
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA convenient and efficient precursor transformation route to well-dispersed, stable, and highly accessible supported Au nanocatalysts with excellent catalytic hydrogenation performances†
Jin-Feng Xie‡
a,
Hai-Tao Li‡a,
Qiang Gao *a,
Hao Wanga and
Yan-Sheng Gong*b
*a,
Hao Wanga and
Yan-Sheng Gong*b
aDepartment of Chemistry, Faculty of Material Science and Chemistry, China University of Geosciences, Wuhan 430074, PR China. E-mail: gaoqiang@cug.edu.cn; Fax: +86 027 6788 3731; Tel: +86 027 6788 3731
bDepartment of Materials Science and Engineering, Faculty of Material Science and Chemistry, China University of Geosciences, Wuhan 430074, PR China. E-mail: gongysh@cug.edu.cn
First published on 26th November 2018
Abstract
A new, convenient, and efficient precursor transformation route for the synthesis of supported Au nanocatalysts was reported. In this strategy, [Au(en)2]3+-riched titanate nanospheres (en: ethylenediamine) with hierarchical flower-like architecture were pre-synthesized via “ammonia etching-ion exchange” processes and then used as the precursors of the objective catalysts. Direct pyrolysis of these precursors, varying in amount of [Au(en)2]3+, led to the formation of Au nanoparticles (AuNPs) with different contents uniformly supported on highly crystalline titania nanoflowers (fTiO2). The fTiO2-supported AuNPs nanocomposites possessed highly open porous structures with large surface areas (142.3–149.3 m2 g−1), which could allow guest molecules to diffuse in and out easily. More interestingly, the formed AuNPs with small size (∼3.8 nm) were well-dispersed and partially embedded into the nanosheets of fTiO2, which was beneficial for achieving high activity while avoiding their detachment from the support during application. Accordingly, the AuNPs/TiO2 catalysts exhibited superior catalytic properties for 4-nitrophenol hydrogenation with significantly higher catalytic efficiencies than many previously reported heterogeneous catalysts. Moreover, the catalytic activity could remain almost unchanged after being recycled several times, demonstrating their high stability. These findings open up a new possibility for rational design and synthesis of supported catalysts for diverse catalytic applications.
1. Introduction
Supported catalysts, composed of active metal nanoparticles (MNPs) well dispersed on porous supports, represent one of the most important classes of catalysts with special interest for a variety of liquid phase reactions such as hydrogenation, oxidation, and other industrial or environmental applications.1 Wetness impregnation is the most widely used method for synthesizing such kinds of catalysts, however, either the aggregation of MNPs or their irreversible detachment from the support surfaces during the catalysis process will reduce the catalytic activity and stability, which is also consistently regarded as a major obstacle in the application of the post-loading method to practical synthesis.2,3 Moreover, the impregnation synthesis of supported MNPs usually requires the use of an excess of reducing reagent (e.g., H2, NaBH4, and hydrazine), which results in a relatively high production cost and/or reagent waste.4 This situation entails efforts to develop novel methodology, potentially more effective and economical, for synthesizing active and stable supported catalysts.Transformations of pre-synthesized solid precursors into morphology-preserved materials has recently emerged as an important new direction in materials chemistry, which will enable possible pathways for the rational design of target materials with a high degree of compositional and structural complexity.5,6 The versatility of precursor transformation method has been well exemplified in the synthesis of various porous carbon-supported MNPs. Specifically, pyrolysis of structurally defined organometallic coordination polymers or metal–organic frameworks (MOFs) under an inert atmosphere can carbonize the organic fractions and generate MNPs on the surface of as-carbonized matrix.7,8 One of the advantages of precursor transformation compared to conventional methods is its ability to preserve the morphology and result in a high level of porosity.9 More importantly, this method can assure homogeneous and firm attachment of MNPs to the carbonaceous matrix, since the MNPs are in situ reduced and deposited on the carbonaceous matrix formed almost simultaneously during pyrolysis.8 However, the resulting supported catalyst typically features small pore sizes (<2 nm) and a significant proportion of MNPs are embedded within the pores,10 which usually makes it difficult for potential reactants to reach the inner active sites and thus leads to a relatively slow reaction rate as compared to exposed catalysts.11 Therefore, the development of alternative transformation routes to fabricate supported catalysts with highly open porous structures toward facilitating fast molecular diffusion as well as promoting the accessibility of active sites, represents a particularly attractive target in this area.
Precursor design is undoubtedly the critical factor for transformation synthesis of catalyst materials, owing to its central role in controlling material composition and structure. Recently, hydrogen or ammonium titanate with three-dimensional (3D) hierarchical porous structure has aroused considerable interests because of its unique structural characteristics and the potential to be controllably transformed into morphology-preserved crystalline TiO2 upon thermal treatment.12–17 For example, Jitputti et al. synthesized monodispersed flower-like ammonium titanate nanospheres via a simple ammonia etching procedure, and demonstrated that the titanate nanospheres were readily transformed into anatase TiO2 nanoflowers by calcination in air.18 Intriguingly, the titanate with highly interconnected 3D hierarchical structures can not only exhibit high specific surface area (>100 m2 g−1), but possess sufficiently open pathways that can favor fast transportation of guest molecules.14 Moreover, it can efficiently adsorb metal ions (Mn+) from aqueous solution according to the following ion exchange reaction: Mn+ + nNH4+-titanate → Mn+-titanate + nNH4+.19 The resulting Mn+-titanate integrates the advantages of hierarchical porous structure and Mn+-riched surface, which urges us to consider the possibility of using it as an alternative precursor for synthesizing supported catalyst. However, direct pyrolysis of Mn+-titanate seems impractical to generate MNPs on the surface of TiO2 matrix due to the absence of reducing agent. Indeed, the Mn+-titanate could be transformed into Mn+-doped TiO2 upon calcination, as demonstrated by our survey and a previous study.20,21
Nevertheless, inspired by the advantages of transformation methodology and as our continuing interest in the development of hierarchically structured catalysts, we hypothesize that if the metal ions adsorbed on titanate are substituted by metal–organic complex ions, homogeneously dispersed MNPs might be in situ fabricated onto the TiO2 matrix, because the organic ligand might act as reducing agent during pyrolysis. To realize this design, organic ligand possessing excellent coordination ability toward metal ions is essentially necessary. It is well known that ethylenediamine (en) is one of the most common organic ligands, which can bind strongly a variety of metal ions to form positively charged [M-(en)m]n+ chelates (m represents the coordination number).22,23 Therefore, it is speculated that the surface NH4+ ions of ammonium titanate should be also replaceable by the [M-(en)m]n+ species via ion exchange, and the resulting [M-(en)m]n+-riched titanates can be further utilized to synthesize supported catalysts with the desired textures, activities, and stabilities.
Herein, we demonstrate for the first time that transformation reactions using [M-(en)m]n+-riched titanates as precursors can be successfully exploited to synthesize high-quality supported catalysts. To illustrate this concept, Au nanoparticles (AuNPs) were chosen as model supported metal component because they are the center of great interest in various catalytic reactions.24–29 Meanwhile, monodispersed flower-like ammonium titanate nanospheres were used as the parent material of hierarchical TiO2 support. It was verified that the ammonium titanate nanospheres could efficiently capture [Au(en)2]3+ species from [Au(en)2]Cl3 solution, and then the [Au(en)2]3+-riched titanate nanospheres were readily transformed into highly crystalline TiO2 nanoflowers-supported AuNPs (AuNPs/fTiO2) via a simple pyrolysis procedure, without the use of any additional reducing reagents. Importantly, benefitting from the inherent advantages of such a precursor, the resulting AuNPs/fTiO2 composites exhibited hierarchical flower-like porous structure and highly exposed active sites, which were previously difficult to achieve using conventional precursors. Moreover, the Au content could be tunable simply by controlling the initial concentration of [Au(en)2]Cl3. For catalytic hydrogenation of 4-nitrophenol (4-NP), the novel supported catalysts exhibited high activity, rapid kinetics, and excellent reusability.
