
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymer semiconductors incorporating head-to-head linked 4-alkoxy-5-(3-alkylthiophen-2-yl)thiazole†
Xin Zhou‡
ab,
Peng Chen‡a,
Chang Woo Kohc,
Sheng Chena,
Jianwei Yua,
Xianhe Zhanga,
Yumin Tanga,
Luca Bianchia,
Han Guo*a,
Han Young Woo *c and
Xugang Guo*a
*c and
Xugang Guo*a
aDepartment of Materials Science and Engineering, The Shenzhen Key Laboratory for Printed Organic Electronics, Southern University of Science and Technology (SUSTech), No. 1088, Xueyuan Road, Shenzhen, Guangdong 518055, China. E-mail: guoh3@sustc.edu.cn
bDepartment of Chemistry, Southern University of Science and Technology (SUSTech), No. 1088, Xueyuan Road, Shenzhen, Guangdong 518055, China. E-mail: guoxg@sustc.edu.cn
cResearch Institute for Natural Sciences, Department of Chemistry, Korea University, Seoul 02841, South Korea. E-mail: hywoo@korea.ac.kr
First published on 18th October 2018
Abstract
Head-to-head linked bithiophenes with planar backbones hold distinctive advantages for constructing organic semiconductors, such as good solubilizing capability, enabling narrow bandgap, and effective tuning of frontier molecular orbital (FMO) levels using minimal thiophene numbers. In order to realize planar backbone, alkoxy chains are typically installed on thiophene head positions, owing to the small van der Waals radius of oxygen atom and accompanying noncovalent S⋯O interaction. However, the strong electron donating alkoxy chains on the electron-rich thiophenes lead to elevated FMO levels, which are detrimental to material stability and device performance. Thus, a new design approach is needed to counterbalance the strong electron donating property of alkoxy chains to bring down the FMOs. In this study, we designed and synthesized a new head-to-head linked building block, 4-alkoxy-5-(3-alkylthiophen-2-yl)thiazole (TRTzOR), using an electron-deficient thiazole to replace the electron-rich thiophene. Compared to previously reported 3-alkoxy-3′-alkyl-2,2′-bithiophene (TRTOR), TRTzOR is a weaker electron donor, which considerably lowers FMOs and maintains planar backbone through the noncovalent S⋯O interaction. The new TRTzOR was copolymerized with benzothiadiazoles with distinct F numbers to yield a series of polymer semiconductors. Compared to TRTOR-based analogous polymers, these TRTzOR-based polymers have broader absorption up to 950 nm with lower-lying FMOs by 0.2–0.3 eV, and blending these polymers with PC71BM leads to polymer solar cells (PSCs) with improved open-circuit voltage (Voc) by ca. 0.1 V and a much smaller energy loss (Eloss) as low as 0.59 eV. These results demonstrate that thiazole substitution is an effective approach to tune FMO levels for realizing higher Vocs in PSCs and the small Eloss renders TRTzOR a promising building block for developing high-performance organic semiconductors.
Introduction
Polymer semiconductors are emerging semiconducting materials, which effectively combine several advantages for applications in various optoelectronic devices, including low-temperature solution processability, tunable physicochemical properties, mechanical durability, and bio-compatibility.1–8 The polymer semiconductors are generally functionalized with appropriate alkyl side chains for achieving good solubility in organic solvents and their attachments must be organized in specific ways to ensure the regioregularity and backbone planarity for optimizing film morphology and device performance.9–12 Taking the most well-known poly(3-hexylthiophene) (P3HT) for example, the head-to-tail (HT) coupling between adjacent thiophene units leads to regioregular arrangement of hexyl side chains and planar polymer backbone, while the undesirable head-to-head (HH) coupling results in unfavorable backbone torsion and much degraded charge transport property, therefore HH linkage is typically avoided in the design of polymer semiconductors.13–15 One feasible approach to overcome this limitation is the adoption of alkoxy chains instead of conventional alkyl ones, which could enable HH-linked bithiophenes with a highly planar backbone conformation due to the smaller van der Waals radius of oxygen atom (versus methylene group in alkyl chain) and the accompanying noncovalent S⋯O coulombic interaction,16–21 exemplified by the well-known conducting polymer poly(3,4-ethylenedioxythiophene) (PEDOT). The planar backbone conformation leads to remarkably improved conductivity of PEDOT after doping.Inspired by the great success of PEDOT, a HH-linked bithophene, 3,3′-dialkoxy-2,2′-bithiophene (BTOR) (Fig. 1a), was designed, demonstrating this concept for realizing high-performance polymer semiconductors.16 The alkoxy chain-enabled planar backbone conformation was verified by density function theory (DFT) calculation and single crystal structures.19,20,22–25 However, the strong electron-donating character of alkoxy chains yielded the elevated energy levels of frontier molecular orbitals (FMOs) for the BTOR-based polymer semiconductors.16 High-lying FMOs are not only detrimental to the materials chemical stability and device performance robustness, which has been one of the key concerns for practical applications of organic electronic devices, but also limit the device performance (i.e., open-circuit voltage, Voc) of polymer solar cells (PSCs) when they are used as donor polymers. In bulk heterojunction solar cells, Voc is proportional to the energy level gap between the lowest unoccupied molecular orbital (LUMO) of acceptor materials and the highest occupied molecular orbital (HOMO) of donor materials, and the elevated HOMO of donor materials can lead to small Vocs.26–29 In attempt to address this issue, we designed 3-alkoxy-3′-alkyl-2,2′-bithiophene (TRTOR, Fig. 1b) which maintains backbone planarity through the S⋯O conformational lock and lower HOMO levels (relative to BTOR) as revealed by DFT calculation and single crystal analysis study.18,30 A remarkable power conversion efficiency (PCE) near 10% was successfully obtained in PSCs using the TRTOR-based polymer donor when blended with PC71BM acceptor.28 However, the Voc (0.66 V) of the TRTOR-based polymer is approximately 0.1–0.2 eV smaller than polymer analogues with alkyl-substituted terthiophene or tetrathiophene.11,31,32 To counterbalance the strong electron-donating effect of alkoxy chains, the alkoxy-functionalized HH bithiophenes should be modified for lowering FMO levels.33–35
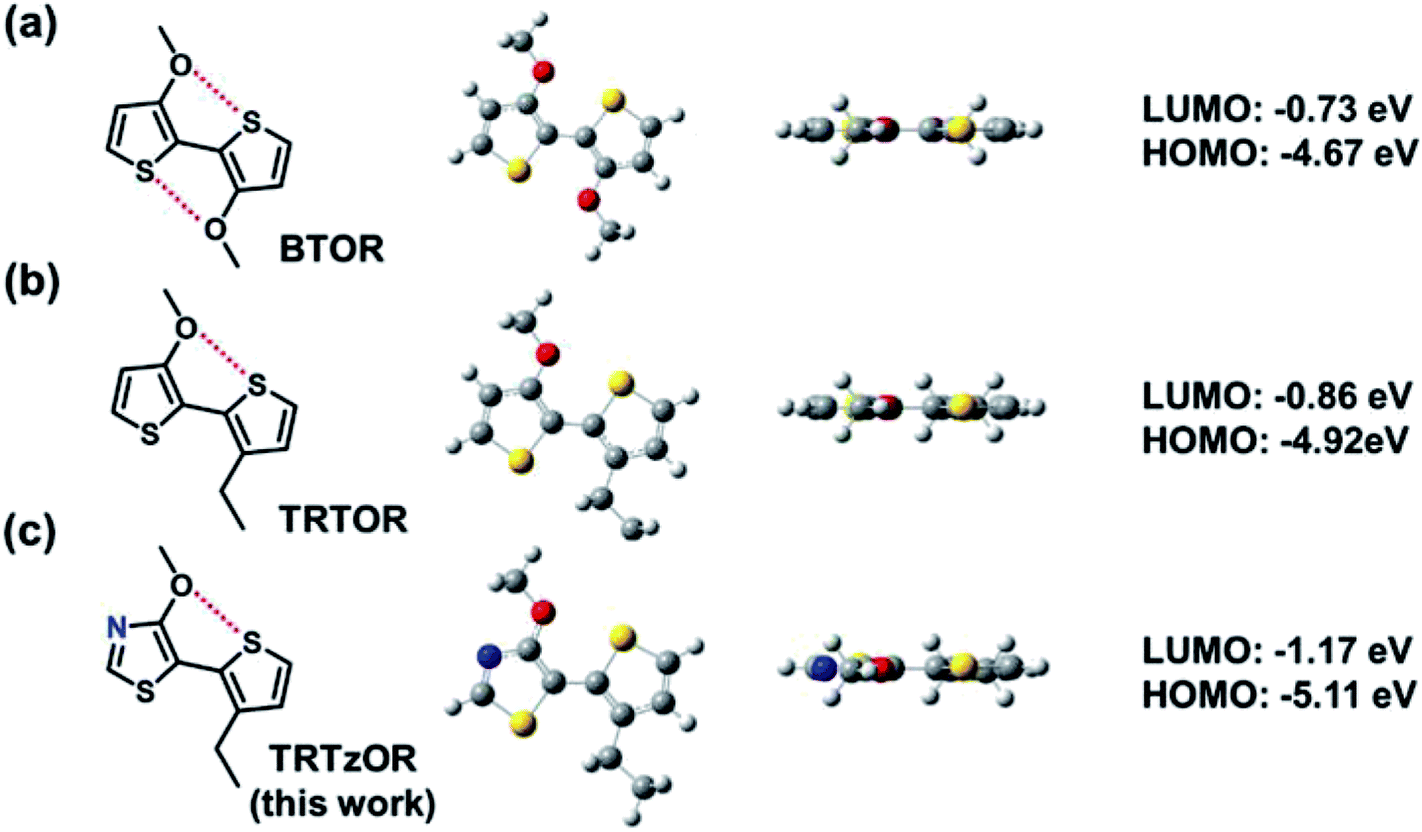 | ||
| Fig. 1 Chemical structures, DFT-optimized geometries (top views and side views), and frontier molecular orbital energy levels of alkoxy chain-based head-to-head linked building blocks: (a) BTOR, (b) TRTOR, and (c) TRTzOR (this work). The intramolecular S⋯O interaction is marked by dotted red line. | ||
Herein, we report the design and synthesis of a novel HH linked building block, 4-alkoxy-5-(3-alkylthiophen-2-yl)thiazole (TRTzOR) (Fig. 1c), in which the electron-deficient thiazole was adopted to replace the electron-rich thiophene unit. As a less electron-rich arene (versus thiophene), thiazole is a commonly used heterocycle along with many of its derivatives that can effectively lower the FMOs of organic semiconductors.36–40 Other possible benefits are the formation of additional intramolecular noncovalent interactions promoted by the N atom of thiazole with other heteroatoms on the adjacent units,36,41 steric hindrance reduction by elimination of the C–H moiety,42 and increased intermolecular coulombic interaction contributed by the large polarity of the thiazole core.36 It was found that the two distinct alkyl chains can well balance the solubility and crystallinity of the previously reported TRTOR-based polymers, yielding highly promising device performance in both OTFTs and PSCs.28 On the basis of our previous work and also for better comparison, these specific side chains, 2-propylheptyl and 2-butyloctyl, were also used in this work. Benzothiadiazole (BT) and its fluorinated derivatives, fluorobenzothiadiazole (fBT) and difluorobenzothiadiazole (ffBT), show strong electron-withdrawing ability and compact geometry, leading to polymer semiconductors with optimized optoelectronic structure and film morphology,43 which were hence chosen here as acceptor co-monomers for these TRTzOR-based polymer semiconductors. A series of semiconductors PTRTzOR-BT (P1), PTRTzOR-fBT (P2), and PTRTzOR-ffBT (P3, Scheme 1), were synthesized by coupling TRTzOR with BT, fBT, and ffBT, respectively. Compared to the TRTOR-based analogue polymers,28 P1–P3 all displayed slightly narrower optical bandgaps (Egs) by 0.05–0.11 eV and lower-lying HOMO levels by 0.2–0.3 eV. PSCs with polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC71BM active layers consistently yielded larger Vocs than the cells using TRTOR-based polymers as donor materials, which are up to 0.76 V for the P3:PC71BM device featuring a 0.1 V improvement versus the cell using TRTOR-based polymer analogue. Therefore, the energy loss (Eloss = Eg − eVoc) in these PSC devices was much smaller as a result of the narrower Egs of 1.31–1.35 eV and the larger Vocs of TRTzOR-based polymers. The estimated Eloss was as low as 0.59 eV for P3:PC71BM device, approaching the lowest value for fullerene-based PSCs.28,44–47 The maximum PCEs of Pn:PC71BM (n = 1–3) devices were quite similar varying between 6% to 6.5% after careful device optimization. The results suggest that TRTzOR is a promising candidate in the family of head-to-head linked electron donating building blocks and thiazole substitution is a promising strategy to generate polymers for applications in PSCs.
:PC71BM active layers consistently yielded larger Vocs than the cells using TRTOR-based polymers as donor materials, which are up to 0.76 V for the P3:PC71BM device featuring a 0.1 V improvement versus the cell using TRTOR-based polymer analogue. Therefore, the energy loss (Eloss = Eg − eVoc) in these PSC devices was much smaller as a result of the narrower Egs of 1.31–1.35 eV and the larger Vocs of TRTzOR-based polymers. The estimated Eloss was as low as 0.59 eV for P3:PC71BM device, approaching the lowest value for fullerene-based PSCs.28,44–47 The maximum PCEs of Pn:PC71BM (n = 1–3) devices were quite similar varying between 6% to 6.5% after careful device optimization. The results suggest that TRTzOR is a promising candidate in the family of head-to-head linked electron donating building blocks and thiazole substitution is a promising strategy to generate polymers for applications in PSCs.
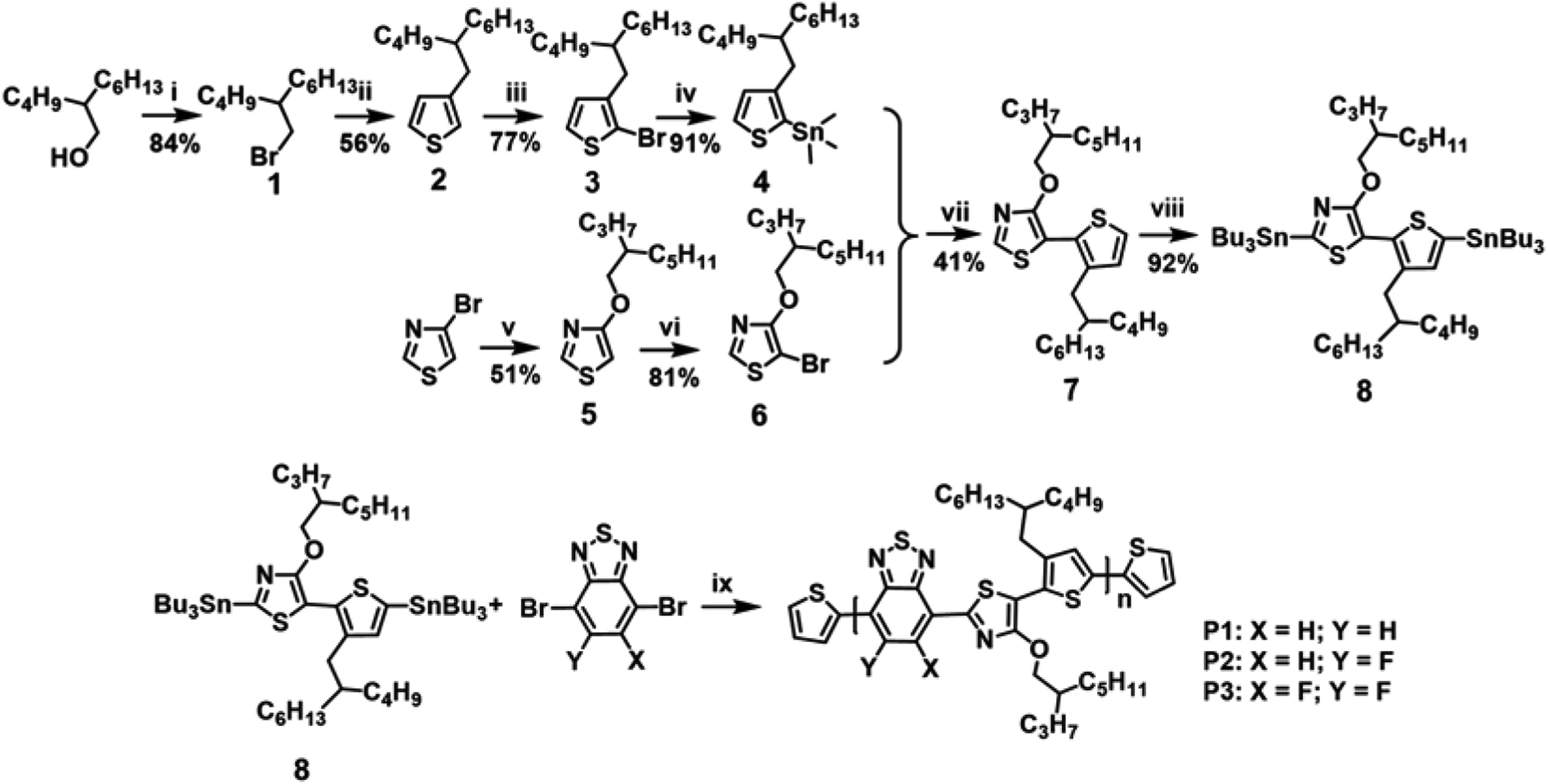 | ||
| Scheme 1 Synthetic routes to the TRTzOR-based polymer semiconductors. Reagent and conditions: (i) NBS, PPh3, DCM, 0 °C; (ii) Mg, I2, Ni(dppp)Cl2, 3-bromothiophene, THF; (iii) NBS, CH3COOH, chloroform; (iv) n-BuLi, SnMe3Cl, THF, −78 °C; (v) 2-propylheptanol, NaH, CuBr, THF; (vi) NBS, chloroform; (vii) Pd(PPh3)2Cl2, toluene, microwave; (viii) n-BuLi, SnBu3Cl, THF, −78 °C; (ix) Pd2(dba)3, P(o-tolyl)3, toluene, microwave irradiation. | ||
Results and discussion
Monomer and polymer synthesis
Scheme 1 depicts the synthetic route to the new building block of TRTzOR. The main consideration in synthesizing the TRTzOR unit is the different chemical reactivity of thiazole and thiophene when combining them through the Stille coupling reaction. Since the first step in the Pd-catalyzed Stille coupling, i.e. the oxidative addition of Pd to the brominated compound, is facilitated by the electron-withdrawing group,48,49 hence the brominated thiazole and the stannylated thiophene were chosen. Compound 3 with a 2-butyloctyl side-chain was first prepared according to a previous literature,28 which was then treated with n-butyl lithium (n-BuLi) and trimethyltin chloride (Me3SnCl) at −78 °C to yield compound 4. Compound 5 was synthesized in a good yield (51%) via a nucleophilic aromatic substitution of the commercially available 4-bromothiazole with sodium 2-propylheptanol.50 Initial attempt to synthesize compound 5 was via transetherification of 4-methoxythiazole that was converted from 4-bromothiazole.33 However, the product (4-methoxythiazole) suffered from a large loss in the first step due to its low boiling point, accordingly the two-step protocol gave compound 5 in a lower yield (33%) than the current procedure. Compound 5 was then treated with N-bromosuccinimide (NBS) to produce compound 6. Then, the new building block TRTzOR was successfully obtained via Stille coupling between stannylated 4 and brominated 6 with a yield of 41%.48 The electron-donating alkoxy chain in thiazole might be responsible for this moderate yield, as it was known to be unfavorable for oxidative addition step in Stille coupling.49To prepare the TRTzOR-based polymer semiconductors, compound 7 was treated with n-BuLi and Bu3SnCl at −78 °C to afford the stannylated monomer 8. After purification through the C18 inverse phase column chromatography, the monomer 8 was obtained in high purity (Fig. S19 and S20†), which was then coupled with different benzothiadiazole-based acceptor co-monomers via the Pd-catalyzed Stille coupling under microwave irradiation to afford the target polymer semiconductors. It should be noted that, due to the asymmetric character of TRTzOR, all polymers are regioirregular, which may negatively affect their electronic properties and self-assemblies.51,52 Polymer chain end-capping was carried out using 2-bromothiophene and 2-(tributylstannyl)thiophene after polymerization. Finally, the polymers were treated under multiple Soxhlet extraction processes with methanol, acetone, hexane, dichloromethane, and chloroform in sequence. All these TRTzOR-based polymers exhibit desirable solubility in organic solvents such as chloroform (CF), chlorobenzene (CB), and dichlorobenzene (DCB). Molecular weights of these TRTzOR-based polymers were determined by high-temperature gel permeation chromatography (GPC), using 1,2,4-trichlorobenzene (1,2,4-TCB) as the eluent at 150 °C relative to a polystyrene standard. The number-average molecular weights (Mns) of P1, P2, and P3 were found to be 21.9, 23.8, and 44.5 kDa with dispersities (ĐMs) of 1.5, 2.6, and 2.2, respectively (Table 1). All other synthetic details could be found in the ESI.† The thermal properties of these TRTzOR-based polymers were measured by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). These polymers show desired thermal stability in nitrogen with the decomposition temperature (Td) of 365, 351, and 350 °C for P1, P2, and P3, respectively (Fig. S1a†), which is defined as the 5% weight loss point. Differing from the TRTOR-based polymer analogues,28 the DSC curves of the TRTzOR-based polymers were mostly featureless within the entire measured temperature range of 30–350 °C (Fig. S1b†).
| Polymer | Mna [kDa] | ĐMa | Tdb [°C] | λmaxc [nm] | λonsetc [nm] | Eoptg c [eV] | Eoptg,DFT d [eV] | EHOMOe [eV] | ELUMOf [eV] |
|---|---|---|---|---|---|---|---|---|---|
| a High-temperature GPC at 150 °C using 1,2,4-trichlorobenzene as the eluent.b 5% Weight loss temperature point.c As-cast films from chloroform solutions.d Converged values for hexamers.e EHOMO = −e(Eonsetox + 4.80) eV, where Eonsetox is determined electrochemically using Fc/Fc+ internal standard.f ELUMO = EHOMO + Eoptg. | |||||||||
| P1 | 21.9 | 1.5 | 365 | 818 | 946 | 1.31 | 1.59 | −5.24 | −3.93 |
| P2 | 23.8 | 2.6 | 351 | 814 | 941 | 1.32 | 1.60 | −5.37 | −4.05 |
| P3 | 44.5 | 2.2 | 350 | 789 | 920 | 1.35 | 1.65 | −5.40 | −4.05 |
Optical and electrochemical properties of polymers
According to the UV-vis absorption spectra (Fig. 2a), these TRTzOR-based polymers show a wide absorption in the range of 500–900 nm in both solution and film states. From solution to film, all polymers display a small bathochromic shift of ca. 30–40 nm accompanied by comparable absorption profile, indicative of their strong aggregation characteristics, which is partially enabled by the highly planar polymer backbones. Density functional theory (DFT) calculation (vide infra) revealed that the introduction of the thiazole reinforces the backbone planarity through the intramolecular noncovalent N⋯H interaction and by eliminating the C–H moiety. Together with the large molecular polarity of the thiazole, these effects promoted stronger intermolecular interactions. The temperature-dependent absorption was also measured to examine the effects of F addition on the polymer aggregation. As shown in their absorption spectra (Fig. S2†), the F addition on the benzothiadiazole leads to intensified aggregation for P2 and P3, as revealed by the least hypochromic shift with a λmax change less than 30 nm as the temperature was raised up to 100 °C. The temperature-dependent aggregation may allow the fine-modulation of disorder/order transitions of polymer chains that is critical for film morphology control when solution-casting the bulk-heterojunction active layer, showing a profound impact on the resulting device performance.53,54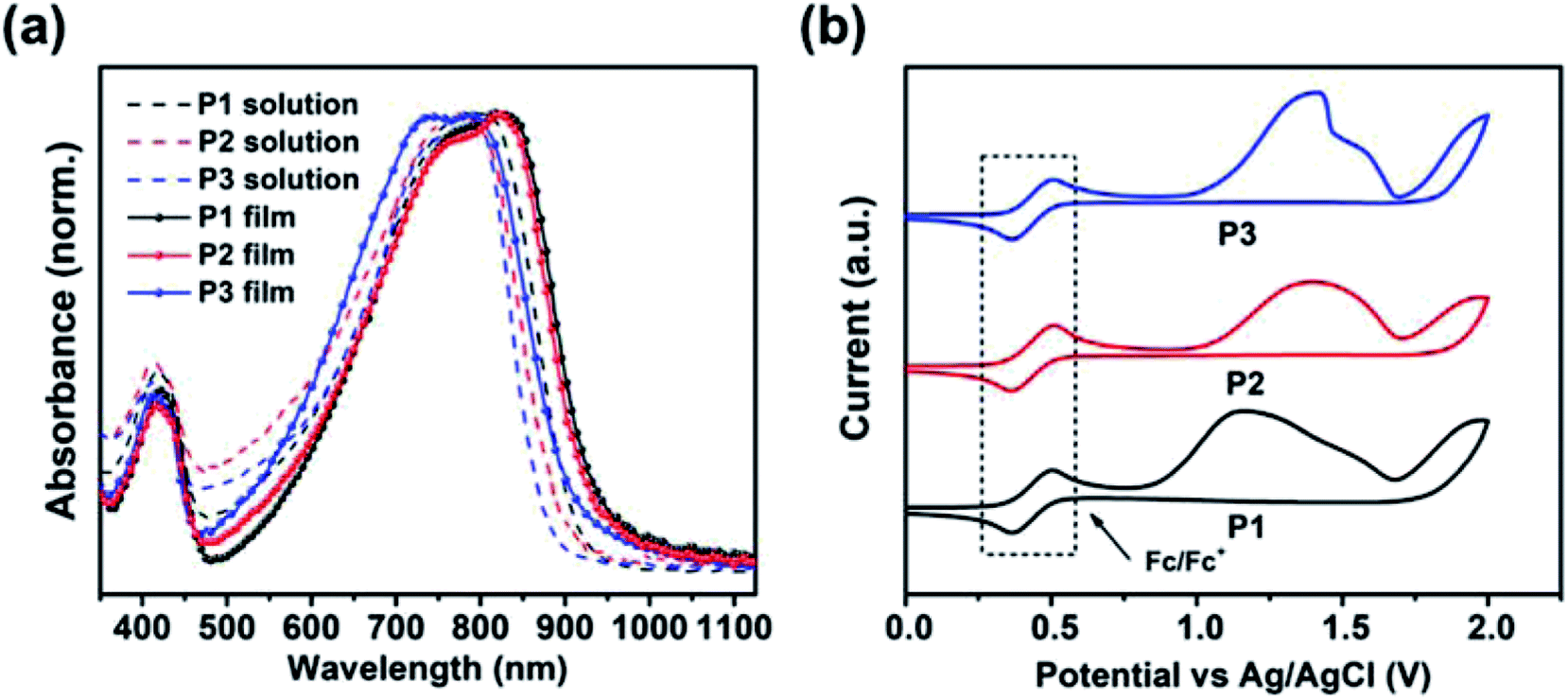 | ||
| Fig. 2 (a) UV-vis absorption spectra of P1–P3 in diluted chloroform solutions (10−5 M) and thin films casted from 5 mg mL−1 chloroform solution; (b) CV curves of P1–P3 films in acetonitrile with a Fc/Fc+ internal standard. | ||
Based on the film absorption onset (λonset), the optical bandgap (Eoptg) was calculated to be 1.31, 1.32, and 1.35 eV for P1, P2, and P3, respectively (Table 1). Compared to the TRTOR-based analogues, the Eoptgs of the TRTzOR-based polymers were slightly reduced by ca. 0.05–0.11 eV, which is likely attributed to the more planar backbone of TRTzOR-based polymers.28 When difluorinated ffBT was used as the comonomer, P3 gave rise to a small blue-shift of ca. 25 nm in λmax and increased Eoptg by 0.03–0.04 eV, compared with the non-/mono-fluorinated polymers P1 and P2.55 A similar phenomenon was observed in the TRTOR-based polymer series,28 and it was in good accordance with findings from DFT excited state calculation (vide infra). Overall, the TRTzOR and TRTOR-based polymers showed comparable optical absorption properties in the solid state, but their FMO levels were quite different as unveiled by the following electrochemical study.
The electrochemical properties of the TRTzOR-based polymers were studied by cyclic voltammetry (CV) using ferrocene/ferrocenium (Fc/Fc+) as the internal standard. Clear oxidation peaks were measured for all three polymers, as shown in Fig. 2b. Based on the oxidation onset (Eonsetox) from the cyclic voltammogram, the HOMO levels (EHOMOs) were determined to be −5.24, −5.37, and −5.40 eV for P1, P2, and P3, respectively. The lower-lying EHOMOs of P2 and P3 were attributed to the additional electron-accepting fluorine atom in the fBT and ffBT acceptor moieties. Due to the weak reduction peaks, the polymer LUMO levels (ELUMOs) were calculated using the equation ELUMO = EHOMO + Eoptg, which were −3.95, −4.05, and −4.05 eV for P1, P2, and P3, respectively. Since the ELUMOs were deduced from this equation instead of ELUMO = EHOMO + Efund (Efund: the fundamental gap),56 the actual ELUMOs should be higher-lying than the currently reported values considering the fact that Efund is typically larger than Eoptg by ∼0.5 eV, which is the difference equaling to the exciton binding energy. Therefore, the actual EHOMOs should be near −3.5 eV, which are in good agreement with their p-type transistor performance (vide infra). Please note that the EHOMOs of TRTzOR-based polymers were deeper by 0.2–0.3 eV than the TRTOR-based analogous polymers,28 owing to the more electron-deficient nature of the thiazole (versus thiophene). The deeper-positioned EHOMO is not only beneficial to the polymer stability, but also increases the Voc when applied in PSCs.
DFT quantum calculation
To gain insights from a theoretical point of view, density functional theory (DFT) calculations were performed with a Gaussian 16 A.03 program.57 We started with molecular backbone geometry study at the B3LYP/6-31(d,p) level, using the hexamers of the repeat units of P1–P3 while the side chains were truncated for calculation simplicity. Highly planar backbone geometry was found for all three polymers after optimization (Fig. S3†). It should be noted that, in DFT calculation, several configurations and conformations of polymer have been tested for the lack of regioregularity. Since TRTzOR motif is expected to be planar via the intramolecular noncovalent S⋯O interaction, the interaction between TRTzOR and its adjacent benzothiadiazole becomes important to understand the geometry of molecular backbone. Thus, we proceeded to investigate the torsion potential between the alkoxy thiazole (TzOR) and the acceptor units (BT, fBT, and ffBT). For this purpose, (tuned)-ωB97X-D functional and 6-31G(d,p) basis set was used, and Fig. 3a plotted the evolution of the torsion potentials with torsion angle at 10° interval, and full structural optimization was performed at each conformation to ensure that energy minimum was reached. It was clear that the 0° conformation was most stable for all three TzOR-acceptor combinations. Interestingly, a higher torsional energy barrier was calculated for TzOR-BT/-fBT than for TzOR-ffBT unit, to deviate from the 0° conformation. This was attributed to the nontraditional C–H⋯N hydrogen-bonding between the nitrogen atom in TzOR and the neighbor hydrogen atom in the BT/fBT units (inset in Fig. 3a).42 Taking TzOR-BT unit for example, the existence of the N⋯H bonding was concluded from the fact that the distance (2.47 Å) between these two atoms was smaller than the sum of the van der Waals radius of N atom (1.6 Å) and H atom (1.2 Å).58 Please note that the DFT calculation results revealed that the F⋯N repulsion is not significant since the hexamer of the P3 repeating unit is also highly planar (Fig. S3†), which is likely attributed to the compact geometry of both ffBT and TRTzOR.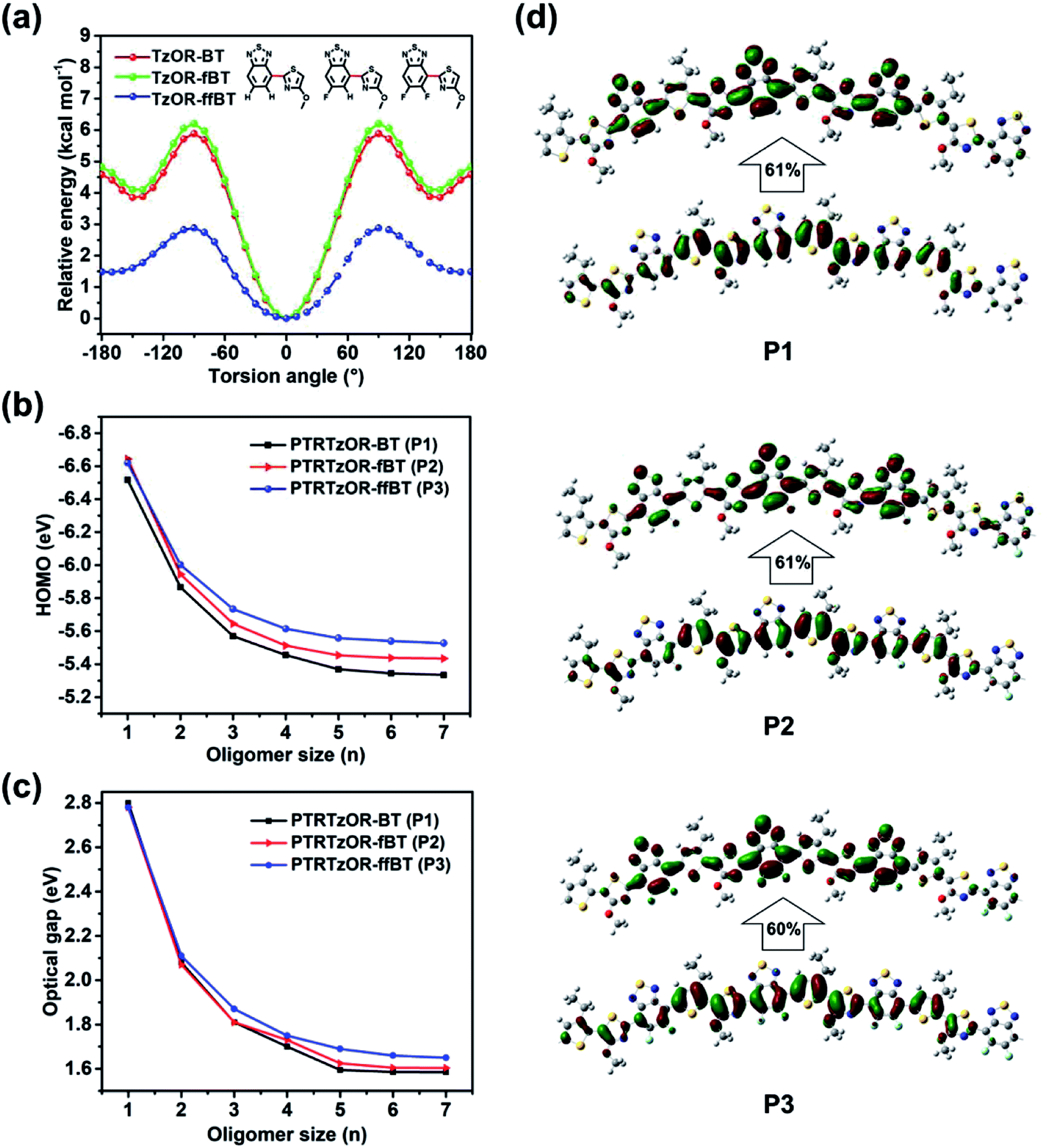 | ||
| Fig. 3 (a) Torsion potentials of TzOR-BT, TzOR-fBT and TzOR-ffBT at different torsional angles, calculated at DFT//tuned-ωB97X-D/6-31G(d,p) level. The 0° conformation was displayed in the graph. (b) Evolution of HOMO levels at tuned-ωB97X-D/6-31G(d,p)//ωB97X-D/6-31G(d,p) level. (c) Evolution of optical bandgaps with the size of the oligomers, calculated at TD-DFT//tuned-ωB97X-D/6-31G(d,p)//ωB97X-D/6-31G(d,p) level of theory. (d) Natural transition orbitals with the largest contribution to the S0 to S1 transitions for P1–P3 (bottom/top: hole/electron wave functions, calculated at TD-DFT//tuned-ωB97X-D/6-31G(d,p)//ωB97X-D/6-31G(d,p) level of theory). In NTO plots, only central parts of the hexamers are displayed while the full views are illustrated in Fig. S4.† | ||
The optoelectronic properties of the polymers were further investigated by time-dependent DFT calculation at a tuned-ωB97X-D level, because this long-range corrected function can provide highly reliable description for electron delocalization by including the dispersion effect.59 Instead of using the default range-separation parameters (ω) of 0.20 in ωB97X-D, we found more appropriate values of 0.09, 0.10, and 0.10 for P1, P2, and P3, respectively, after specialized search procedures.60–62 Subsequently, time-dependent DFT calculations were conducted to search the natural transition orbitals (NTOs)63 with the largest contribution to the S0 to S1 optical transitions (Fig. 3d and S4†). For all three polymers, the hole wave functions were delocalized across the chain over ca. 5 repeating units, while the electron wave functions were mostly localized onto the acceptor units within similar range. The EHOMOs appeared to be dependent on the size of the oligomers, and they gradually converged as the oligomer size increased to heptamer (Fig. 3b). The converged EHOMOs were −5.33, −5.43, and −5.53 eV for P1, P2, and P3, respectively. A similar trend was observed for the Eoptg calculation (Fig. 3c), and the converged Eoptgs were determined to be 1.59, 1.60, and 1.65 eV for P1, P2, and P3 respectively, by the excited states calculation using the same function and basis set. The DFT calculated EHOMOs and Eoptgs were well consistent with the experimental results, validating the planar backbone geometry of the TRTzOR-based polymers predicted by the DFT calculation.
Organic thin-film transistors and polymer solar cells
A bottom-gate/top-contact (BGTC) device with a configuration of p+-Si/SiO2 (300 nm)/semiconductor/Au (40 nm) was adopted for the fabrication of organic thin-film transistors (OTFTs). The SiO2 dielectric layer was treated with octadecyltrimethoxysilane (OTMS) self-assembled monolayer (SAM) to reduce the surface energy and passivate the charge carrier traps.64 Table S1† summarizes the OTFT performance parameters of polymers P1–P3 and the representative transistor characteristics are given in Fig. S5.† All three polymers showed unipolar p-type transport characteristics, and it was found that thermal annealing can improve transistor performance. Under their optimal annealing temperatures (200 or 250 °C), the maximum saturation hole mobility was 0.13, 0.05, and 0.09 cm2 V−1 s−1, for P1, P2, and P3, respectively. These mobility values were lower than those of the TRTOR-based analogous polymers, but were still sufficiently high for these TRTzOR-based polymers to be applied in polymer solar cells (PSCs).PSCs with a conventional structure65 were fabricated for photovoltaic device study. The PSCs were fully optimized under a variety of conditions (Tables S2–S4†) by varying processing solvents, additives and electron injection layers, and the best-performing solar cells were obtained with a device architecture of ITO/PEDOT:PSS/polymer:PC71BM/PDINO/Al, where PDINO is a perylene diimide derivative with amino N-oxide terminal substituent. The polymer:PC71BM active layers were spin-casted from chloroform solutions with 3% diphenyl ether (DPE)66 as the processing additive. The current density–voltage (J–V) curves of the best-performing devices are illustrated in Fig. 4a and their device performance parameters are summarized in Table 2.
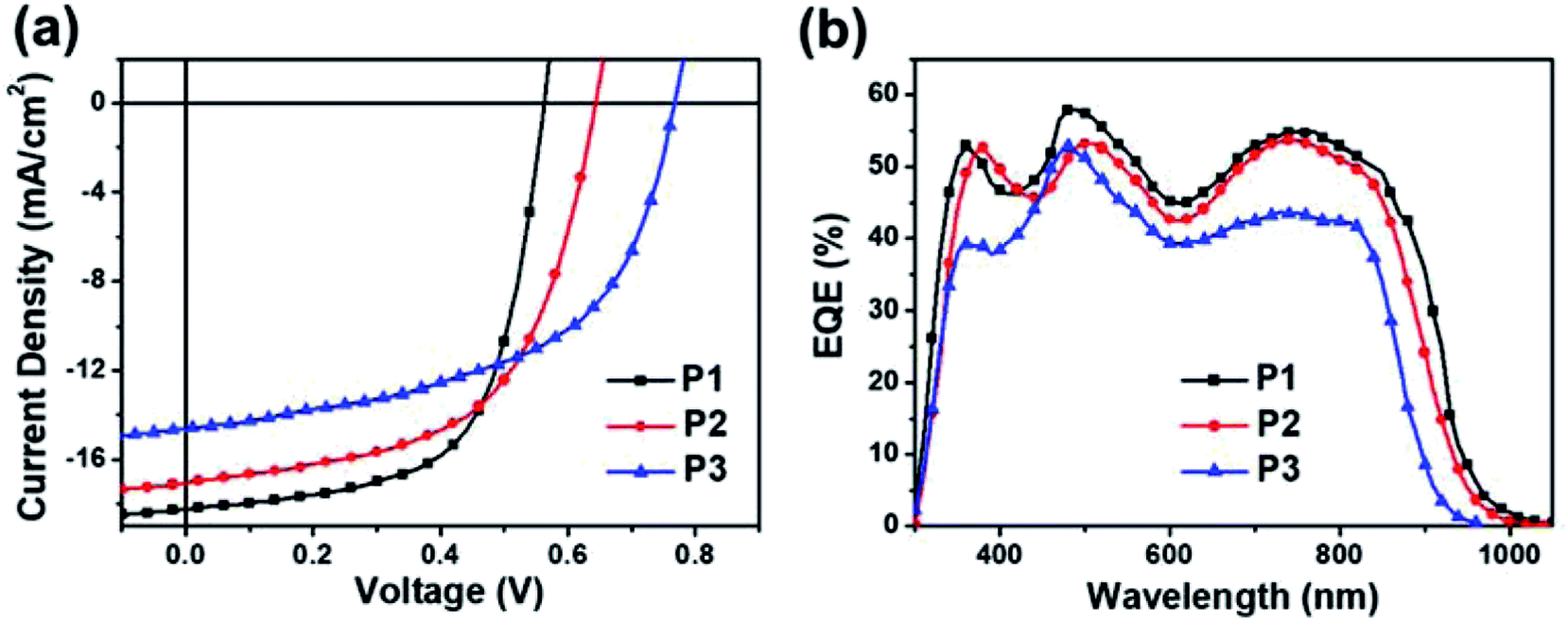 | ||
| Fig. 4 (a) J–V curves and (b) EQE spectra of the best-performing conventional PSCs with PC71BM under AM1.5 light (100 mW cm−2). | ||
:PC71BM/PDINO/Al
| Active layer | Thickness [nm] | μh,SCLC [cm2 V−1 s−1] | μe,SCLC [cm2 V−1 s−1] | Voc [V] | Jsc [mA cm−2] | FF [%] | PCEa [%] | Elossb [eV] |
|---|---|---|---|---|---|---|---|---|
| a Data represented the best performing devices with the average PCEs from eight devices shown in the parentheses.b Eloss = Eoptg − eVoc. | ||||||||
| P1:PC71BM |
160 | 8.14 × 10−4 | 7.79 × 10−4 | 0.56 | 18.22 | 63.07 | 6.46 (6.41) | 0.75 |
| P2:PC71BM |
140 | 7.40 × 10−4 | 2.89 × 10−4 | 0.64 | 17.04 | 57.24 | 6.27 (6.22) | 0.68 |
| P3:PC71BM |
200 | 3.65 × 10−4 | 7.60 × 10−4 | 0.76 | 14.55 | 54.47 | 6.09 (5.92) | 0.59 |
From P1 to P3, the Vocs gradually increase, showing a good agreement with the trend of deepening EHOMOs. The largest Voc of 0.76 V was obtained for P3:PC71BM, which was 0.1 V larger than that of the PSC using TRTOR-based analogue.28 A similar trend was also observed in PSCs based on the P1 and P2 donor polymers, demonstrating the effectiveness of thiazole substitution in Voc enhancement by lowering of HOMO levels compared to thiophene. Hence, a reduction of energy loss, which is defined as Eloss = Eoptg − eVoc, was observed relative to the devices using TRTzOR-based donor polymers. The estimated Eloss was as low as 0.59 eV in the P3:PC71BM based device, which is among the lowest Eloss values with regard to the PSCs based on thiazole-based donor polymers.44 However, the larger Voc and smaller Eloss were compromised by their relatively low short-circuit current density (Jsc) and fill factor (FF) values, probably due to the non-ideal blend film morphology with limited charge transport and significant charge recombination.67,68 After systematic device optimization, the maximum PCEs achieved in these PSCs were actually pretty comparable for all three donor polymers, varying only from 6.46% for P1 to 6.09% for P3. Please note that these PCE values were reasonably high within the results based on the donor polymers containing thiazole or thiazole derivatives,36,39,44,69–72 exceeded only by a few reports with higher PCEs but with larger Elosss.69–72 Considering the limited number of reports of low Elosss in PSCs, this materials system might be of great interest for further in-depth study on the underlying mechanism and physics.73
The external quantum efficiency (EQE) spectra of the best-performing PSCs were shown in Fig. 4b. The Jscs integrated from the EQE spectra were 17.75, 16.43, and 13.83 mA cm−2 for PSCs using P1, P2, and P3, respectively. They matched well with the Jscs obtained from the J–V measurements. Unlike TRTOR-based polymers, the Jsc values decreased as the number of fluorine atoms (on the benzothiadiazole unit) increased in these TRTzOR-based polymers. The photocurrent response was more or less uniform across the entire absorption range for all three polymer:PC71BM films, exhibiting the highest EQE value of 58% in P1:PC71BM film. The broad absorption range (300–1000 nm) and the large Jsc of 18.22 mA cm−2 achieved by P1:PC71BM PSCs indicate their great potentials as the rear cell active materials for complementary light absorption in tandem PSC devices.74
The charge transport properties of the blend films were investigated by space charge limited current (SCLC) method for both hole-only devices with a ITO/PEDOT:PSS/polymer:PC71BM/MoO3/Ag architecture and electron-only devices with a ITO/ZnO/polymer:PC71BM/PDINO/Al architecture. The hole mobility (μh,SCLC) and electron mobility (μe,SCLC) results are summarized in Table 2, and the corresponding J1/2–V plots are presented in Fig. S6.† The μh,SCLC and the μe,SCLC of P1:PC71BM film were 8.14 × 10−4 and 7.79 × 10−4 cm2 V−1 s−1, respectively, showing a well-balanced hole and electron transport with the μh/μe ratio of 1.04, which is consistent with its highest FF in PSCs. However, the hole and electron transport became less balanced in P2:PC71BM (μh/μe = 2.56) and P3:PC71BM (μh/μe = 0.48) blend films, which likely led to reduced FFs.75,76 Not surprisingly, the μh/μe ratio became even more imbalanced in blend films without using processing additive (Table S5†), as a result of non-optimal film microstructures revealed by morphology study (vide infra).
Film morphologies and their correlations to device performance
To understand the film morphology and PSC performance evolution, tapping-mode atomic force microscope (AFM) and transmission electron microscopy (TEM) measurements were carried out for polymer:PC71BM blend films prepared with and without the DPE additive (Fig. 5, S7–S8 and Table S6†). In addition, two-dimensional grazing incidence wide-angle X-ray scattering (2D-GIWAXS) technique was used to investigate their detailed molecular packing structures (Fig. 6, S9, and Table S7–S8†).
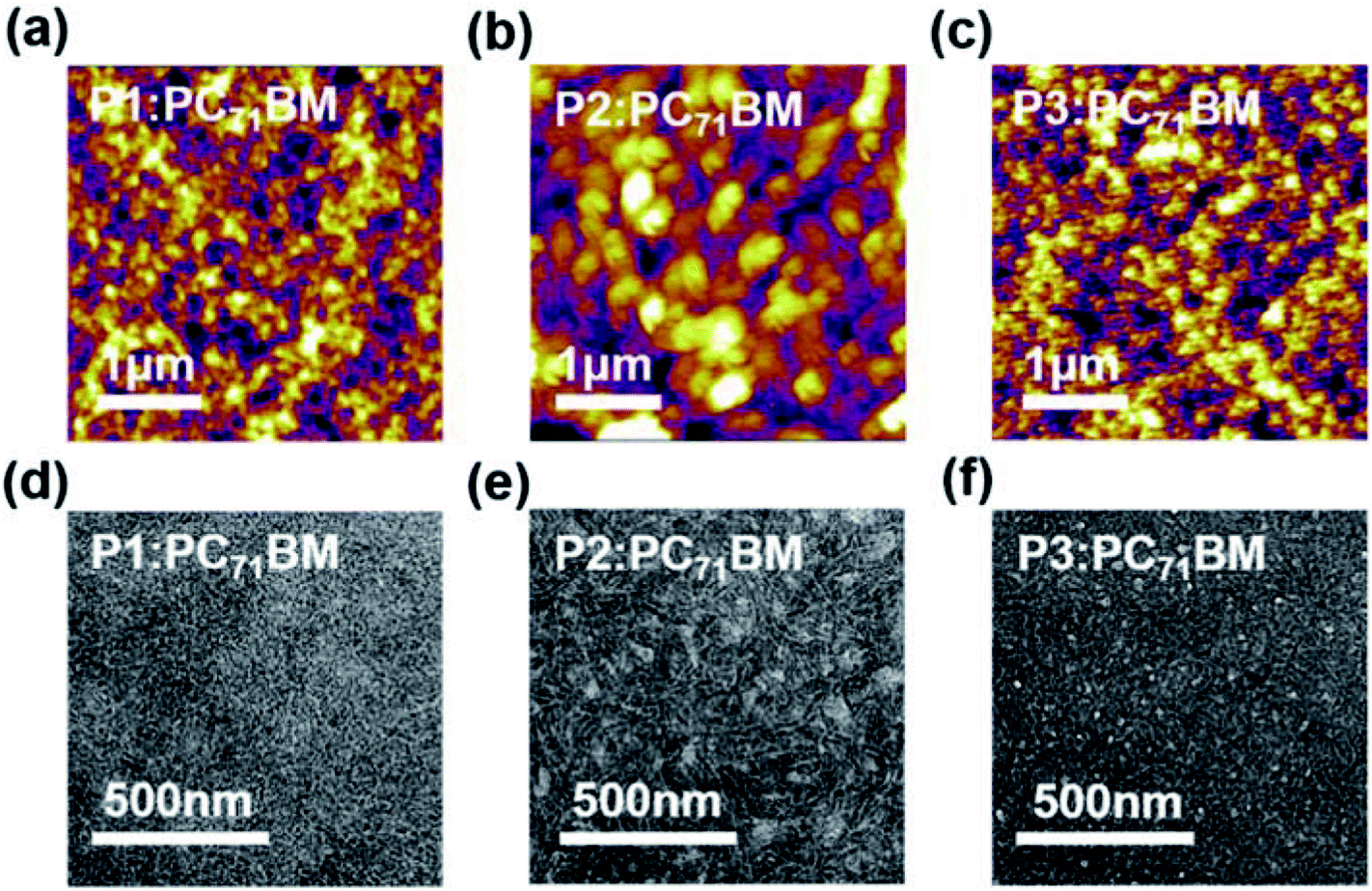 | ||
| Fig. 5 (a–c) Tapping-mode AFM height images and (d–f) TEM images of polymer:PC71BM blend films prepared under the same conditions for the optimal PSC devices. | ||
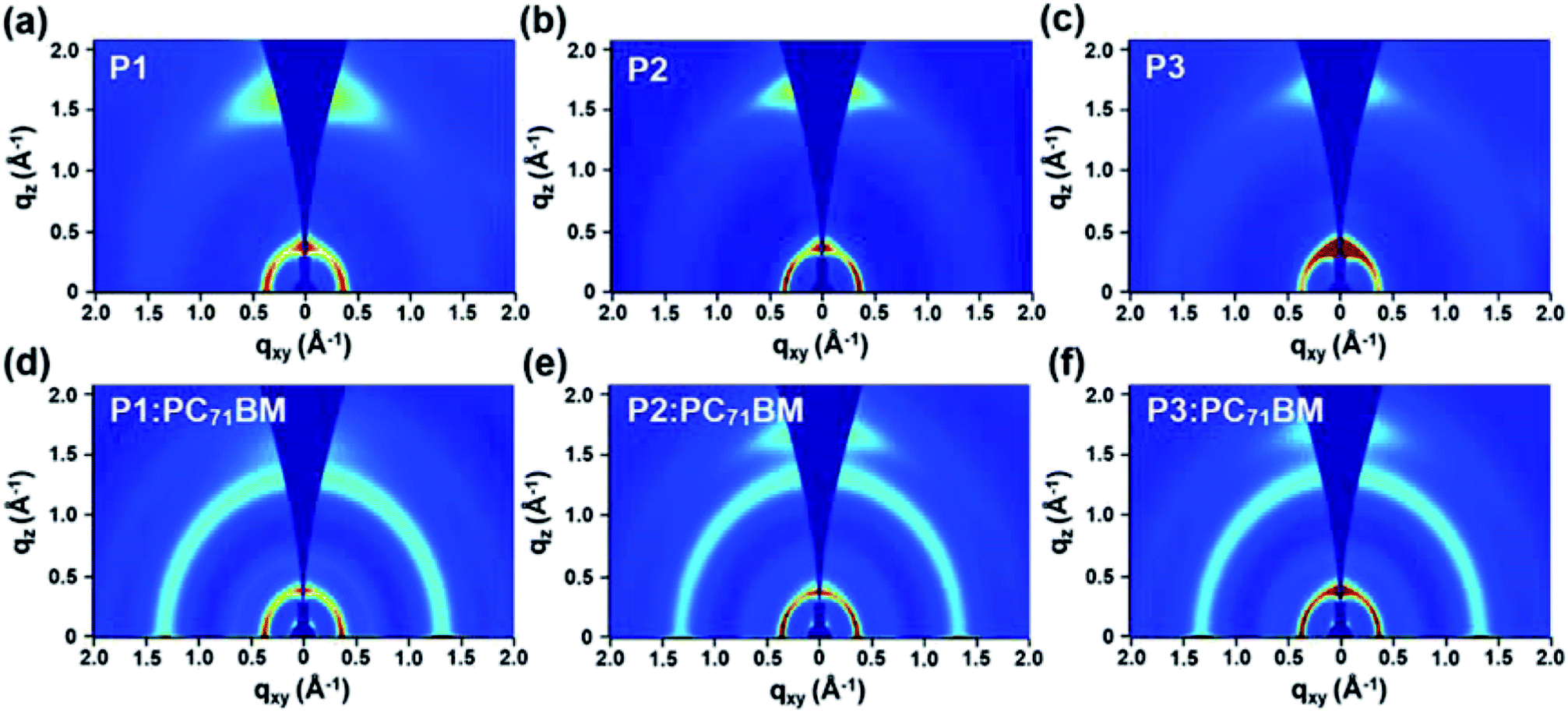 | ||
| Fig. 6 GIWAXS images of (a–c) neat polymer films and (d–f) polymer:PC71BM blend films prepared under same conditions for the optimal PSC devices. | ||
For P1:PC71BM and P2:PC71BM blend films without using DPE additive, aggregates of several hundred nanometers in size were clearly observed in both AFM and TEM images, implying poor miscibility between the donor and acceptor materials. The DPE additive led to improved miscibility and finer phase separation especially for P1:PC71BM film, which appeared to be most homogeneous one among the three blend films, thus explaining its highest Jsc and FF in PSC devices. Despite apparent morphology improvement and reduced root-mean-square (RMS) roughness for the P2:PC71BM film upon DPE addition, large amount of inhomogeneous aggregates were still observable in the TEM images (Fig. 5e). A similar trend was observed for P3:PC71BM with DPE addition. This is likely related to the stronger self-aggregation of P2 and P3 revealed by the temperature-dependent absorption spectra (Fig. S2†), yielding poor miscibility and coarse phase separation between the donor and acceptor materials, thus resulting in smaller Jscs and FFs in PSC devices.77,78
The GIWAXS study revealed that all three polymers exhibited a predominant face-on packing structure in the neat films, showing the pronounced (010) diffraction peak in the out-of-plane (OOP) direction together with the in-plane (IP) (100) scattering (Fig. 6). From P1 to P2 and to P3, their π–π stacking distance was calculated to be 3.98, 3.80 and 3.78 Å, respectively (Table S7†). The decreasing trend in the π–π stacking distance was in line with the gradually increasing aggregation tendency from P1 to P3, as a result of enhanced acceptor strength by the fluorine substituents. Owing to their higher aggregation tendency, the OOP (010) diffraction peak still remained clearly for P2:PC71BM and P3:PC71BM blend films, while it became much weaker for P1:PC71BM. In-depth analysis of the crystal coherence length (CCL) was performed on the line-cut profiles plotted in Fig. S9† using the Scherrer equation.79 As summarized in Table S8,† the larger CCL values based on the IP (100) scattering were calculated for P2 and P3 over P1, in both neat films and polymer:PC71BM blend films. For instance, the CCL values were determined to be 84.1, 124.9, and 123.1 Å for P1:PC71BM, P2:PC71BM, and P3:PC71BM, respectively, which show a good agreement with the TEM results. The strong aggregating nature of P2 and P3 disrupts the formation of optimal film morphology when blended with PC71BM acceptor, resulting in a decrease of Jsc and FF in PSC devices and limiting the achievable PCE values. The modulation of aggregation tendency of donor polymers therefore is an important aspect for PSC applications, and requires a careful optimization of the material structures.54
Conclusions
In summary, we have designed and synthesized a new head-to-head linked thienylthiazole building block TRTzOR. By replacing thiophene with electron-deficient thiazole, TRTzOR showed the lower-lying FMO energy levels than the previously reported TRTOR, while preserving a highly planar backbone enabled by intramolecular noncovalent S⋯O interaction. Compared with TRTOR-based analogues, the TRTzOR-based polymers showed the narrower Eoptgs of 1.31–1.35 eV and HOMO level lowering by 0.2–0.3 eV. In addition, the thiazole incorporation also promotes other type of noncovalent coulombic interactions and further enhances the chain planarity due to the less steric hindrance by replacing the C–H moiety in thiophene with N atom. Such highly planar backbone in combination with the large molecular polarity of thiazole contributed to their strong self-aggregation. Applying these TRTzOR-based polymers into PSCs increased the Voc by 0.1 V, and more importantly much smaller Eloss as low as 0.59 eV was achieved compared to TRTOR-based analogues, which approaches the lowest value for fullerene-based PSCs. The maximum PCE of ∼6.5% with the Jsc of 18.22 mA cm−2 was reasonably high for PSCs based on the thiazole-containing donor polymers. These results demonstrated that TRTzOR is a promising building block for constructing high-performance polymer semiconductors. On the basis of its electronic property and molecular geometry of TRTzOR, optimizing the acceptor comonomers is expected to yield the polymer semiconductors with further improved device performance.Conflicts of interest
There are no conflicts of interest to declareAcknowledgements
X. Guo is grateful to the National Science Foundation of China (NSFC, 21774055), Shenzhen Basic Research Fund (JCYJ20170817105905899), Shenzhen Peacock Plan Project (KQTD20140630110339343). H. Guo acknowledges the support from Basic Research Fund of Shenzhen City (JCYJ20160530190226226). X. Zhou is grateful to the Undergraduate Student Innovation Training Program (2016X24) and technical supports provided by High Performance Computer of SUSTech. X. Zhang is grateful to the Undergraduate Student Innovation Training Program (2016X14). H. Y. W. is grateful to the financial support from the NRF of Korea (2016M1A2A2940911 and 2015M1A2A2057506).References
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- T. M. Swager, Macromolecules, 2017, 50, 4867–4886 CrossRef CAS.
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- G.-J. N. Wang, A. Gasperini and Z. Bao, Adv. Electron. Mater., 2018, 4, 1700429 CrossRef.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- R. Steyrleuthner, R. Di Pietro, B. A. Collins, F. Polzer, S. Himmelberger, M. Schubert, Z. Chen, S. Zhang, A. Salleo, H. Ade, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2014, 136, 4245–4256 CrossRef CAS PubMed.
- H. Hu, K. Jiang, G. Yang, J. Liu, Z. Li, H. Lin, Y. Liu, J. Zhao, J. Zhang, F. Huang, Y. Qu, W. Ma and H. Yan, J. Am. Chem. Soc., 2015, 137, 14149–14157 CrossRef CAS PubMed.
- M. Saito and I. Osaka, J. Mater. Chem. C, 2018, 6, 3668–3674 RSC.
- T.-A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233–244 CrossRef CAS.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409–419 RSC.
- X. Guo and M. D. Watson, Org. Lett., 2008, 10, 5333–5336 CrossRef CAS PubMed.
- X. Guo, F. S. Kim, S. A. Jenekhe and M. D. Watson, J. Am. Chem. Soc., 2009, 131, 7206–7207 CrossRef CAS PubMed.
- X. Guo, Q. Liao, E. F. Manley, Z. Wu, Y. Wang, W. Wang, T. Yang, Y.-E. Shin, X. Cheng, Y. Liang, L. X. Chen, K.-J. Baeg, T. J. Marks and X. Guo, Chem. Mater., 2016, 28, 2449–2460 CrossRef CAS.
- W. Shi, T. Zhao, J. Xi, D. Wang and Z. Shuai, J. Am. Chem. Soc., 2015, 137, 12929–12938 CrossRef CAS PubMed.
- M. J. Marsella and T. M. Swager, J. Am. Chem. Soc., 1993, 115, 12214–12215 CrossRef CAS.
- Y. Liu, Z. Zhang, S. Feng, M. Li, L. Wu, R. Hou, X. Xu, X. Chen and Z. Bo, J. Am. Chem. Soc., 2017, 139, 3356–3359 CrossRef CAS PubMed.
- L. Robitaille, M. Leclerc and C. L. Callender, Chem. Mater., 1993, 5, 1755–1761 CrossRef CAS.
- R. Cloutier and M. Leclerc, J. Chem. Soc., Chem. Commun., 1991, 1194–1195 RSC.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS PubMed.
- S. V. Meille, A. Farina, F. Bezziccheri and M. C. Gallazzi, Adv. Mater., 1994, 6, 848–851 CrossRef CAS.
- L. Huo and J. Hou, Polym. Chem., 2011, 2, 2453–2461 RSC.
- Y. Liang and L. Yu, Acc. Chem. Res., 2010, 43, 1227–1236 CrossRef CAS PubMed.
- S. Shi, Q. Liao, Y. Tang, H. Guo, X. Zhou, Y. Wang, T. Yang, Y. Liang, X. Cheng, F. Liu and X. Guo, Adv. Mater., 2016, 28, 9969–9977 CrossRef CAS PubMed.
- S. Zhang, Y. Qin, J. Zhu and J. Hou, Adv. Mater., 2018, 30, 1800868 CrossRef PubMed.
- J. Chen, Q. Liao, G. Wang, Z. Yan, H. Wang, Y. Wang, X. Zhang, Y. Tang, A. Facchetti, T. J. Marks and X. Guo, Macromolecules, 2018, 51, 3874–3885 CrossRef CAS.
- Z. Chen, P. Cai, J. Chen, X. Liu, L. Zhang, L. Lan, J. Peng, Y. Ma and Y. Cao, Adv. Mater., 2014, 26, 2586–2591 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- X. Guo, J. Quinn, Z. Chen, H. Usta, Y. Zheng, Y. Xia, J. W. Hennek, R. P. Ortiz, T. J. Marks and A. Facchetti, J. Am. Chem. Soc., 2013, 135, 1986–1996 CrossRef CAS PubMed.
- J. Huang, Y. Tang, K. Gao, F. Liu, H. Guo, T. P. Russell, T. Yang, Y. Liang, X. Cheng and X. Guo, Macromolecules, 2017, 50, 137–150 CrossRef CAS.
- J. Huang, H. Guo, A. Uddin Mohammad, J. Yu, Y. Woo Han and X. Guo, Adv. Electron. Mater., 2018, 4, 1700519 CrossRef.
- Y. Lin, H. Fan, Y. Li and X. Zhan, Adv. Mater., 2012, 24, 3087–3106 CrossRef CAS PubMed.
- I. Osaka, R. Zhang, G. Sauvé, D.-M. Smilgies, T. Kowalewski and R. D. McCullough, J. Am. Chem. Soc., 2009, 131, 2521–2529 CrossRef CAS PubMed.
- J. Jäger, N. Tchamba Yimga, M. Urdanpilleta, E. von Hauff and F. Pammer, J. Mater. Chem. C, 2016, 4, 2587–2597 RSC.
- C. Duan, J. J. van Franeker, M. M. Wienk and R. A. J. Janssen, Polym. Chem., 2016, 7, 5730–5738 RSC.
- H. Bronstein, M. Hurhangee, E. C. Fregoso, D. Beatrup, Y. W. Soon, Z. Huang, A. Hadipour, P. S. Tuladhar, S. Rossbauer, E.-H. Sohn, S. Shoaee, S. D. Dimitrov, J. M. Frost, R. S. Ashraf, T. Kirchartz, S. E. Watkins, K. Song, T. Anthopoulos, J. Nelson, B. P. Rand, J. R. Durrant and I. McCulloch, Chem. Mater., 2013, 25, 4239–4249 CrossRef CAS.
- Y. Shi, H. Guo, M. Qin, J. Zhao, Y. Wang, H. Wang, Y. Wang, A. Facchetti, X. Lu and X. Guo, Adv. Mater., 2018, 30, 1705745 CrossRef PubMed.
- N. E. Jackson, B. M. Savoie, K. L. Kohlstedt, M. Olvera de la Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, J. Am. Chem. Soc., 2013, 135, 10475–10483 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 2231–2234 CrossRef CAS PubMed.
- D. Baran, T. Kirchartz, S. Wheeler, S. Dimitrov, M. Abdelsamie, J. Gorman, R. S. Ashraf, S. Holliday, A. Wadsworth, N. Gasparini, P. Kaienburg, H. Yan, A. Amassian, C. J. Brabec, J. R. Durrant and I. McCulloch, Energy Environ. Sci., 2016, 9, 3783–3793 RSC.
- J. Zhang, Y. Li, J. Huang, H. Hu, G. Zhang, T. Ma, P. C. Y. Chow, H. Ade, D. Pan and H. Yan, J. Am. Chem. Soc., 2017, 139, 16092–16095 CrossRef CAS PubMed.
- K. Kawashima, Y. Tamai, H. Ohkita, I. Osaka and K. Takimiya, Nat. Commun., 2015, 6, 10085 CrossRef CAS PubMed.
- Z. Bao, W. K. Chan and L. Yu, J. Am. Chem. Soc., 1995, 117, 12426–12435 CrossRef CAS.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS PubMed.
- Q. Wei, S. Miyanishi, E. Zhou, K. Hashimoto and K. Tajima, Synth. Met., 2014, 196, 139–144 CrossRef CAS.
- W. Wen, L. Ying, B. B. Y. Hsu, Y. Zhang, T.-Q. Nguyen and G. C. Bazan, Chem. Commun., 2013, 49, 7192–7194 RSC.
- H. Zhong, C.-Z. Li, J. Carpenter, H. Ade and A. K. Y. Jen, J. Am. Chem. Soc., 2015, 137, 7616–7619 CrossRef CAS PubMed.
- H. Hu, P. C. Y. Chow, G. Zhang, T. Ma, J. Liu, G. Yang and H. Yan, Acc. Chem. Res., 2017, 50, 2519–2528 CrossRef CAS PubMed.
- P. Chen, S. Shi, H. Wang, F. Qiu, Y. Wang, Y. Tang, J.-R. Feng, H. Guo, X. Cheng and X. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 21481–21491 CrossRef CAS PubMed.
- Y. Zhang, S.-C. Chien, K.-S. Chen, H.-L. Yip, Y. Sun, J. A. Davies, F.-C. Chen and A. K. Y. Jen, Chem. Commun., 2011, 47, 11026–11028 RSC.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- S. Liu, X. Song, S. Thomas, Z. Kan, F. Cruciani, F. Laquai, J.-L. Bredas and M. Beaujuge Pierre, Adv. Energy Mater., 2017, 7, 1602574 CrossRef.
- T. Stein, L. Kronik and R. Baer, J. Am. Chem. Soc., 2009, 131, 2818–2820 CrossRef CAS PubMed.
- T. Körzdörfer and J.-L. Brédas, Acc. Chem. Res., 2014, 47, 3284–3291 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2011, 33, 580–592 CrossRef PubMed.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- Y. Ito, A. A. Virkar, S. Mannsfeld, J. H. Oh, M. Toney, J. Locklin and Z. Bao, J. Am. Chem. Soc., 2009, 131, 9396–9404 CrossRef CAS PubMed.
- T. Kim, J. Y. Lee, J. Heo, B. Lim and J. Y. Kim, Polym. Chem., 2018, 9, 1216–1222 RSC.
- H.-C. Liao, C.-C. Ho, C.-Y. Chang, M.-H. Jao, S. B. Darling and W.-F. Su, Mater. Today, 2013, 16, 326–336 CrossRef CAS.
- C. J. Brabec, M. Heeney, I. McCulloch and J. Nelson, Chem. Soc. Rev., 2011, 40, 1185–1199 RSC.
- D. Mo, H. Wang, H. Chen, S. Qu, P. Chao, Z. Yang, L. Tian, Y.-A. Su, Y. Gao, B. Yang, W. Chen and F. He, Chem. Mater., 2017, 29, 2819–2830 CrossRef CAS.
- I. Osaka, M. Saito, T. Koganezawa and K. Takimiya, Adv. Mater., 2013, 26, 331–338 CrossRef PubMed.
- D. Zhu, X. Bao, Q. Zhu, C. Gu, M. Qiu, S. Wen, J. Wang, B. Shahid and R. Yang, Energy Environ. Sci., 2017, 10, 614–620 RSC.
- X. Bao, Y. Zhang, J. Wang, D. Zhu, C. Yang, Y. Li, C. Yang, J. Xu and R. Yang, Chem. Mater., 2017, 29, 6766–6771 CrossRef CAS.
- D. Zhu, X. Bao, D. Ouyang, J. Wang, X. Yuan, Q. Wang, D. Zhou, S. Wen and R. Yang, Nano Energy, 2017, 40, 495–503 CrossRef CAS.
- P. W. M. Blom, V. D. Mihailetchi, L. J. A. Koster and D. E. Markov, Adv. Mater., 2007, 19, 1551–1566 CrossRef CAS.
- T. Ameri, N. Li and C. J. Brabec, Energy Environ. Sci., 2013, 6, 2390–2413 RSC.
- M.-H. Jao, H.-C. Liao and W.-F. Su, J. Mater. Chem. A, 2016, 4, 5784–5801 RSC.
- X. Guo, N. Zhou, S. J. Lou, J. Smith, D. B. Tice, J. W. Hennek, R. P. Ortiz, J. T. L. Navarrete, S. Li, J. Strzalka, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Nat. Photonics, 2013, 7, 825 CrossRef CAS.
- Y. Huang, E. J. Kramer, A. J. Heeger and G. C. Bazan, Chem. Rev., 2014, 114, 7006–7043 CrossRef CAS PubMed.
- Z. Yang, H. Chen, H. Wang, D. Mo, L. Liu, P. Chao, Y. Zhu, C. Liu, W. Chen and F. He, Polym. Chem., 2018, 9, 940–947 RSC.
- D.-M. Smilgies, J. Appl. Crystallogr., 2009, 42, 1030–1034 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08360f |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |