 MHz (2 nuclei), a(1H) = 16.5 MHz (1 nucleus) and a(1H) = 15.6 MHz (6 nuclei). | ||
We turned to mono(methine)cyanine 9 featuring electron-richer benzimidazole patterns.19 However, the reaction of 9 with m-CPBA afforded known20 1,3-dimethyl- benzimidazolium 10, and not the expected di(benzimidazolium)ketone 132+ (Scheme 2). Note that uncompleted, but clean, formation of 10 was still observed when adding sub-stoichiometric (one equivalent) m-CPBA over one hour at −78 °C. Importantly, dication 132+ could be finally synthesized by the oxidation of di(imine)methane 1119 by m-CPBA, followed by di(alkylation) of the resulting di(imine)ketone 12. Given that 132+ was found almost unreactive towards water, the formation of 10 from 9 results from further oxidative cleavages of 132+, and not its hydrolysis.21 According to cyclic voltammetry experiments, 132+ undergoes successive reductions at E1/2 = −0.12 V vs. Fc/Fc+ (reversible) and Epc = −0.9 V (with further chemical evolution), which we attributed to the formation of radical cation 13˙+ and zwitterionic oxyallyl 13, respectively (see ESI†). We performed the electrochemical reduction of a solution of 132+ in acetonitrile at E = −0.5 V. The stoichiometry (one coulomb per mole of substrate), the observation of a strong EPR signal, as well as an excellent fit between experimental and theoretical hyperfine coupling constants, confirmed the formation of persistent 13˙+ (Fig. 2b), which ultimately decayed at room temperature after several hours.
The formation of di(imine)ketone 12 from 11 was so clean that it prompted us to explore further the direct oxidation of NacNac precursors. To our delight, treatment of 1a22 afforded 2a in 98% yield. In contrast with the former long and tedious syntheses from literature, the one-step reaction was completed after one hour at room temperature on multigram scales. EPR monitoring of the reaction showed the formation of a paramagnetic intermediate. Simulation of the hyperfine structure of the spectra required significant coupling with a single proton, in addition to two equivalent nitrogen atoms and six protons (Fig. 2c). This suggested the transient formation of N-protonated radical 1a·H˙+, parented to 4˙+, 7˙+ and 13˙+, thus implying closely related pathways for the m-CPBA oxidation of vinamidiniums and vinamidine 1a.
The only few reported β-di(imine)ketones were derivatives of acetylacetone and ortho-substituted anilines. Apart from 1a, which can be stored for several days, they were described as unstable ligands, to be used as soon as synthesized.6d We applied our protocole to vinamidine 1b21 with 2,4,6-trimethylaryl N-substituents and, indeed, the resulting ketone 2b decayed into a complex mixture within hours. Fast work-up allowed for its isolation in 75% yield (Scheme 3). However, even freshly crystalized 2b contained an impurity with similar NMR chemical shifts, except for a 13C NMR signal (quarternary carbon) at 94 ppm in place of the CO band of 2b at 194 ppm. Although the instability of 2b limited further investigations, drying crystals in vacuo in presence of P2O5 decreased the amount of impurity, allowing us to assign this latter to the corresponding hydrated gem-diol 2b·H20.23,24
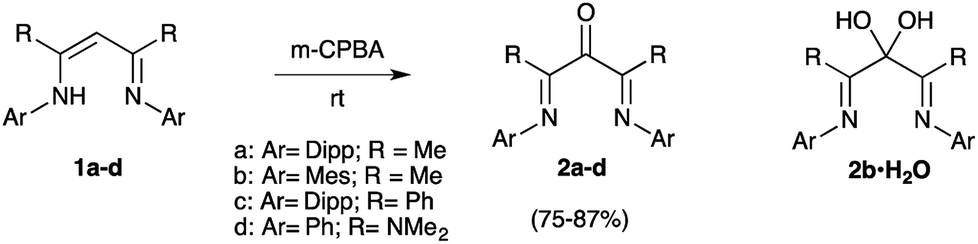 | ||
| Scheme 3 One-step synthesis of α-keto-β-diimines 2a–d from vinamidines 1a–d. | ||
Finally, we considered vinamidines 1c25 and 1d,4b,d with phenyl and di(methyl)amino R groups, respectively. The corresponding di(imine)ketones 2c,d, which are out of reach of previous methods, were isolated in 86–87% yield. They features similar key structural data (IRATR: ν = 1700 cm−1; 13C NMR δCO = 194–191 ppm). Their structures were asserted by a structural X-ray diffraction study (Fig. 1). Importantly, in ketones 2c,d were found remarkably bench stable and have been stored for month with no noticeable degradation.
In conclusion, the synthesis and characterization of radicals 7˙+ and 13˙+ are further evidences that introducing 1,3-di(amino)oxyallyl patterns is a robust principle for the design of persistent radical cations. However, the outcome of the reaction of vinamidiniums with m-CPBA is too dependent of the substitution pattern to constitute a general route and over-oxidation is only manageable with extra electron-donating amino groups. In contrast, when applied to vinamidines, this protocol allows for a straightforward synthesis of α-keto-β-diimines. In addition to its simplicity, stable derivatives were isolated, with unprecedented bulky or electron-donating R groups. We are now evaluating these new ligands for nickel-initiated polymerization of ethylene.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the French National Agency for Research (ANR-14-CE06-0013-01 and ANR-17-ERC2-0015). University of Grenoble-Alpes contributed through ICMG Chemistry Nanobio Platform, the LabEx ARCANE (ANR-11-LABX-0003-01), the “Centre de Calcul Intensif en Chimie de Grenoble” and AGIR-POLE (grant for MT).Notes and references
- (a) L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031 CrossRef CAS PubMed; (b) D. J. Emslie and W. E. Piers, Coord. Chem. Rev., 2002, 233–234, 131 Search PubMed; (c) H. W. Roesky, S. Singh, V. Jancik and V. Chandrasekhar, Acc. Chem. Res., 2004, 37, 969 CrossRef CAS PubMed; (d) D. J. Mindiola, Acc. Chem. Res., 2006, 39, 813 CrossRef CAS PubMed; (e) C. J. Cramer and W. B. Tolman, Acc. Chem. Res., 2007, 40, 601 CrossRef CAS PubMed; (f) P. L. Holland, Acc. Chem. Res., 2008, 41, 905 CrossRef CAS PubMed; (g) Y. C. Tsai, Coord. Chem. Rev., 2012, 256, 722 CrossRef CAS.
- For examples with R = tert-butyl, see: (a) J. Vela, J. M. Smith, Y. Yu, N. A. Ketterer, C. J. Flaschenriem, R. J. Lachicotte and P. L. Holland, J. Am. Chem. Soc., 2005, 127, 7857 CrossRef CAS PubMed; (b) Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961 CrossRef CAS PubMed; (c) G. Zhao, F. Basuli, U. J. Kilgore, H. Fan, H. Aneetha, J. C. Huffman, G. Wu and D. J. Mindiola, J. Am. Chem. Soc., 2006, 128, 13575 CrossRef CAS PubMed.
- For some representative examples, see: (a) D. S. Laitar, C. J. N. Mathison, W. M. Davis and J. P. Sadighi, Inorg. Chem., 2003, 42, 7354 CrossRef CAS PubMed; (b) S. Hong, L. M. R. Hill, A. K. Gupta, B. D. Naab, J. B. Gilroy, R. G. Hicks, C. J. Cramer and W. B. Tolman, Inorg. Chem., 2009, 48, 4514 CrossRef CAS PubMed; (c) D. T. Carey, E. K. Cope-Eatough, E. Vilaplana-Mafé, F. S. Mair, R. G. Pritchard, J. E. Warren and R. J. Woods, Dalton Trans., 2003, 1083 RSC; (d) H. A. Chiong and O. Daugulis, Organometallics, 2006, 25, 4054 CrossRef CAS; (e) A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Angew. Chem., Int. Ed., 2007, 46, 6339 CrossRef CAS PubMed; (f) S. Kernbichl, M. Reiter, F. Adams, S. Vagin and B. Rieger, J. Am. Chem. Soc., 2017, 139, 6787 CrossRef CAS PubMed; (g) S. Kundu, W. Y. Kim, J. A. Bertke and T. H. Warren, J. Am. Chem. Soc., 2017, 139, 1045 CrossRef CAS PubMed; (h) K. S. Choung, M. D. Islam, R. W. Gou and T. S. Teets, Inorg. Chem., 2017, 56, 14326 CrossRef CAS PubMed.
- (a) V. Regnier, Y. Planet, C. E. Moore, J. Pecaut, C. Philouze and D. Martin, Angew. Chem., Int. Ed., 2017, 56, 1031 CrossRef CAS PubMed; (b) D. C. H. Do, A. Keyser, A. V. Protchenko, B. Maitland, I. Pernik, H. Niu, E. L. Kolychev, A. Rit, D. Vidovic, A. Stasch, C. Jones and S. Aldridge, Chem.–Eur. J., 2017, 23, 5830 CrossRef CAS PubMed; (c) I. Pernik, B. Maitland, A. Stasch and C. Jones, Can. J. Chem., 2018, 96, 513 CrossRef CAS; (d) M. Tripathi, V. Regnier, C. Lincheneau and D. Martin, New J. Chem., 2017, 41, 15016 RSC.
- (a) C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404 CrossRef CAS PubMed; (b) R. Jiao, X. Shen, M. Xue, Y. Zhang, Y. Yao and Q. Shen, Chem. Commun., 2010, 46, 4118 RSC; (c) M. M. Rodriguez, E. Bill, W. W. Brennessel and P. L. Holland, Science, 2011, 334, 780 CrossRef CAS PubMed; (d) L. A. Harris, E. C. Y. Tam, M. P. Coles and J. R. Fulton, Dalton Trans., 2014, 43, 13803 RSC; (e) K. Grubel, W. W. Brennessel, B. Q. Mercado and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 16807 CrossRef CAS PubMed; (f) A. Kalita, V. Kumar and B. Mondal, RSC Adv., 2015, 5, 643 RSC.
- (a) J. D. Azoulay, R. S. Rojas, A. V. Serrano, H. Ohtaki, G. B. Galland, G. Wu and G. C. Bazan, Angew. Chem., Int. Ed., 2009, 48, 1089 CrossRef CAS PubMed; (b) J. D. Azoulay, Y. Schneider and G. C. Bazan, US20090299020A1, 2009; (c) J. D. Azoulay, G. C. Bazan and G. B. Galland, Macromolecules, 2010, 43, 2794 CrossRef CAS; (d) A. Sokolohorskyj, O. Zeleznik, I. Cisarova, J. Lenz, A. Lederer and J. Merna, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2440 CrossRef CAS.
- (a) J. D. Azoulay, Y. Schneider, G. B. Galland and G. C. Bazan, Chem. Commun., 2009, 6177 RSC; (b) J. D. Azoulay, H. Y. Gao, Z. A. Koretz, G. Kehr, G. Erker, F. Shimizu, G. B. Galland and G. C. Bazan, Macromolecules, 2012, 45, 4487 CrossRef CAS.
- For the reaction of dioxygen with NacNac–cobalt complexes, see: (a) M. C. Weiss and V. L. Goedken, J. Am. Chem. Soc., 1976, 98, 3389 CrossRef CAS; (b) B. Durham, T. J. Anderson, J. A. Switzer, J. F. Endicott and M. D. Glick, Inorg. Chem., 1977, 16, 271 CrossRef CAS; (c) J. A. Switzer and F. J. Endicott, J. Am. Chem. Soc., 1980, 102, 1181 CrossRef CASNacNac-iron complexes: (d) D. P. Riley and D. H. Busch, Inorg. Chem., 1983, 22, 4141 CrossRef CAS; (e) D. B. MacQueen, C. Lange, M. Calvin, J. W. Otvos, L. O. Spreer, C. B. Allan, A. Ganse and R. B. Frankel, Inorg. Chim. Acta, 1997, 263, 125 CrossRef CASFor lanthanides: (f) J. Zhang, Z. Zhang, Z. Chen and X. Zhou, Dalton Trans., 2012, 41, 357 RSCFor alkaline earths: (g) B. Liu, V. Dorcet, L. Maron, J. F. Carpentier and Y. Sarazin, Eur. J. Inorg. Chem., 2012, 3023 CrossRef CAS.
- S. Yokota, Y. Tachi and S. Itoh, Inorg. Chem., 2002, 41, 1342 CrossRef CAS PubMed.
- V. Regnier, F. Molton, C. Philouze and D. Martin, Chem. Commun., 2016, 52, 11422 RSC.
- (a) D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7014 CrossRef CAS PubMed; (b) J. K. Mahoney, D. Martin, F. Thomas, C. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519 CrossRef CAS PubMed.
- For the reaction of stable carbenes with CO, see also: (a) V. Lavallo, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2006, 45, 3488 CrossRef CAS PubMed; (b) T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039 CrossRef CAS PubMed; (c) U. Siemeling, C. Färber, C. Bruhn, M. Leibold, D. Selent, W. Baumann, M. von Hopffgarten, C. Goedecke and G. Frenking, Chem. Sci., 2010, 1, 697 RSC; (d) T. Schulz, C. Färber, M. Leibold, C. Bruhn, W. Baumann, D. Selent, T. Porsch, M. C. Holthausen and U. Siemeling, Chem. Commun., 2013, 49, 6834 RSC; (e) T. Schulz, C. Färber, M. Leibold, C. Bruhn, P. Prochnow, J. E. Bandow, T. Schneider, T. Porsch, M. C. Holthausen and U. Siemeling, Chem. Commun., 2014, 50, 2341 RSC.
- For the quest on stable 1,3-(diamino)oxyallyl derivatives, see: (a) V. Regnier and D. Martin, Org. Chem. Front., 2015, 2, 1536 RSC; (b) M. Devillard, V. Regnier, M. Tripathi and D. Martin, J. Mol. Struct., 2018, 1172, 3 CrossRef CAS.
- (a) H. Ahlbrecht, W. Raab and C. Vonderheid, Synthesis, 1979, 127 CrossRef CAS; (b) R. Pajkert, T. Böttcher, M. Ponomarenko, M. Bremer and G.-V. Röschenthaler, Tetrahedron, 2013, 69, 8943 CrossRef CAS.
- For the reaction of m-CPBA with halides see: (a) N. J. Bunce and D. D. Tanner, J. Am. Chem. Soc., 1969, 91, 6096 CrossRef CAS; (b) M. Srebnik, Synth. Commun., 1989, 19, 197 CrossRef CAS.
- Experimental EPR spectra were fitted with the EasySpin simulation package: S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- DFT calculations were performed using the program package Gaussian09: Gaussian 09, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al., Gaussian Inc., Wallingford CT, 2009. See ESI† for complete citation and further details.
- The trione is usually isolated as a mixture with its hydrate: Z. L. Wang, X. L. An, L. S. Ge, J.-H. Jin, X. Luo and W.-P. Deng, Tetrahedron, 2014, 70, 3788 CrossRef CAS.
- (a) C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206 CrossRef CAS PubMed; (b) W.-C. Chen, Y.-C. Hsu, C.-Y. Lee, G. P. A. Yap and T.-G. Ong, Organometallics, 2013, 32, 2435 CrossRef CAS.
- A. K. Diba, C. Noll, M. Richter, M. T. Gieseler and M. Kalesse, Angew. Chem., Int. Ed., 2010, 49, 8367 CrossRef PubMed.
- We propose that further reaction of 132+ with m-CPBA yields Bayer-Villiger type products, either (imidazolyl)-carbonates or carboxylate derivatives. These electrophiles being highly activated, they would readily undergo hydrolysis, affording 1,3-dimethylbenzimidazolium salts.
- For a convenient protocole for the condensation of anilines with acetylacetone, see: L.-M. Tang, Y.-Q. Duan, X.-F. Li and Y.-S. Li, J. Organomet. Chem., 2006, 691, 2023 CrossRef CAS.
- Such compounds have been mentioned in patents: Y. Tang, Z. Chen, G. Ji, X. Sun, C. Xu and J. Li, WO 2015024517, 2015.
- In parented Pt-complexes the 13C chemical shift of the central carbon is in the 80–110 ppm range: M. L. Scheuermann, U. Fekl, W. Kaminsky and K. I. Goldberg, Organometallics, 2010, 29, 4749 CrossRef CAS.
- M. Arrowsmith, M. R. Crimmin, M. S. Hill and G. Kociok-Köhn, Dalton Trans., 2013, 42, 9720 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C spectra of all new compounds, computational and crystallographic (CCDC 1866491–1866494) details. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra08220k |
| This journal is © The Royal Society of Chemistry 2018 |