2. Experimental
2.1 Chemicals and reagents
Hydrogen tetrachloroaurate(III)hydrate (HAuCl4·4H2O), ethylenediamine (en), concentered ammonium hydroxide (NH3·H2O, 28 wt%), ethanol, sodium tetraborohydrate (NaBH4), 4-nitrophenol (4-NP) and were all purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Titanium isopropoxide (Ti(OC3H7)4, 98%) was obtained from J&K Scientific Ltd. (Beijing, China). Deionized water was used throughout our experiments.2.2 Synthesis
2.3 Characterization
The hierarchical structure and sizes of as-prepared samples were observed by scanning electron microscope (SEM, SU8010, Hitachi, Japan) and transmission electron microscopy (TEM, Philips CM12). X-ray diffraction (XRD) measurements were performed on an X-ray diffractometer (D8-FOCUS, Bruker, Germany) using Cu Kα radiation (λ = 0.15405 nm). X-ray photoelectron spectroscopy (XPS) analysis was conducted on a VG Scientific ESCALAB Mark II spectrometer equipped with two ultra-high vacuum (UHV) chambers. Optical absorbance spectra were obtained using a UV-vis diffuse reflectance spectrometer (UV-vis DRS, UV-2550PC, Shimadzu Co., Japan). N2 adsorption/desorption measurements were performed on a Micromeritics ASAP2020 surface area analyzer at a liquid nitrogen temperature (77 K). The AuNPs loading amounts were determined with inductively coupled plasma atomic emission spectrometry (ICP-AES, Leeman, USA).2.4 Catalytic procedures
Catalytic performances of AuNPs/fTiO2 catalysts were evaluated by catalyzing conversion of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) in the presence of NaBH4 as the reducing agent. Typically, 0.2 mL of 4-NP solution (5 mmol L−1), 3 mL of NaBH4 solution (0.2 mol L−1), and 6.8 mL of deionized water were added to a standard quartz curette. Subsequently, 5 mg of AuNPs/fTiO2 powders were introduced to start the reaction under shaking conditions (200 rpm). The reaction temperature was set at 20, 30, or 40 °C, and UV-vis spectra of the solution were recorded at predetermined time intervals to monitor the progress of the reduction reaction.3. Results and discussion
Scheme 1 depicts the synthetic strategy of well-dispersed, stable, and highly accessible fTiO2-supported Au nanoparticles (AuNPs/fTiO2). The flower-like ammonium titanate nanospheres (etched TiO2) are first synthesized via ammonia etching by employing monodispersed amorphous titania nanospheres (TiO2) as the starting material.18 Once the ammonium titanate nanospheres are dispersed in [Au(en)2]Cl3 solution, the surface NH4+ ions of ammonium titanate can be easily replaced by the [Au(en)2]3+ species via ion exchange. Then, pyrolysis of the [Au(en)2]3+-riched titanate nanospheres can lead to formation of the objective product AuNPs/fTiO2 with homogeneous distribution of AuNPs on the surface of flower-like titania (fTiO2), where the ethylenediamine (en) ligand acts as reducing reagent. It is worth noting that the Au content of the AuNPs/fTiO2 is tunable simply by controlling the initial concentration of [Au(en)2]Cl3 in solution. At low [Au(en)2]Cl3 concentration (≤0.24 mg L−1), it was found that all the [Au(en)2]3+ species in solution were completely captured by the ammonium titanate (monitored by ICP-AES), so the actual loading amounts of Au in AuNPs/fTiO2-I (0.42 wt%) and AuNPs/fTiO2-II (0.84 wt%) were exactly identical to the theoretical values. When the concentration of [Au(en)2]Cl3 exceeded 0.36 mg L−1, the solution [Au(en)2]3+ species were found to be incompletely adsorbed by ammonium titanate, which resulted in a slightly lower loading amounts in AuNPs/fTiO2-III (1.66 wt%) and AuNPs/fTiO2-IV (3.33 wt%) than the theoretical values (1.68 wt% and 3.36 wt%). To probe whether the physicochemical characteristics of the as-synthesized AuNPs/fTiO2 composites with different Au contents were satisfactorily developed, multiple analytical techniques including SEM, TEM, XRD, XPS, and N2 adsorption/desorption measurements were applied.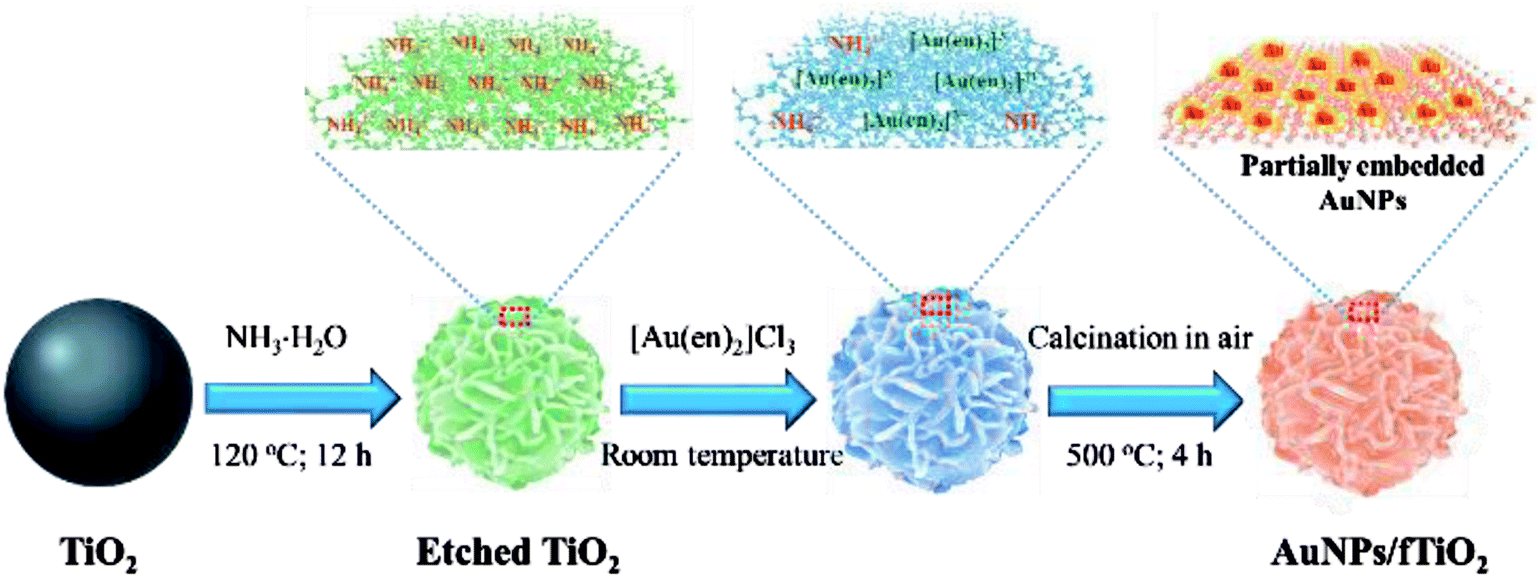 | ||
| Scheme 1 Synthetic route of the flower-like titania-supported AuNPs (AuNPs/fTiO2). | ||
SEM images were collected for TiO2, etched TiO2, fTiO2 (pure titania without AuNPs loading), and AuNPs/fTiO2 samples with different Au contents to distinguish their morphological difference. As seen from Fig. 1a, the TiO2 sample almost consists of monodispersed and uniform spherical particles with diameters of about 400–500 nm. Upon hydrothermal treatment in ammonia solution at 120 °C for 12 h, these TiO2 nanospheres were readily etched by ammonia due to their amorphous nature and converted to ammonium titanate ((NH4)2Ti2O5·H2O) according to the findings of similar experiments in the literature.21 The resulting ammonium titanate (i.e., etched titania) shows a unique flower-like architecture that is well organized by many nanosheets (Fig. 1b). Moreover, these nanoflowers also exhibit a high monodispersity, and their diameter is not changed visibly as compared to TiO2 precursor. Fig. 1c–g show the SEM images of samples (fTiO2, AuNPs/fTiO2-I, AuNPs/fTiO2-II, AuNPs/fTiO2-III, and AuNPs/fTiO2-IV) after calcination at 500 °C for 4 h without and with AuNPs loading. Little morphology difference can be found between the five samples and they are also similar to the etched titania. These results verify that the nanosheets-assembled flower-like structure can be well maintained during pyrolysis processes. Furthermore, the existence of Au in the four AuNPs/fTiO2 samples was identified by EDS, and the results is shown in Fig. 1h. Evidently, besides Ti and O elements, the Au element can be also found in each AuNPs/fTiO2 sample. Moreover, as expected, the peak intensity of Au element increases with the increase of AuNPs loading amount.
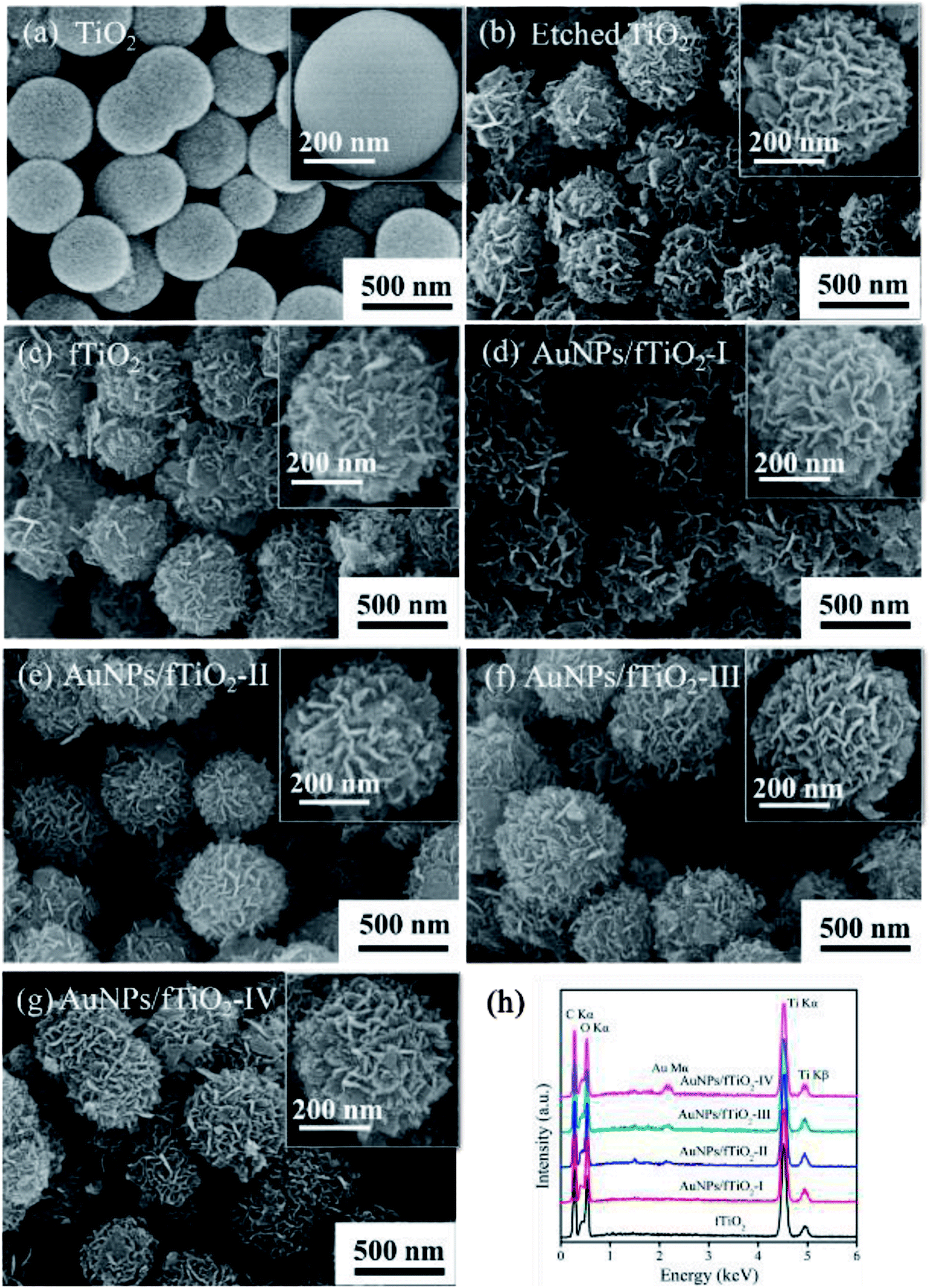 | ||
| Fig. 1 (a–g) SEM images of the samples involved in this study, and (h) EDS spectra of the flower-like titania samples without and with AuNPs loading. | ||
To better illustrate the structure of the supported Au nanocatalysts, representative TEM images taken from the AuNPs/fTiO2-III sample is given in Fig. 2. From Fig. 2a, it can be seen that the AuNPs/fTiO2 particle exhibits a highly open hierarchical flower-like structure assembled by nanosheets. The magnified TEM image (Fig. 2b) clearly shows that ultrafine AuNPs with well-dispersed distribution are uniformly located on the surfaces of nanosheets and no large irregular Au particle is found. The size distribution histogram of AuNPs calculated from the corresponding TEM image is given in Fig. 2c. Clearly, the AuNPs exhibit very small particle sizes (∼3.8 nm), which should be active enough for possibly achieving a high catalytic activity.31,32 More interestingly, the AuNPs do not only decorate the titania nanosheets but are partially embedded within them; thus, the AuNPs/fTiO2 composite is expected to be highly stable during catalytic process. Further analysis of TEM image was conducted using selected area electron diffraction (SAED) (Fig. 2d). The diffraction spots can be assigned to anatase-phased titania ((101), (200), (105), and (004) faces) and cubic-phased Au ((111) and (200) faces),33,34 respectively, which confirms the successful transformation of [Au(en)2]3+-riched titanate precursor to highly crystalline AuNPs/fTiO2.
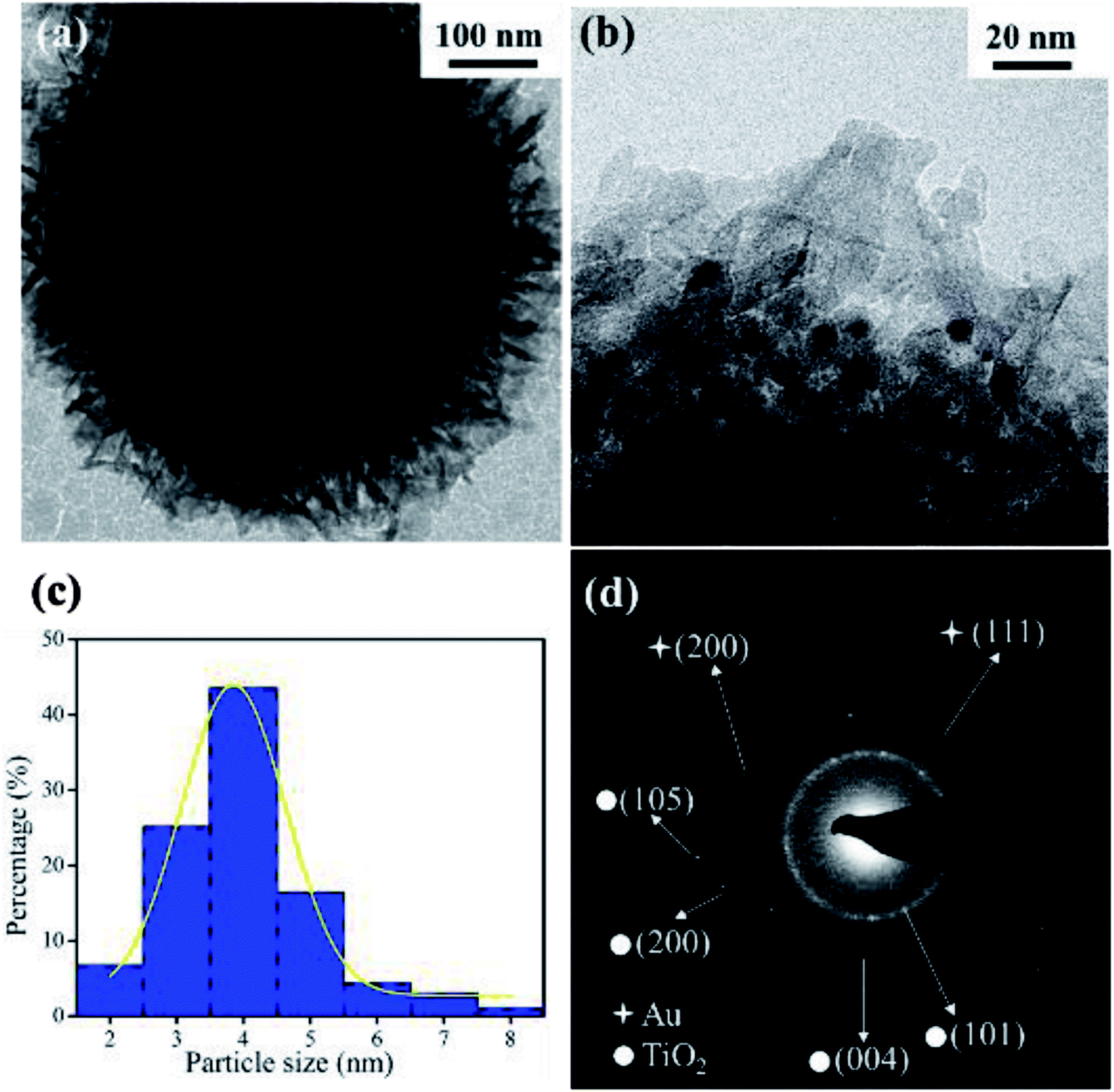 | ||
| Fig. 2 (a) Low and (b) high resolution TEM images of AuNPs/fTiO2-III, (c) particle size distribution curve, and (d) selected-area electron diffraction (SAED) pattern. | ||
Phase structures of fTiO2 and AuNPs/fTiO2 samples were investigated by XRD, and the results are shown in Fig. 3. In pattern of fTiO2 sample, the peaks at 25.3°, 37.8°, 47.9°, 53.9°, 55.0°, and 62.6° can be assigned to the diffractions of (101), (004), (200), (105), (211), and (204) crystalline planes of anatase (JCPDS card no. 04-0477),35 respectively, and no impurity peaks are found, indicating that the fTiO2 is in a single anatase phase. For each AuNPs/fTiO2 sample, the anatase peaks are still well-resolved and nearly identical to those of fTiO2 sample, which indicates that the crystalline structure of fTiO2 is not influenced by the loading of AuNPs. Besides, the peaks at 38.14°, 44.5°, and 64.7° also appear in their XRD patterns, which can be indexed to the (111), (200), and (220) planes of the Au phase, respectively, according to JCPDS 04-0784.36 In agreement with above-discussed EDS result, the intensity of the characteristic peaks of Au phase significantly increases as the loading amount of Au increases. The XRD data further confirm the successful preparation of the AuNPs/fTiO2 catalysts.
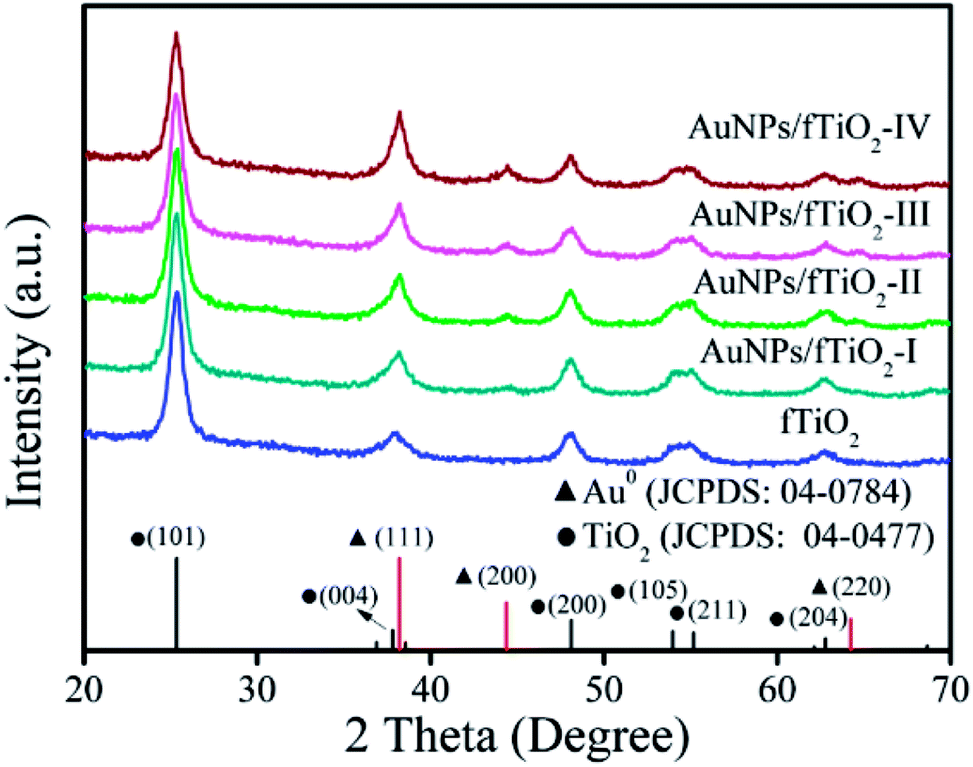 | ||
| Fig. 3 XRD patterns of the flower-like titania samples without and with AuNPs loading. | ||
The XPS analysis on AuNPs/fTiO2-III was performed to obtain information about the chemical composition of this kind of composites. For comparison, pure titania sample (fTiO2) was also investigated. The elements identified in the survey spectra are Ti, O, and C for both samples, and Au for AuNPs/TiO2-III, where the C emission peak should be due to the ex situ preparation process or the transfer process of the sample into the UHV chamber (Fig. 4a).35 The high-resolution Ti XPS spectra of TiO2 and AuNPs/fTiO2-III exhibit two peaks at 464.4 eV (2p1/2) and 458.7 eV (2p3/2) (Fig. 4b), which can be assigned to the Ti4+ oxidation state according to the reported results.37 Fig. 4c shows that the binding energies of Au 4f7/2 and Au 4f5/2 in AuNPs/TiO2-III are 83.4 and 87.0 eV, respectively, and the interval value between the two peaks is 3.6 eV, indicating the metallic nature of AuNPs.38 Compared to the reported results from pure unsupported gold (Au 4f7/2 = 84.0 eV and Au 4f5/2 = 87.7 eV),39 both Au 4f peaks of AuNPs/TiO2-III shows negative shifts, reflecting the strong interaction of AuNPs with fTiO2 that changes their electronic environment.38,40 For a supported catalyst, one common disadvantage is that the supported nanoparticles easily come away from the surface of support during use in aqueous media. The presence of strong interaction between AuNPs and fTiO2 might be able to overcome that disadvantage and make the AuNPs/fTiO2 highly stable in catalytic processes.41–43
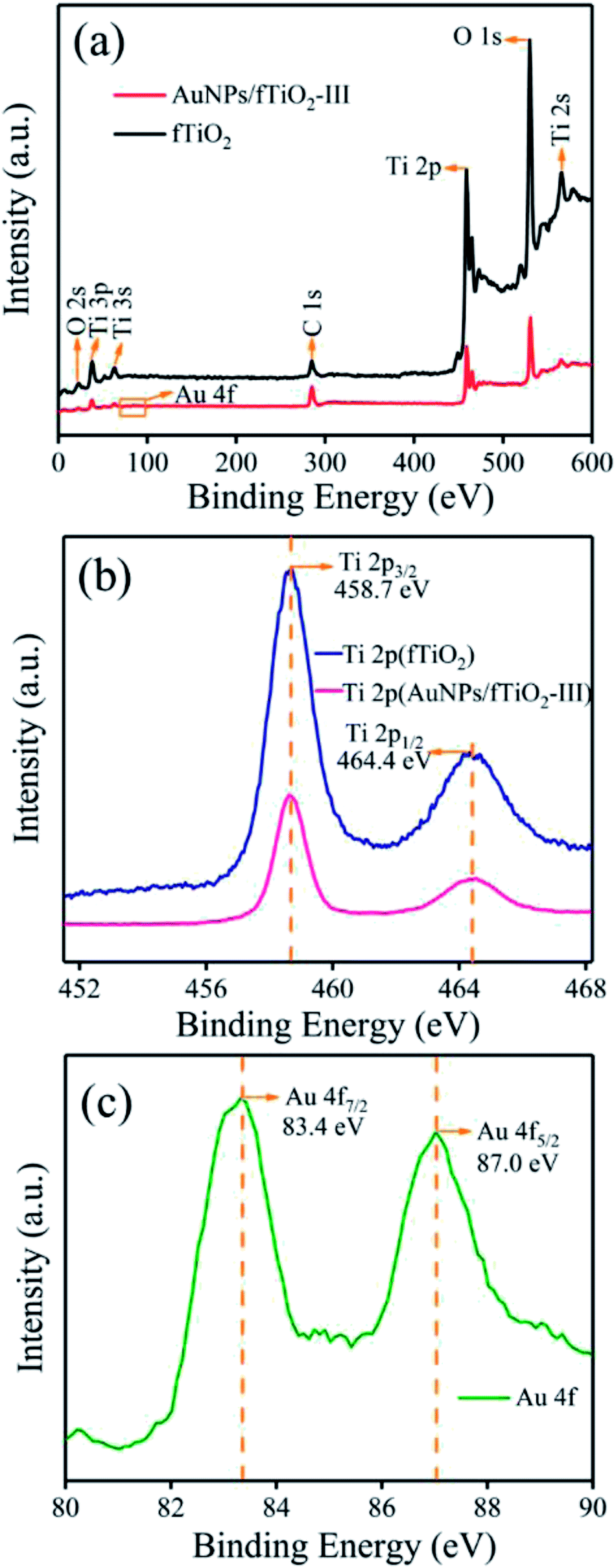 | ||
| Fig. 4 (a) XPS spectra of fTiO2 and AuNPs/fTiO2-III, (b) Ti 2p spectra of both samples, and (c) Au 4f spectrum of AuNPs/fTiO2-III. | ||
The fTiO2 and AuNPs/fTiO2 samples were further analyzed by Raman and UV-vis DRS spectroscopies. As shown in Fig. 5a, the fTiO2 exhibits modes at 143 cm−1 (Eg), 197 cm−1 (Eg), 397 cm−1 (B1g), 517 cm−1 (A1g), and 637 cm−1 (Eg), which are characteristic for titania anatase.44 From the inset of Fig. 5a, it is evident that the anatase Eg absorption mode at 144 cm−1 is gradually shifted to higher energies with the increase of Au content, suggesting increased crystalline defects within the anatase crystal structure.40 Such defects should be attributed to the partial embedment of AuNPs into titania framework,45 as demonstrated by TEM image. Fig. 5b shows UV-vis DRS spectra for fTiO2 and four AuNPs/fTiO2 samples. All these samples exhibit strong absorption below 400 nm due to band gap excitation of anatase matrixes.46 Besides, the UV-vis DRS spectra of four AuNPs/fTiO2 samples exhibit a significantly enhanced absorption in the visible range (500–600 nm) owing to the plasmon resonance effect of metallic Au, which also indicates the existence of AuNPs.40 The absorbance intensifies and red-shifts with increasing Au loading, which suggests the formation of more and bigger particles.40
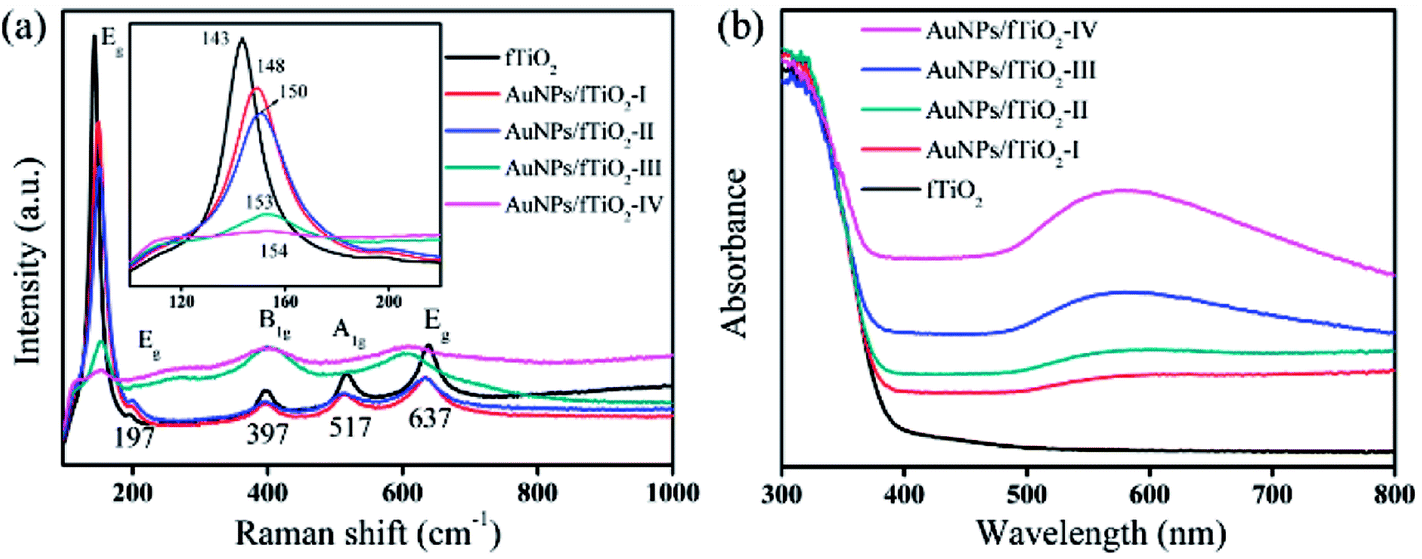 | ||
| Fig. 5 (a) Raman and (b) UV-vis DRS spectra of the flower-like titania samples without and with AuNPs loading. | ||
N2 adsorption/desorption measurement was conducted to determine the textural parameters of fTiO2 and AuNPs/fTiO2 samples. As shown in Fig. S1a,† the five samples exhibit a type II isotherm pattern with an obvious hysteresis loop, indicating their porous characteristics.20,47 Moreover, all these samples display an almost identical pore size distribution (obtained by the Barrett–Joyner–Halenda (BJH) method) (Fig. S1b†), which mainly centers at about 6.86 nm, implying that the AuNPs in AuNPs/fTiO2 samples should be largely embedded into the titania skeletons, thereby avoiding pore blocking. The detailed texture parameters are further calculated, and it is found that the surface areas (142.3–149.3 m2 g−1) and pore volumes (0.39–0.43 cm3 g−1) of these samples are also very close to each other (Table 1). Compared to some previously developed TiO2-supported AuNPs catalysts (Table S1, ESI†), the AuNPs/fTiO2 catalysts have a larger surface areas and pore volumes. In general, the catalytic performance of a heterogeneous catalyst is positively correlated with its surface area.48 The large surface area of theses flower-like composites, in conjugation with their highly open structures, distinct crystallinities, and high monodispersity of AuNPs, should be considerably beneficial to their catalytic performances.
| Sample | Surface area (m2 g−1) | Pore size(nm) | Pore volume (cm3 g−1) |
|---|---|---|---|
| fTiO2 | 145.1 | 6.86 | 0.41 |
| AuNPs/fTiO2-I | 148.9 | 6.86 | 0.43 |
| AuNPs/fTiO2-II | 149.3 | 6.84 | 0.43 |
| AuNPs/fTiO2-III | 146.9 | 6.86 | 0.41 |
| AuNPs/fTiO2-IV | 142.3 | 6.88 | 0.39 |
The reduction of 4-nitrophenol (4-NP) in the presence of NaBH4 was used as the model reaction to evaluate the catalytic performances of the AuNPs/fTiO2 samples. For comparison, control experiments were first carried out. As shown in Fig. 6a, it can be found that the concentration of 4-NP remains nearly unchanged in the absence of AuNPs/fTiO2, indicating that NaBH4 alone cannot reduce 4-NP under our experiment conditions. When fTiO2 is added to the 4-NP/NaBH4 aqueous solution, the concentration change of 4-NP is still negligible over the investigated period (Fig. 6b), indicating the pure titania material is basically inefficient in catalyzing NaBH4 to generate active hydrogen. In contrast, addition of fTiO2-supported AuNPs into 4-NP/NaBH4 aqueous solution could cause the fading and ultimate bleaching of the yellow color of the 4-NP solution. The processes were monitored by the UV-vis spectrophotometry. Fig. 6c presents the typical spectral results of 4-NP/NaBH4 in the presence of AuNPs/fTiO2-III. It is found that the absorption of 4-NP at 400 nm decreases quickly, along with a concomitant increase of a new peak at 315 nm that can be assigned to 4-aminophenol (4-AP).49,50 The conversion rate of 4-NP can reach 49.2% within 0.5 min, 80.3% within 2 min, and nearly 100% within 6 min. Moreover, the UV-vis spectra show an isosbestic point between the two absorption bands, indicating that the 4-NP has been specifically converted to 4-AP without any side reaction.49,50
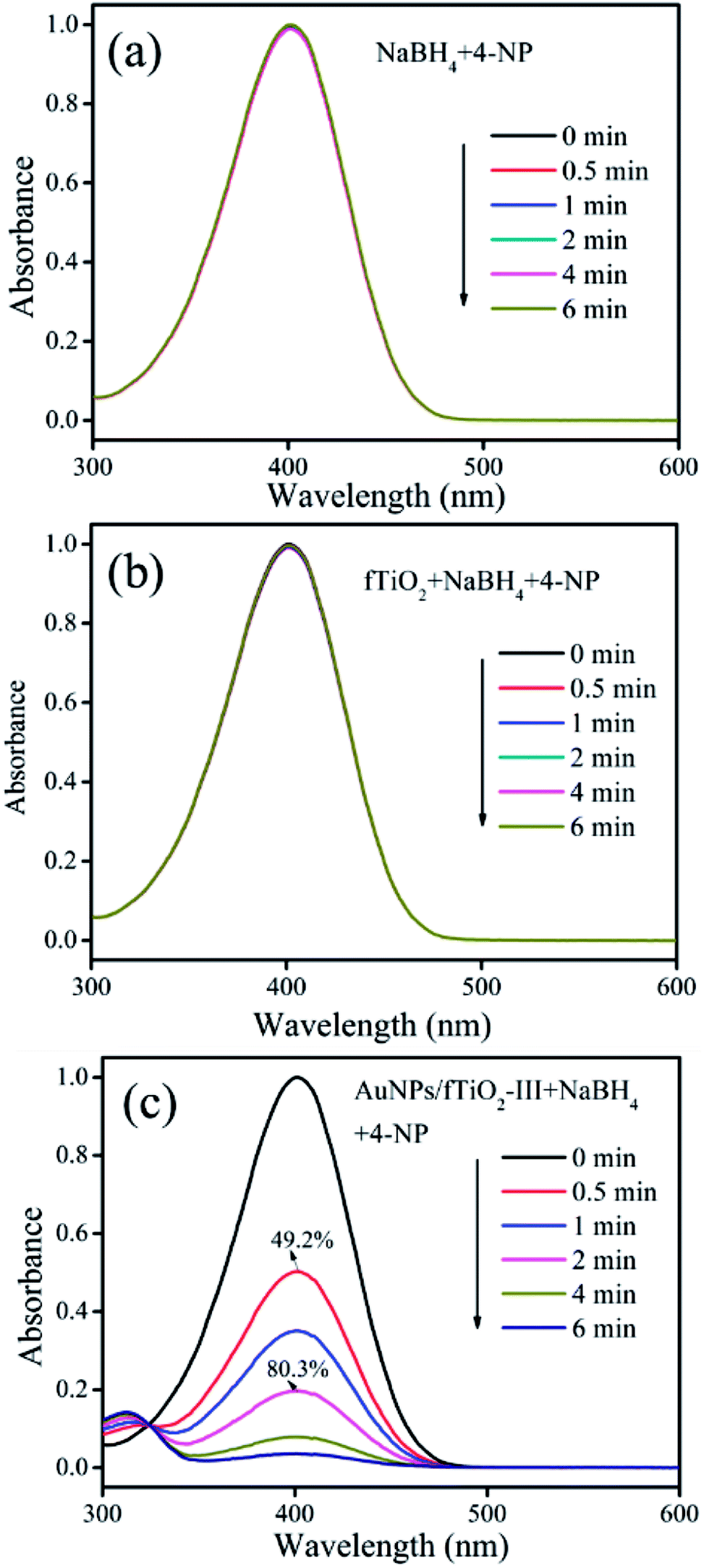 | ||
| Fig. 6 UV-vis absorption spectra of 4-NP during reduction reactions in the presence of (a) NaBH4 alone, (b) fTiO2/NaBH4, and (c) AuNPs/fTiO2/NaBH4, respectively. | ||
To gain insight about the difference of catalytic activities between different AuNPs/fTiO2 catalysts and their temperature-dependent catalytic kinetic behaviors, a series of comparative experiments were further conducted. Considering the concentration of NaBH4 used in our experiments largely exceeds that of 4-NP, the reaction rate can be assumed to be independent of NaBH4 concentration. Therefore, a pseudo-first-order kinetic equation can be applied to evaluate the catalytic rate, which is written as −dct/dt = kct or −ln(ct/c0) = kt, where c0 and ct are the initial concentration of 4-NP and its concentration at time t, respectively, and k is the rate constant (min−1).35 Fig. S2† show the plots of −ln(ct/c0) as a function of t for the reactions respectively catalyzed by AuNPs/fTiO2-I (Fig. S2a†), AuNPs/fTiO2-II (Fig. S2b†), AuNPs/fTiO2-III (Fig. S2c†), and AuNPs/fTiO2-IV (Fig. S2d†) catalysts under different temperatures (20, 30, and 40 °C). According to the linear relationship between −ln(ct/c0) and t, we calculated the rate constants k values from the slopes of the straight lines. In each case, it is found that the rate constant k exhibits an increasing trend with increasing temperature. For example, the k value of AuNPs/fTiO2-III-catalyzed reaction is 0.3972 min−1 at 20 °C, increases to 0.8887 min−1 at 30 °C, and further to 1.2139 min−1 at 40 °C (Fig. S2c†), clearly demonstrating that the catalytic efficiency is positively correlated with the reaction temperature. This observation should be understandable because the reducibility of NaBH4 can be enhanced by increasing the reaction temperature, as widely demonstrated in many previous studies.51
By comparing the kinetic data obtained from different AuNPs/fTiO2 catalysts under identical temperature condition (Fig. S2†), it can be found that the k value increases firstly and then decreases slightly with the increased content of AuNPs, i.e., AuNPs/fTiO2-I < AuNPs/fTiO2-II < AuNPs/fTiO2-IV < AuNPs/fTiO2-III. For instance, the k values at 30 °C are 0.5630 min−1 for AuNPs/fTiO2-I, 0.6084 min−1 for AuNPs/fTiO2-II, 0.8887 min−1 for AuNPs/fTiO2-III, and 0.7937 min−1 for AuNPs/fTiO2-IV, respectively. For the first three samples (i.e., AuNPs/fTiO2-I, AuNPs/fTiO2-II, and AuNPs/fTiO2-III), the persistent increase in catalytic efficiency should be due to the increased loading amount of AuNPs which can provide more active sites for the hydrogenation reaction. When the loading amount of AuNPs further increase (for AuNPs/fTiO2-IV), the catalytic efficiency is slightly poorer than that of AuNPs/fTiO2-III, which should be ascribed to the formation of relatively bigger particles in such case, as revealed by UV-vis DRS results. These findings indicate that a suitable loading amount of AuNPs is crucial for achieving a high catalytic efficiency.
Interestingly, the k value (0.8887 min−1) for AuNPs/fTiO2-III obtained at a common temperature (e.g., 30 °C) is significantly higher than those for many recently reported nanocatalysts, as summarized in Table 2.52–59 This somewhat indicates the AuNPs/fTiO2-III has outstanding catalytic performance for reduction of 4-NP. Furthermore, the activation energy (Ea) was also calculated from the plots of ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k against 1/T according to the Arrhenius equation.35 The Ea values were estimated to be 40.5 kJ mol−1 for AuNPs/fTiO2-I, 41.7 kJ mol−1 for AuNPs/fTiO2-II, 42.8 kJ mol−1 for AuNPs/fTiO2-III, and 43.6 kJ mol−1 for AuNPs/fTiO2-IV, respectively. In general, the Ea values of ordinary chemical reactions are usually between 60 and 250 kJ mol−1.60 The results presented in this study imply that the catalytic reduction reactions of 4-NP in aqueous solution by our heterogeneous catalytic systems require a lower activation energy and can be easily achieved. Moreover, these values are also significantly lower than those of many previously reported AuNPs-based catalysts, such as magnetically recoverable Au nanocatalyst (51.2 kJ mol−1),56 Au-based nanoboxes (55.44 ± 3.15 kJ mol−1),61 and Au/PDDA/NCC (69.2 kJ mol−1),62 further confirming high catalytic abilities of AuNPs/fTiO2-type catalysts. These results of both activation energies and rate constants indicate the excellent catalytic reduction activities of AuNPs/fTiO2 composites, which should be related closely to their unique features such as highly open structures, high surface areas, large pore volumes, and high dispersion of AuNPs.
k against 1/T according to the Arrhenius equation.35 The Ea values were estimated to be 40.5 kJ mol−1 for AuNPs/fTiO2-I, 41.7 kJ mol−1 for AuNPs/fTiO2-II, 42.8 kJ mol−1 for AuNPs/fTiO2-III, and 43.6 kJ mol−1 for AuNPs/fTiO2-IV, respectively. In general, the Ea values of ordinary chemical reactions are usually between 60 and 250 kJ mol−1.60 The results presented in this study imply that the catalytic reduction reactions of 4-NP in aqueous solution by our heterogeneous catalytic systems require a lower activation energy and can be easily achieved. Moreover, these values are also significantly lower than those of many previously reported AuNPs-based catalysts, such as magnetically recoverable Au nanocatalyst (51.2 kJ mol−1),56 Au-based nanoboxes (55.44 ± 3.15 kJ mol−1),61 and Au/PDDA/NCC (69.2 kJ mol−1),62 further confirming high catalytic abilities of AuNPs/fTiO2-type catalysts. These results of both activation energies and rate constants indicate the excellent catalytic reduction activities of AuNPs/fTiO2 composites, which should be related closely to their unique features such as highly open structures, high surface areas, large pore volumes, and high dispersion of AuNPs.
| Sample | Type | k (min−1) | Ref. |
|---|---|---|---|
| Pt NFs | Nanoflowers | 0.042 | 52 |
| Pt–Au pNDs/RGOs | Supported bimetallic nanodendrites | 0.228 | 53 |
| Silver/iron oxide | Nanocomposite | 0.351 | 54 |
| H40-PEI-PEG-stabilized AuNPs | Core–shell | 0.196 | 55 |
| Fe3O4–Au | Supported nanocomposite | 0.381 | 56 |
| Au–Fe3O4 | Nanodumbbells | 0.63 | 57 |
| Au@C | Yolk–shell | 0.312 | 58 |
| Fe3O4@SiO2–Au@mSiO2 | Core–shell | 0.42 | 59 |
| AuNPs/fTiO2-I | Nanoflowers | 0.5630 | This work |
| AuNPs/fTiO2-II | Nanoflowers | 0.6084 | This work |
| AuNPs/fTiO2-III | Nanoflowers | 0.8887 | This work |
| AuNPs/fTiO2-IV | Nanoflowers | 0.7937 | This work |
Another advantage of AuNPs/fTiO2-type catalysts developed in this work is their high stability. Here, we investigated the cyclic stability of AuNPs/fTiO2-III by reusing the catalyst under identical reaction conditions. As shown in Fig. 7, the catalytic efficiency of AuNPs/fTiO2-III for the reduction of 4-NP remains almost unchanged after five cycles. Compared to the recently reported catalysts such as Ni/SNTs (∼20% activity loss after 5 cycles),63 Cu2O@h-BN (50% activity loss after 4 cycles),64 PVPh-Ni3Co1 (55% activity loss after 7 cycles),65 and Ni nanoparticles in hydrogel network (25% activity loss after 5 cycles),66 our catalyst shows a significantly better durability. The excellent stability of AuNPs/fTiO2-type catalyst should be due to the partial embedment of AuNPs into the titania nanosheets, which can effectively avoid detachment from support and prevent their agglomeration under external stimulus.
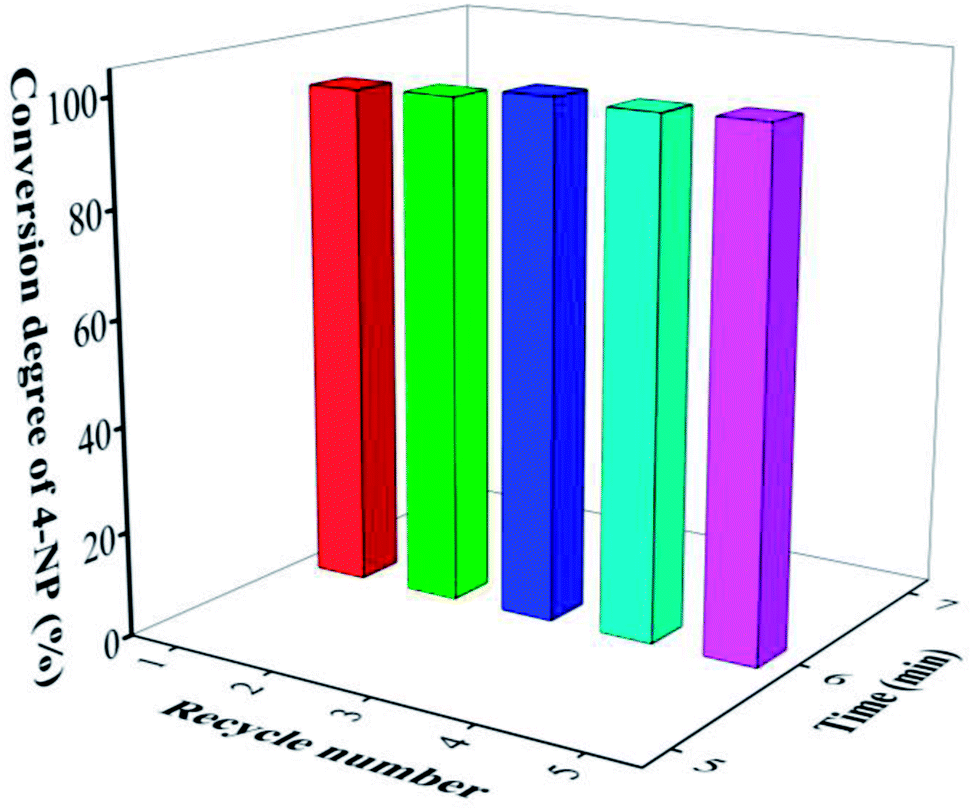 | ||
| Fig. 7 The reusability of AuNPs/fTiO2-III catalyst in reduction of 4-NP. | ||
4. Conclusions
In summary, synthesis of well-dispersed, stable, and highly accessible supported Au nanocatalysts was achieved for the first time via a simple and efficient pyrolytic transformation route using [Au(en)2]3+-riched titanate nanoflowers as precursor. The as-prepared AuNPs/fTiO2 nanocatalysts, composed of AuNPs supported on flower-like titania (fTiO2), exhibited highly open porous structures with distinct crystallinities and large surface areas (142.3–149.3 m2 g−1), where the AuNPs were highly dispersed and their loading amount could be predetermined prior to pyrolysis by controlling the [Au(en)2]3+ contents in precursors. More interestingly, the AuNPs did not only decorate the nanosheets of fTiO2 support but were partially embedded within them, which could avoid pore blocking and prevent their detachment from support. Due to these important characteristics, the AuNPs/fTiO2 nanocomposites showed excellent catalytic efficiency and high stability for reduction of 4-NP. Through replacing [Au(en)2]3+ with other metal–organic complex ions, we believe that the interesting precursor transformation might be extendable to the fabrication of different supported nanocatalysts with unique structures as well as advanced physical and/or chemical properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21303170) and the Fundamental Research Funds for the Central Universities, China University of Geosciences (Wuhan) (No. CUGL150414, CUGL140413 and CUG120115).Notes and references
- G. Prieto, J. Zečević, H. Friedrich, K. Jong and P. Jongh, Nat. Mater., 2013, 12, 34 CrossRef CAS PubMed
.
- H. Liu, L. Zhang, N. Wang and D. Su, Angew. Chem., Int. Ed., 2014, 53, 12634 CAS
- Z. Li, J. Liu, Z. Huang, Y. Yang, C. Xia and F. Li, ACS Catal., 2013, 3, 839 CrossRef CAS
- T. Ji, L. Chen, M. Schmitz, F. Bao and J. Zhu, Green Chem., 2015, 17, 2515 RSC
- R. Murugavel, M. Walawalkar, M. Dan, H. Roesky and C. Rao, Acc. Chem. Res., 2004, 37, 763 CrossRef CAS PubMed
- J. Lee, Y. Sa, T. Kim, H. Moon and S. Joo, J. Mater. Chem. A, 2014, 2, 10435 RSC
- Y. Yang, Y. Ren, C. Sun and S. Hao, Green Chem., 2014, 16, 2273 RSC
- Y. Yang, G. Gao, X. Zhang and F. Li, ACS Catal., 2014, 4, 1419 CrossRef CAS
- X. Xu, R. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988 CrossRef CAS PubMed
- X. Zhang, G. Ji, W. Liu, B. Quan, X. Liang, C. Shang, Y. Cheng and Y. Du, Nanoscale, 2015, 7, 12932 RSC
- M. Wang, C. Ye, S. Bao, Z. Chen, Y. Yu, Y. Zhang and M. Xu, Chem. Commun., 2016, 52, 12992 RSC
- D. Fattakhova-Rohlfing, A. Zaleska and T. Bein, Chem. Rev., 2014, 114, 9487 CrossRef CAS PubMed
- N. Sutradhar, S. Pahari, M. Jayachandran, A. Stephan, J. Nair, B. Subramanian, H. Bajaj, H. Mody and A. Panda, J. Mater. Chem. A, 2013, 1, 9122 RSC
- M. Feng, W. You, Z. Wu, Q. Chen and H. Zhan, ACS Appl. Mater. Interfaces, 2013, 5, 12654 CrossRef CAS PubMed
- C. Lin, D. Wong and S. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 16669 CrossRef PubMed
- H. Wu, X. Lou and H. Hng, Chem.–Eur. J., 2012, 18, 2094 CrossRef CAS PubMed
- Y. Takezawa and H. Imai, Small, 2006, 2, 390 CrossRef CAS PubMed
- J. Jitputti, T. Rattanavoravipa, S. Chuangchote, S. Pavasupree, Y. Suzuki and S. Yoshikawa, Catal. Commun., 2009, 10, 378 CrossRef CAS
- W. Liu, X. Zhao, T. Wang, J. Fu and J. Ni, J. Mater. Chem. A, 2015, 3, 17676 RSC
- H. Li, Q. Gao, B. Han, Z. Ren, K. Xia and C. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 371 CrossRef CAS PubMed
- W. Zhang, W. Zhou, J. Wright, Y. Kim, D. Liu and X. Xiao, ACS Appl. Mater. Interfaces, 2014, 6, 7292 CrossRef CAS PubMed
- R. Hancock and A. Martell, Chem. Rev., 1989, 89, 1875 CrossRef CAS
- B. Zawisza, A. Baranik, E. Malicka, E. Talik and R. Sitko, Microchim. Acta, 2016, 183, 231 CrossRef CAS PubMed
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096 RSC
- M. Rudolph and A. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC
- W. Zi and F. Toste, Chem. Soc. Rev., 2016, 45, 4567 RSC
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1 CrossRef CAS
- M. Hülsey, J. Zhang and N. Yan, Adv. Mater., 2018, 1802304 CrossRef PubMed
- Q. Yao, X. Yuan, T. Chen, D. Leong and J. Xie, Adv. Mater., 2018, 1802751 CrossRef PubMed
- H. Zhu, C. Liang, W. Yan, S. Overbury and S. Dai, J. Phys. Chem. B, 2006, 110, 10842 CrossRef CAS PubMed
- M. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed
- S. Cao, J. Chang, L. Fang and L. Wu, Chem. Mater., 2016, 28, 5596 CrossRef CAS
- Z. Jiang, W. Wei, D. Mao, C. Chen, Y. Shi, X. Lv and J. Xie, Nanoscale, 2015, 7, 784 RSC
- G. Darabdhara, M. Amin, G. Mersal, E. Ahmed, M. Das, M. Zakaria, V. Malgras, S. Alshehri, Y. Yamauchi, S. Szunerits and R. Boukherroub, J. Mater. Chem. A, 2015, 3, 20254 RSC
- Z. Ren, H. Li, Q. Gao, H. Wang, B. Han, K. Xia and C. Zhou, Mater. Des., 2017, 121, 167 CrossRef CAS
- J. Zhang, Y. Tang, K. Lee and M. Ouyang, Science, 2010, 327, 1634 CrossRef CAS PubMed
- G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R. Fitzmorris, C. Wang, J. Zhang and Y. Li, Nano Lett., 2011, 11, 3026 CrossRef CAS PubMed
- Y. Yu, C. Cao, Z. Chen, H. Liu, P. Li, Z. Dou and W. Song, Chem. Commun., 2013, 49, 3116 RSC
- A. Luna, E. Novoseltceva, E. Louarn, P. Beaunier, E. Kowalska and B. Ohtani, Appl. Catal., B, 2016, 191, 18 CrossRef CAS
- H. Li, Z. Bian, J. Zhu, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 4538 CrossRef CAS PubMed
- J. Fang, J. Li, B. Zhang, X. Yuan, H. Asakura, T. Tanaka, K. Teramura, J. Xie and N. Yan, Nanoscale, 2015, 7, 6325 RSC
- R. Nasaruddin, T. Chen, N. Yan and J. Xie, Coord. Chem. Rev., 2018, 368, 60 CrossRef CAS
- R. Nasaruddin, T. Chen, J. Li, N. Goswami, J. Zhang, N. Yan and J. Xie, ChemCatChem, 2018, 10, 395 CrossRef CAS
- A. Sandoval, R. Zanella and T. Klimova, Catal. Today, 2017, 282, 140 CrossRef CAS
- S. Farsinezhad, S. Banerjee, B. Rajeeva, B. Wiltshire, H. Sharma, A. Sura, A. Mohammadpour, P. Kar, R. Fedosejevs and K. Shankar, ACS Appl. Mater. Interfaces, 2017, 9, 740 CrossRef CAS PubMed
- X. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018 CrossRef CAS PubMed
- R. Xiong, C. Lu, Y. Wang, Z. Zhou and X. Zhang, J. Mater. Chem. A, 2013, 1, 14910 RSC
- Y. Wang, H. Arandiyan, J. Scott, A. Bagheri, H. Dai and R. Amal, J. Mater. Chem. A, 2017, 5, 8825 RSC
- P. Zhang, Y. Sui, G. Xiao, Y. Wang, C. Wang, B. Liu, G. Zhou and B. Zou, J. Mater. Chem. A, 2013, 1, 1632 RSC
- Y. Han, X. Wu, X. Zhang, Z. Zhou and C. Lu, ACS Sustainable Chem. Eng., 2016, 4, 6322 CrossRef CAS
- S. Saha, A. Pal, S. Kundu, S. Basu and T. Pal, Langmuir, 2010, 26, 2885 CrossRef CAS PubMed
- S. Mourdikoudis, T. Altantzis, L. Liz-Marzán, S. Bals, J. Pastoriza-Santos and J. Pérez-Juste, CrystEngComm, 2016, 18, 3422 RSC
- J. Lv, A. Wang, X. Ma, R. Xiang, J. Chen and J. Feng, J. Mater. Chem. A, 2015, 3, 290 RSC
- J. Chiou, B. Lai, K. Hsu and D. Chen, J. Hazard. Mater., 2013, 248–249, 394 CrossRef CAS PubMed
- Y. Dai, P. Yu, X. Zhang and R. Zhuo, J. Catal., 2016, 337, 65 CrossRef CAS
- Y. Chang and D. Chen, J. Hazard. Mater., 2009, 165, 664 CrossRef CAS PubMed
- F. Lin and R. Doong, J. Phys. Chem. C, 2011, 115, 6591 CrossRef CAS
- B. Guan, X. Wang, Y. Xiao, Y. Liu and Q. Huo, Nanoscale, 2013, 5, 2469 RSC
- Y. Deng, Y. Cai, Z. Sun, J. Liu, C. liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8466 CrossRef CAS PubMed
- Y. Yao, L. Wang, L. Sun, S. Zhu, Z. Huang, Y. Mao, W. Lu and W. Chen, Chem. Eng. J., 2013, 101, 424 CrossRef CAS
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2010, 10, 30 CrossRef CAS PubMed
- E. Lam, S. Hrapovic, E. Majid, J. Chong and J. Luong, Nanoscale, 2012, 4, 997 RSC
- S. Zhang, S. Gai, F. He, S. Ding, L. Li and P. Yang, Nanoscale, 2014, 6, 11181 RSC
- C. Huang, W. Ye, Q. Liu and X. Qiu, ACS Appl. Mater. Interfaces, 2014, 6, 14469 CrossRef CAS PubMed
- M. Raula, M. Rashid, S. Lai, M. Roy and T. Mandal, ACS Appl. Mater. Interfaces, 2012, 4, 878 CrossRef CAS PubMed
- N. Sahiner, H. Ozay, O. Ozay and N. Aktas, Appl. Catal., A, 2010, 385, 201 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08379g |
| ‡ The two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |