
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic solid-phase extraction using a mixture of two types of nanoparticles followed by gas chromatography-mass spectrometry for the determination of six phthalic acid esters in various water samples†
Hui Lia,
Zhen Cao *a,
Xiaolin Caoa,
Zejun Jianga,
A. M. Abd El-Atybc,
Yan Qia,
Hua Shaoa,
Fen Jina,
Lufei Zhenga and
Jing Wanga
*a,
Xiaolin Caoa,
Zejun Jianga,
A. M. Abd El-Atybc,
Yan Qia,
Hua Shaoa,
Fen Jina,
Lufei Zhenga and
Jing Wanga
aInstitute of Quality Standard and Testing Technology for Agro-products, Chinese Academy of Agricultural Sciences, Key Laboratory of Agro-product Quality and Safety, Ministry of Agriculture, Beijing, 100081, P. R. China. E-mail: caozhen01@caas.cn; Fax: +86-10-82106568; Tel: +86-010-82106513
bDepartment of Pharmacology, Faculty of Veterinary Medicine, Cairo University, 12211 Giza, Egypt
cDepartment of Medical Pharmacology, Medical Faculty, Ataturk University, Erzurum 25240, Republic of Turkey
First published on 27th November 2018
Abstract
Two types of magnetic microspheres (Fe3O4@MIL-100 and Fe3O4@SiO2@polythiophene) were prepared and characterized as mixed sorbents for magnetic solid-phase extraction (MSPE) of six phthalic acid esters (PAEs), including dimethyl phthalate (DMP), diethyl phthalate (DEP), di-n-butyl phthalate (DBP), benzyl butyl phthalate (BBP), di-2-ethylhexyl phthalate (DEHP), and di-n-octyl phthalate (DnOP) from water samples prior to gas chromatography-mass spectrometry (GC-MS) analysis. The synthetic magnetic nanocomposites exhibited good repeatability and chemical stability, and improved extraction efficiency for the tested PAEs. The mixture of the two types of nanoparticles substantially improved the extraction efficiency of both DMP and DEP. The key parameters affecting the extraction efficiency, such as the type and the amount of sorbent, eluent (desorption solvent), adsorption and desorption time, pH of sample solution, and sample volume, were investigated and optimized, respectively. Under optimized conditions, the developed method showed satisfactory linearity in the range of 5–5000 μg L−1 with coefficients of determination (R2) > 0.9935. The method detection limits (MDLs) and limits of quantitation (LOQs) were between 0.35–0.91 μg L−1 and 1.1–2.9 μg L−1, respectively. At three fortification levels (1.0, 10.0, and 50.0 μg L−1), the mean recoveries ranged from 76.9–109.1% with favorable relative standard deviations (RSDs) < 9%. The feasibility of the method was evaluated by analysis of water samples from various sources (tap, drinking, and mineral water). The results show that the developed method is suitable for determination of trace level PAEs in water samples.
1. Introduction
Phthalic acid esters (PAEs) are primarily used as plasticizers in the manufacture of plastics, children's toys, medical devices, food packaging materials, and other plastic products, to improve their flexibility, elasticity, and processability.1–3 PAEs are not tightly bound to the polymers1 and they can be easily leached into food or water. A series of studies have revealed that PAEs might cause teratogenic, carcinogenic, reproductive and developmental effects, and endocrine disruption in animals.2,4,5 With their extensive uses, PAEs might pose a potential risk to human health. It is therefore important to develop an effective and reliable quantitative method for determination of PAEs at trace level.Considering the relatively low concentration of PAEs in real samples, sample pretreatment and enrichment procedures are normally necessary prior to their chromatographic analysis. To date, solid-phase extraction (SPE)6–8 and solid-phase microextraction (SPME)9–11 are commonly used as prior sample pretreatment for PAEs detection. However, SPE is somewhat time-consuming and may suffer from blockage of the cartridges2,12 that may lead to extraction failure. High cost, fragility, and poor chemical and thermal stabilities of SPME fibers limit its extensive application in the SPME method.1,12 To overcome the aforementioned pitfalls, a new mode of SPE, namely magnetic solid-phase extraction (MSPE), has been introduced as a promising extraction approach. Magnetic sorbents, the core of MSPE, exhibit excellent extraction efficiency and can be easily and rapidly isolated from the mixture solution using an external magnetic field. Various functionalized magnetic nanomaterials have been fabricated and used as an appropriate sorbent in MSPE techniques. For instance, magnetic covalent triazine frameworks/Ni (CTFs/Ni) composites with good thermal and chemical stability were prepared and used to develop a MSPE method for the analysis of 6 PAEs (DMP, DEP, DBP, BBP, DEHP and DnOP) in plastic packaging material.1 The developed method was simple, sensitive, and consumed less organic solvents. Meanwhile, Xu et al.12 synthesized Fe3O4@silica@triblock-copolymer magnetic microspheres as MSPE sorbents for detecting the same 6 PAEs analytes in apparel textile. The aforementioned studies fail to extract both DMP and DEP with satisfactory extraction efficiency because of their weaker hydrophobic interaction with synthesized composites, which makes them unsuitable for better quantification of both DMP and DEP.
Recently, metal–organic frameworks (MOFs) combined with the magnetic core (Fe3O4) have successfully been synthesized and attracted much attention as efficient sorbents in MSPE. It has to be noted that Fe3O4@MIL-100 has a promising application in analysis of environmental organic pollutants. In this context, Chen and co-workers13 designed and synthesized magnetic Fe3O4@MIL-100 (Fe) core–shell microspheres as sorbents for the MSPE of polychlorinated biphenyls (PCBs) from environmental water samples. Their results demonstrated that the prepared composites exhibited good extraction efficiency for the tested analytes owing to π–π interaction. Considering PAEs having benzene rings (same as PCBs), the Fe3O4@MIL-100 (Fe) composite might be applicable for the extraction of PAEs with the same mode of π–π interaction.
Except for magnetic MOFs composites, magnetic nanoparticles (NPs) modified and functionalized by polymers are considered attractive alternative sorbents for MSPE of organic pollutants. Among functionalized polymer sorbents, much attention has been recently paid to polythiophene (PT) due to its high hydrothermal stability.14 PT has several double bonds that can interact with pollutants containing benzene rings through π–π interaction. Mehdinia et al.14 synthesized graphene/Fe3O4@polythiophene (G/Fe3O4@PT) nanocomposite as a MSPE sorbent for determination of five polycyclic aromatic hydrocarbons (PAHs) in environmental water samples. Furthermore, Tahmasebi and co-workers15 prepared polythiophene-coated Fe3O4 nanoparticles (Fe3O4@PT NPs) to extract three type of PAEs, including DBP, DEHP, and dioctyl adipate (DOA) from environmental water samples. However, the synthetic procedure of G/Fe3O4@PT was complicated and time-consuming.
The current study was aimed to synthesize both Fe3O4@MIL-100 and Fe3O4@SiO2@PT and use the mixture of magnetic NPs as MSPE sorbents for the extraction of 6 PAEs, including DMP, DEP, DBP, BBP, DEHP, and DnOP, prior to GC-MS analysis. This mixture was used to improve the extraction efficiency of both DMP and DEP. The principal parameters affecting extraction efficiencies were optimized, and the performance parameters of the developed method were also evaluated. To prove the feasibility, the developed method was finally applied to determine the target PAEs in various water samples. To the best of our knowledge, the stable and effective silica-coated NPs with surface grafting by PT (Fe3O4@SiO2@PT) were synthesized and characterized for the first time herein for the extraction of PAEs.
2. Materials and methods
2.1. Reagents and materials
Ferric chloride hexahydrate (FeCl3·6H2O) and benzene-1,3,5-tricarboxylic acid (H3BTC) were purchased from Sigma Aldrich (St. Louis, MO, US). Ferrous chloride tetrahydrate (FeCl2·4H2O) was obtained from Xi Long Chemical Co., Ltd. (Guangdong, China). Methylacrylic acid (MAA), tetraethyl orthosilicate (TEOS), and thiophene were supplied by Aladdin Industrial Corporation (Shanghai, China). Ammonia solution (25%, w/w) was purchased from Macklin Biochemical Co., Ltd. (Shanghai, China). Analytical grade ethanol and trichloromethane were provided by Beijing Chemical Works (Beijing, China). LC grade acetonitrile (ACN) and hexane were acquired from Fisher Scientific (Pittsburgh, USA). DMP (99.5% purity), DEP (99.5% purity), DBP (99.4% purity), BBP (98.4% purity), DEHP (99.6% purity), and DnOP (99.5% purity) were supplied by Dr Ehrenstorfer GmbH (Augsburg, Germany).The standard stock solutions (1.0 mg mL−1) of the tested PAEs were prepared by reconstituting the appropriate amount of each tested analyte in ACN and stored at 4 °C in dark. Working solutions were prepared daily by appropriate dilution with ACN. To minimize the risk of xenobiotic phthalate contamination, all solvents were examined for the presence of phthalates before use.
2.2. GC-MS
A SHIMADZU TQ-8040 (Kyoto, Japan) GC-MS system equipped with an auto-injector (AOC-20i) was used for analysis. Chromatographic separation was conducted on a DB5-MS capillary column (30 m × 0.25 mm i.d., 0.25 μm thickness; J&W Scientific, CA, USA). The temperature of the inlet and MS transfer line was maintained at 280 °C. The injection was performed in a splitless mode. The oven temperature was initially set at 60 °C for 1 min, programmed to 240 °C at 30 °C min−1, held for 2 min, and finally increased to 270 °C at 5 °C min−1 and held for 5 min. Helium carrier gas (purity ≥ 99.999%) was maintained at a constant flow rate of 1.03 mL min−1. A sample injection volume of 1 μL was used for each run and the solvent delay was set to 5 min. Quantitative analysis was performed in selected ion monitoring (SIM) mode. Detailed analytical parameters such as retention time and SIM transition are compiled in Table 1.| Compound | Retention time (min) | Target ion | Qualifier ions |
|---|---|---|---|
| DMP | 7.098 | 163 | 133, 77 |
| DEP | 8.450 | 149 | 177, 176 |
| DBP | 9.035 | 149 | 205, 150 |
| BBP | 12.490 | 149 | 206, 91 |
| DEHP | 14.520 | 149 | 167, 57 |
| DnOP | 17.275 | 149 | 279, 150 |
2.3. Synthesis of magnetic microspheres
Fe3O4@MIL-100 core–shell was synthesized as reported previously.13 Briefly, 0.10 g of Fe3O4 NPs was dispersed in 20 mL of MAA ethanol solution (0.58 mM) with ultrasonication for 5 min and then shaking for 24 h at room temperature. The precipitate was isolated from the solution by an external magnetic field and washed several times with deionized water and ethanol. Then, the NPs (0.10 g) synthesized above was dissolved in 5 mL of FeCl3·6H2O ethanol solution (10 mM) for 15 min and then in 5 mL of H3BTC ethanol solution (10 mM) for 30 min at 70 °C. Finally, the product was washed several times with ethanol and dried at 60 °C for 24 h.
The Fe3O4@SiO2@PT nanocomposites were prepared via adding 0.3 g of Fe3O4@SiO2, 1.0 g of anhydrous FeCl3, and 0.25 g of thiophene to 55 mL of chloroform, respectively. The mixtures were stirred for 6 h at room temperature. Thereafter, the microspheres were separated by an ordinary magnet, washed several times with ethanol, and then dried for 24 h at 60 °C.
2.4. MSPE procedure
Thirty microgram of magnetic nanoparticles (15 mg of Fe3O4@MIL-100 and 15 mg of Fe3O4@SiO2@PT) were added into a glass vial containing 10 mL of water solution (pH = 6). Using ultrasonication, the mixture was homogenously dispersed in the aqueous phase for 1 min, and then the solution was shaken in an oscillator at 200 rpm for 15 min. The sorbents were separated from the aqueous phase by an external magnet and the supernatant was discarded. Afterward, 1 mL of ACN was added and shaken for 10 min. The solution was collected by magnetic separation and then evaporated to dryness under a mild nitrogen stream at room temperature. At last, the residue was dissolved in 200 μL of hexane and filtered through a 0.22 μm glass membrane into a glass autosampler vial for GC-MS analysis.3. Results and discussion
3.1. Characterization of the magnetic Fe3O4@MIL-100 and Fe3O4@SiO2@PT composites
The morphologies of synthesized microspheres were characterized by transmission electron microscopy (TEM) (JEOL-JEM-2100F, Japan). As shown in Fig. 1a, the Fe3O4 core was coated by a SiO2 shell with approximately 10 nm in thickness. Fig. 1b showed evenly distributed transparent materials on the surface of Fe3O4@SiO2 composites, which indicated the formation of Fe3O4@SiO2@PT core–shell composites. The synthetic magnetic composites were composed of a Fe3O4 core and a MOF layer in 10 nm of thickness as shown in Fig. 1c. The mixture of the two sorbents Fe3O4@MIL-100 and Fe3O4@SiO2@PT (recycled after one use) dispersed uniformly as shown in the TEM image (Fig. 1d).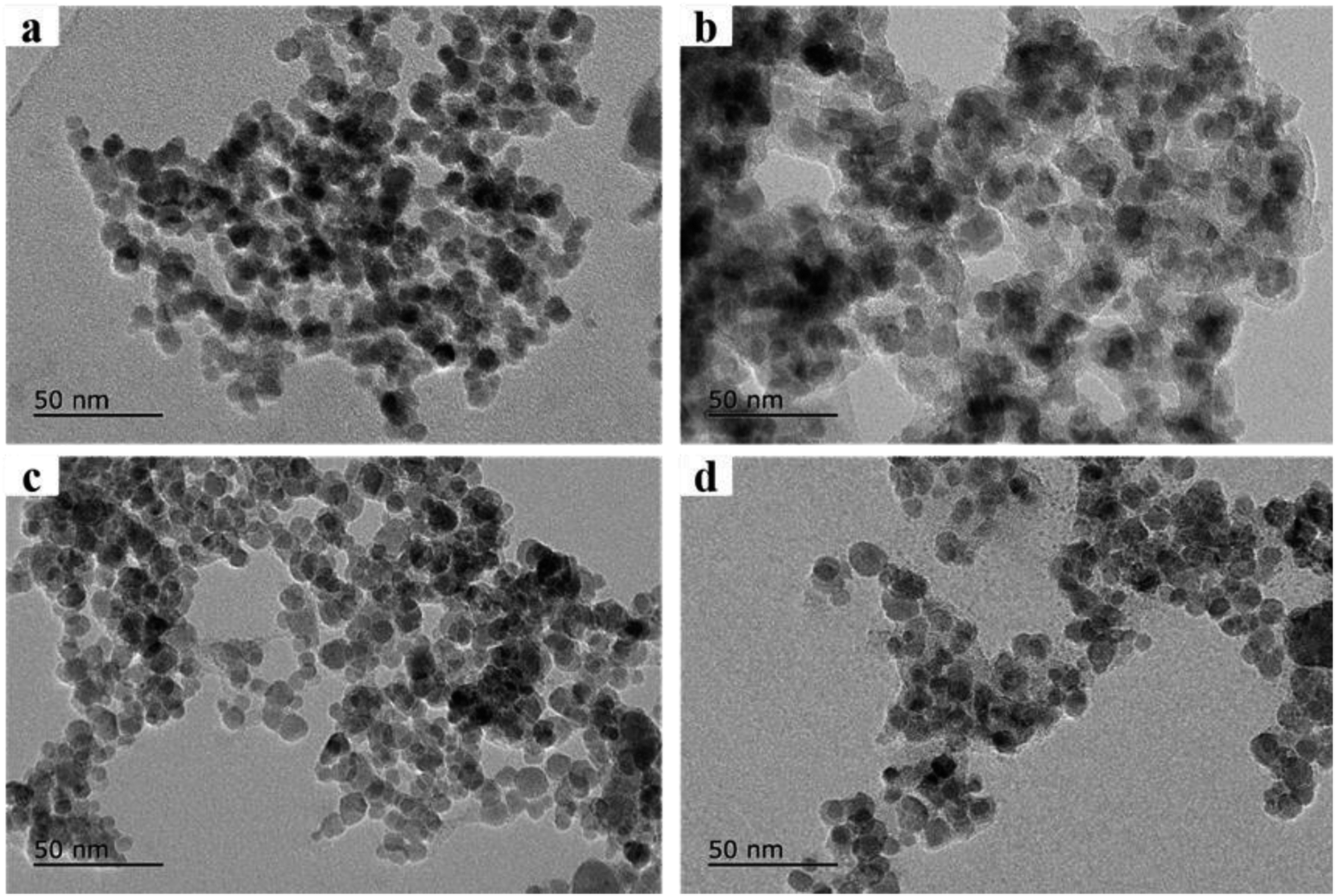 | ||
| Fig. 1 Transmission Electron Microscope images of prepared nano-materials. (a) Fe3O4@SiO2, Fe3O4 core was coated by a SiO2 shell with approximately 10 nm in thickness, (b) Fe3O4@SiO2@PT, (c) Fe3O4@MIL-100, the MOF layer was about 10 nm of thickness, (d) the mixture of Fe3O4@SiO2@PT and Fe3O4@MIL-100 recycled after one use. | ||
Sufficient magnetic properties are crucial for magnetic sorbents to be effectively separated in aqueous solutions. The magnetic curves of Fe3O4@MIL-100 and Fe3O4@SiO2@PT composites (Fig. 2A) were assessed by vibrating sample magnetometer (VSM). The curve of Fe3O4@MIL-100 exhibited no magnetic hysteresis loop, which indicates superparamagnetic properties of Fe3O4@MIL-100,13 while the curve of Fe3O4@SiO2@PT showed slight hysteresis (Fig. 2A). Both composites clearly display strong magnetic properties with saturation magnetization values of 62.0 and 38.2 emu g−1, respectively. And both values of the saturation magnetization are sufficient for separation of the composites by a magnet.18 In this study, the obtained magnetic composites were collected by a magnet within 5 s in aqueous solutions. This results indicate that the mixed magnetic microspheres are suitable for magnetic separation.
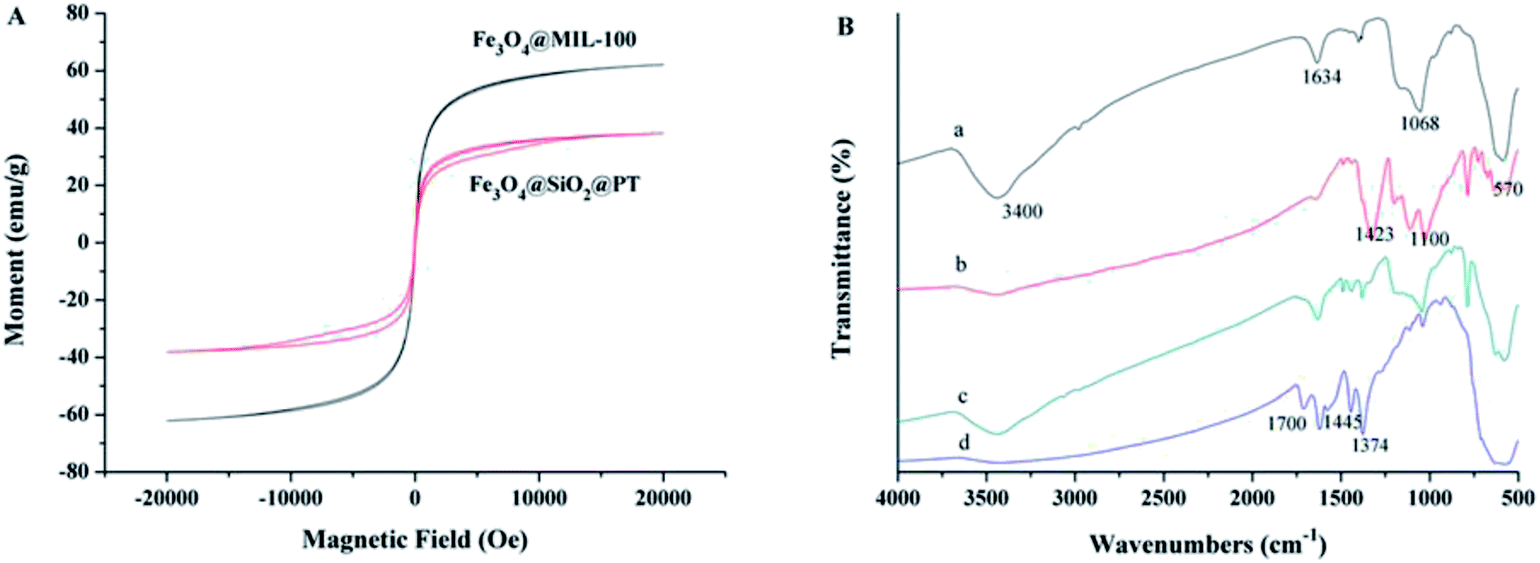 | ||
| Fig. 2 Characterization of prepared nano-materials, (A) magnetic curves of prepared two composites, and (B) the FTIR spectra of (a) Fe3O4@SiO2, (b) Fe3O4@SiO2@PT, (c) the mixture recycled after one use, and (d) Fe3O4@MIL-100. | ||
Fourier transform infrared (FTIR) spectra of the synthesized microspheres were obtained over the wave number range from 500 to 4000 cm−1 on a PerkinElmer Spectrum 100 FTIR spectrometer (Waltham, MA, US) using the nujol mull method. The characteristic peaks at 3400 and 1634 cm−1 are attributed to the O–H stretching bands (Fig. 2B-a), while the band at 1068 cm−1 was correspondent to the Si–O–Si vibration,17 and the peak at 570 cm−1 was assigned to the Fe–O–Fe stretching of Fe3O4. These indicate that the SiO2 shell has been successfully coated on the surface of Fe3O4. In contrast to Fe3O4@SiO2, the spectrum of Fe3O4@SiO2@PT exhibits additional peaks, which are related to PT coating (Fig. 2B-b). More specifically, the adsorption peaks at 1423 cm−1 and 1100 cm−1 were assigned to the vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching and C–H aromatic bonds of thiophene ring, respectively (Fig. 2B-b).15 For Fe3O4@MIL-100 (Fig. 2B-d), a band at 570 cm−1 was associated with Fe–O–Fe stretching of Fe3O4, whereas the other adsorption peaks are linked with the structure of MIL-100. For instance, a peak appeared at 1700 cm−1 was attributed to CO stretching vibration in H3BTC. The characteristic peaks at 1445 cm−1 and 1374 cm−1 were allocated to both asymmetric and symmetric stretching of carboxyl groups in H3BTC, respectively.16 This result further confirms the formation of a MIL-100 layer on the surface of Fe3O4. Fig. 2B-c showed the spectra of the mixture of Fe3O4@MIL-100 and Fe3O4@SiO2@PT (recycled after one use), with all characteristic peaks for both NPs.
C stretching and C–H aromatic bonds of thiophene ring, respectively (Fig. 2B-b).15 For Fe3O4@MIL-100 (Fig. 2B-d), a band at 570 cm−1 was associated with Fe–O–Fe stretching of Fe3O4, whereas the other adsorption peaks are linked with the structure of MIL-100. For instance, a peak appeared at 1700 cm−1 was attributed to CO stretching vibration in H3BTC. The characteristic peaks at 1445 cm−1 and 1374 cm−1 were allocated to both asymmetric and symmetric stretching of carboxyl groups in H3BTC, respectively.16 This result further confirms the formation of a MIL-100 layer on the surface of Fe3O4. Fig. 2B-c showed the spectra of the mixture of Fe3O4@MIL-100 and Fe3O4@SiO2@PT (recycled after one use), with all characteristic peaks for both NPs.
3.2. Optimization of the MSPE procedure
To obtain satisfactory extraction efficiencies for the tested PAEs using MSPE, the main factors, including the type and amount of sorbents, eluents, adsorption and desorption time, pH conditions, and sample volume, were systematically evaluated by spiking 10 μg L−1 of each tested PAEs in blank drinking water samples.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe3O4@SiO2@PT = 1:1, w/w) was used conjointly for the extraction of the tested PAEs compounds, satisfactory extraction efficiencies (over 90%) were obtained for each analyte (Fig. 3). As the results, the mixture of Fe3O4@MIL-100 and Fe3O4@SiO2@PT was finally chosen for the MSPE process.
:Fe3O4@SiO2@PT = 1:1, w/w) was used conjointly for the extraction of the tested PAEs compounds, satisfactory extraction efficiencies (over 90%) were obtained for each analyte (Fig. 3). As the results, the mixture of Fe3O4@MIL-100 and Fe3O4@SiO2@PT was finally chosen for the MSPE process.
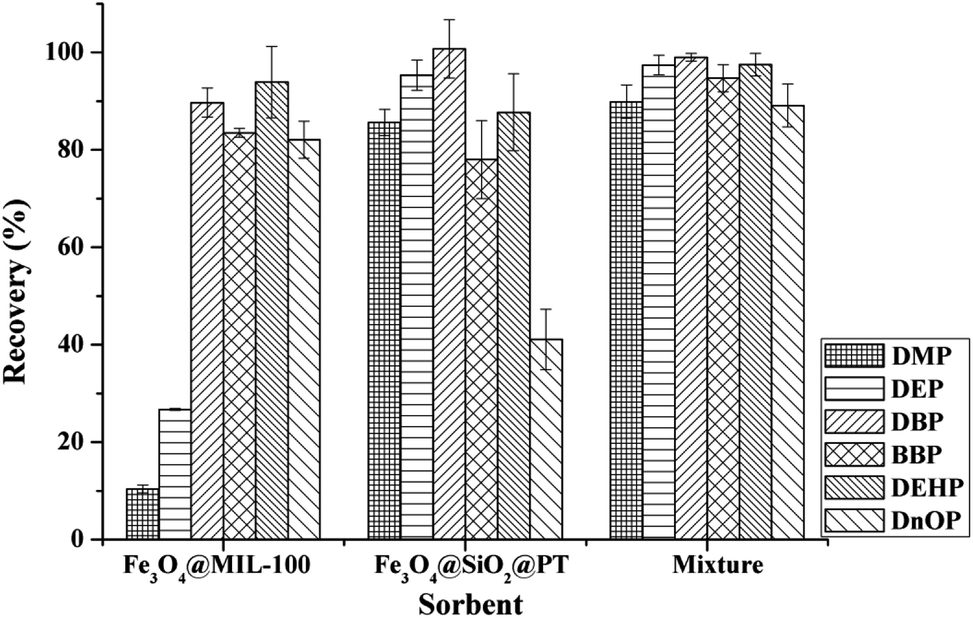 | ||
| Fig. 3 Extraction efficiency of six phthalic acid esters with composites Fe3O4@SiO2@PT, Fe3O4@MIL-100 and the mixture of Fe3O4@SiO2@PT and Fe3O4@MIL-100, respectively. Extraction conditions: 10 mL of spiked drinking water at 10.0 μg L−1, 30 mg of sorbents, 1 mL of ACN as elution solvent, pH 6.0, adsorption time 15 min, and desorption time 10 min. | ||
:1, w/w) and the total amount of the mixed sorbents of 10, 20, 30, 40 and 50 mg when 10 mL of drinking water at pH 6.0 was extracted for 25 min (adsorption time 15 min and desorption time 10 min), and eluted with 1 mL of ACN. As shown in Fig. 4, the adsorption efficiency of DEP and BBP showed little change with increasing sorbents amount. This finding might indicate that the mixed sorbents had good adsorption performance for the extraction of both DEP and BBP, even small amounts of sorbents achieved satisfactory adsorption. The most satisfactory adsorption efficiencies (for DBP, BBP, DEHP, and DnOP) were achieved when 30 mg of sorbents were used. However, the adsorption efficiency of DnOP decreased dramatically with further increment of the amount of the mixed sorbents, which might be attributed to the incomplete elution.19 Therefore, 30 mg of mixed sorbents (15 mg of Fe3O4@MIL-100 and 15 mg of Fe3O4@SiO2@PT) were selected as the optimal amount for the extraction of the tested PAEs. It's worth noting that higher extraction recovery may be obtained with other mixture ratio of Fe3O4@MIL-100/Fe3O4@SiO2@polythiophene sorbents.
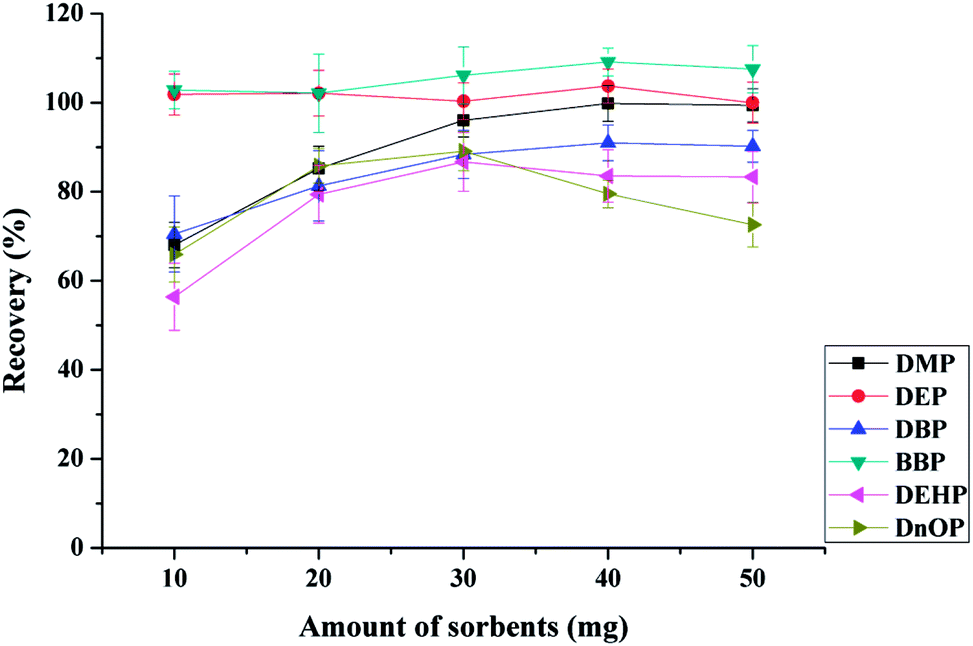 | ||
| Fig. 4 Extraction efficiency of six phthalic acid esters with different amount of the mixed sorbents. Extraction conditions: 10 mL of spiked drinking water at 10.0 μg L−1, 1 mL of ACN as elution solvent, pH 6.0, adsorption time 15 min, desorption time 10 min, and mixed sorbents (Fe3O4@MIL-100:Fe3O4@SiO2@PT = 1:1, w/w) as the MSPE sorbents. | ||
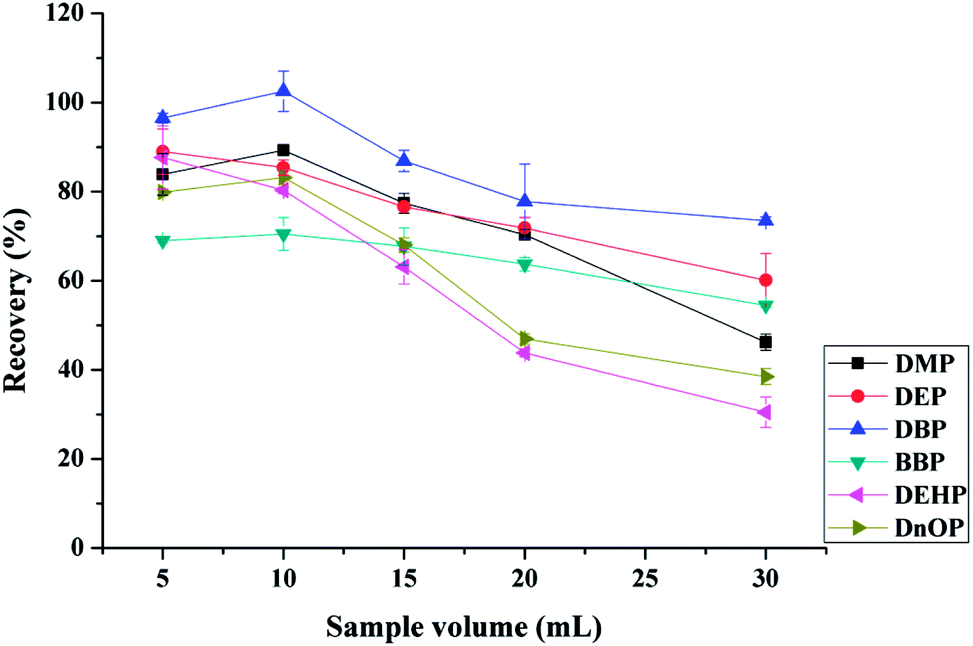 | ||
| Fig. 5 Extraction efficiency of six phthalic acid esters (10 μg L−1) with different amount of sample volume. Extraction conditions: 30 mg of mixed sorbents (15 mg of Fe3O4@MIL-100 and 15 mg of Fe3O4@SiO2@PT), 1 mL of ACN as elution solvent, pH 6.0, adsorption time 15 min, and desorption time 10 min. | ||
3.3. Reusability evaluation
The reusability of the mixed magnetic sorbents was investigated under optimal conditions for several consecutive MSPE cycles. After each cycle, the mixed sorbents were regenerated by washing twice with 2 mL of ACN each time. The negative detection of the target analytes in the washing solution indicates the absence of carry-over during the extraction procedure. No obvious changes of the extraction efficiency for the tested analytes were observed within 7 cycles (Fig. S5†). After 7 cycles, the sorbents still demonstrated strong extraction ability for both DEP and DBP, but less extraction efficiency for the rest of the analytes. Then, the prepared mixed adsorbents can be recycled for 7 times for enrichment and extraction of the tested PAEs in water samples.3.4. Control of the xenobiotic phthalate contamination
In the present study, several attempts have been made to minimize the secondary contamination. First, before usage, all solvents were checked for the presence of phthalates. Second, glass flasks and beakers were used during magnetic microspheres synthesis, whereas glass vials, pipettes and glass membranes were used necessarily during sample preparation and analysis. Third, the glassware was thoroughly rinsed with acetone and hexane and then dried at 60 °C for 6 h, and was finally placed in a furnace oven at 400 °C for 6 h. Forth, a new DB5-MS capillary column was used for phthalate analysis. The injection port seals, liners, and septa were replaced regularly. As contamination is common issue in phthalate analysis, all efforts were put to minimize the xenobiotic phthalate contamination. We had evaluated the contamination of blank samples, the used blank pure solvent and the GC-MS instrumental blank. The measured values were all below the MDLs. All these results indicated that the xenobiotic phthalate contamination from our laboratory might have little influence on this study.3.5. Method validation
| Compound | Linear range (μg L−1) | Coefficient of determination R2 | MDL (μg L−1) | LOQ (μg L−1) | RSD (%, n = 6) |
|---|---|---|---|---|---|
| DMP | 5–5000 | 0.9960 | 0.75 | 2.4 | 5.4 |
| DEP | 5–5000 | 0.9935 | 0.56 | 1.8 | 7.5 |
| DBP | 5–5000 | 0.9994 | 0.91 | 2.9 | 3.6 |
| BBP | 5–5000 | 0.9994 | 0.72 | 2.3 | 3.5 |
| DEHP | 5–5000 | 0.9993 | 0.75 | 2.4 | 5.7 |
| DnOP | 5–5000 | 0.9992 | 0.35 | 1.1 | 4.3 |
| PAEs | Spiked concentration (μg L−1) | Tap water | Drinking water | Mineral water | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Found (μg L−1) | R (%) | RSD (%) | Found (μg L−1) | R (%) | RSD (%) | Found (μg L−1) | R (%) | RSD (%) | ||
| a ND: not detected. MDL: method detection limit. | ||||||||||
| DMP | 0 | <MDL | <MDL | <MDL | ||||||
| 1 | 0.83 | 82.8 | 5.2 | 0.90 | 90.3 | 2.7 | 0.88 | 87.7 | 4.7 | |
| 10 | 8.94 | 89.4 | 3.1 | 9.47 | 94.7 | 3.5 | 9.05 | 90.5 | 1.5 | |
| 50 | 44.8 | 89.6 | 3.6 | 41.1 | 82.2 | 7.8 | 45.5 | 90.9 | 8.0 | |
| DEP | 0 | <MDL | <MDL | <MDL | ||||||
| 1 | 0.84 | 83.7 | 4.2 | 0.89 | 88.6 | 3.3 | 0.77 | 76.9 | 3.8 | |
| 10 | 8.06 | 80.6 | 4.6 | 8.06 | 80.6 | 3.1 | 9.47 | 94.7 | 4.2 | |
| 50 | 45.3 | 90.6 | 3.9 | 48.0 | 96.0 | 2.2 | 48.8 | 97.6 | 2.5 | |
| DBP | 0 | <MDL | <MDL | <MDL | ||||||
| 1 | 0.99 | 99.2 | 1.4 | 0.95 | 94.7 | 2.9 | 1.09 | 109.1 | 3.5 | |
| 10 | 10.3 | 103.2 | 5.3 | 10.1 | 101.2 | 3.9 | 10.7 | 107.3 | 2.9 | |
| 50 | 46.3 | 92.5 | 1.9 | 42.5 | 84.9 | 7.3 | 41.6 | 83.2 | 5.3 | |
| BBP | 0 | ND | <MDL | <MDL | ||||||
| 1 | 0.99 | 99.9 | 2.4 | 0.97 | 96.8 | 2.4 | 1.03 | 103.0 | 2.9 | |
| 10 | 9.03 | 90.3 | 2.0 | 10.4 | 103.5 | 2.2 | 9.59 | 95.9 | 2.7 | |
| 50 | 53.2 | 106.4 | 5.2 | 51.7 | 103.3 | 4.6 | 51.5 | 102.9 | 3.7 | |
| DEHP | 0 | <MDL | <MDL | <MDL | ||||||
| 1 | 0.83 | 82.8 | 5.8 | 0.81 | 81.4 | 3.1 | 0.93 | 93.3 | 2.5 | |
| 10 | 10.7 | 107.0 | 2.4 | 0.90 | 90.4 | 2.3 | 8.36 | 83.6 | 4.4 | |
| 50 | 45.8 | 91.6 | 3.6 | 45.3 | 90.6 | 1.8 | 47.6 | 95.3 | 1.9 | |
| DnOP | 0 | ND | ND | ND | ||||||
| 1 | 0.87 | 86.5 | 5.4 | 0.92 | 91.6 | 2.7 | 0.86 | 85.8 | 3.3 | |
| 10 | 8.28 | 82.8 | 7.5 | 9.60 | 96.0 | 2.2 | 9.04 | 90.4 | 6.3 | |
| 50 | 47.2 | 94.4 | 2.1 | 46.1 | 92.2 | 2.6 | 46.7 | 93.5 | 3.5 | |
3.6. Analysis of real samples
To demonstrate the potential applicability of the developed method, various water samples, including tap, drinking and mineral water were monitored for the tested PAEs. Tap water (not for drinking) was obtained from our laboratory, while drinking and mineral water were purchased from local big market. It has to be noted that DnOP was not detected in any of the monitored samples, whereas DMP, DEP, DBP, BBP, and DEHP were detected in various samples with concentrations being lower than their MDLs (Table 3). The selected ion chromatograms of the tested PAEs in tap water analyzed by the proposed MSPE-GC-MS method are shown in Fig. 6.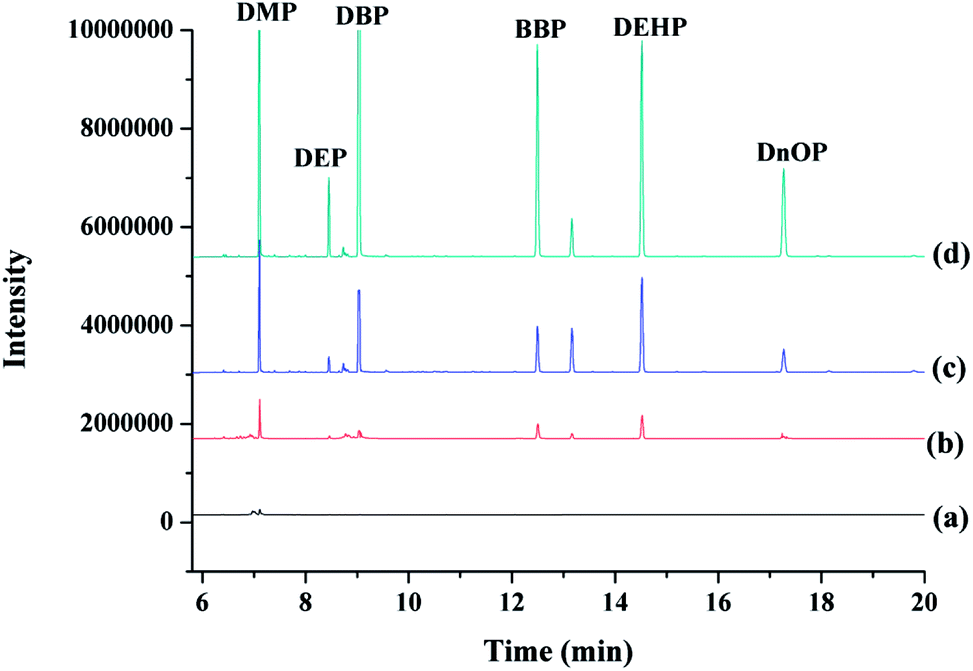 | ||
| Fig. 6 Typical selected ion chromatogram of six phthalic acid esters in tap water: non-spiked tap water (a), tap water spiked at 1.0 μg L−1 (b), 10.0 μg L−1 (c) and 50.0 μg L−1 (d). Extraction conditions: 30 mg of mixed sorbents (15 mg of Fe3O4@MIL-100 and 15 mg of Fe3O4@SiO2@PT), 1 mL of ACN as elution solvent, pH 6.0, adsorption time 15 min, desorption time 10 min, and sample volume 10 mL. | ||
3.7. Method comparison
The performance parameters of the developed method in terms of extraction time (including adsorption and desorption time), the amount of sorbents and reusability of sorbents were compared with other methods for PAEs analysis. As compiled in Table 4, the total extraction time of the developed method herein was 25 min, which is shorter than that recorded in Fe@SiO2-PEI-HPLC-UV (44 min)19 and Mag-graphene@mSiO2-C18-GC-MS (35 min).22 The amount of adsorbents used in this study was lower than that used in Fe3O4@PT-GC-FID method15 and Mag-graphene@mSiO2-C18-GC-MS method,22 and was equal to that used in Fe3O4@silica@triblockcopolymer-GC-MS method.12 In the present study, the synthetic magnetic nanocomposites can be reused for 7 times, which was superior to the previous report.1 Several reports1,12,22 have recorded poor extraction efficiency of DMP or DEP. In this study, the conjoint magnetic composites (Fe3O4@MIL-100 and Fe3O4@SiO2@PT) used here significantly improve the extraction efficiency of both DMP and DEP.| Analyte | Method | Extraction time (min) | Adsorbent amount (mg) | Reusability (times) | Ref. |
|---|---|---|---|---|---|
| a CTF: Covalent triazine frameworks; GC-FID: gas chromatography-flame ionization detector; ZIF-8: Zeolitic Imidazolate Framework-8; HPLC-DAD: high-performance liquid chromatography-diode array detection; PEI: polyethyleneimine; HPLC-UV: high-performance liquid chromatography-ultraviolet detector; DIBP: diisobutyl phthalate; DOA: dioctyl adipate; DPP: diphenyl phthalate; DPIP: diphenyl isophthalate; DCHP: dicyclohexyl phthalate. | |||||
| DMP DEP DBP BBP DEHP DnOP | Magnetic CTFs/Ni-GC-FID | 25 | 10 | 3 | 1 |
| DMP DEP DBP BBP DnOP | Fe3O4@ZIF-8-HPLC-DAD | 16 | 20 | 10 | 2 |
| DMP DEP DIBP DBP BBP DEHP DnOP | Fe3O4@silica@triblockcopoly-mer-GC-MS | 20 | 30 | — | 12 |
| DBP DEHP DOA | Fe3O4@PT-GC-FID | 12 | 100 | — | 15 |
| DPP DBP BBP DPIP DCHP | Fe@SiO2-PEI-HPLC-UV | 44 | 25 | — | 19 |
| DMP DEP DBP BBP DEHP DnOP | Mag-graphene@mSiO2-C18-GC-MS | 35 | 35 | — | 22 |
| DMP DEP DBP BBP DEHP DnOP | Fe3O4@MIL-100 & Fe3O4@SiO2@PT-GC-MS | 25 | 30 | 7 | This work |
4. Conclusions
In the present work, two types of magnetic nanocomposites (Fe3O4@MIL-100 and Fe3O4@SiO2@PT) were successfully prepared and characterized, and used as efficient mixed sorbents for the extraction and enrichment of 6 PAEs (DMP, DEP, DBP, BBP, DEHP, and DnOP) in water samples, owing to π–π and the hydrophobic interactions. Samples were quantified using GC-MS. The mixed sorbents exhibited good chemical stability and reusability (at least 7 times). Under the optimal conditions, satisfactory extraction efficiencies and high sensitivities were obtained for all tested analytes. The developed method was successfully applied for monitoring real water samples. The developed method is simple, rapid and effective, and may be extrapolated for analysis of other hydrophobic aromatic compounds at trace levels in water samples.Conflicts of interest
The authors have declared no conflicts of interest.Acknowledgements
This work was supported by the Special Program for Basic Work of the Ministry of Science and Technology of China (2013FY110100), and Guangxi Innovation-driven Development Project (AA17204043-2).References
- Z. J. Yan, M. He, B. B. Chen, B. Gui, C. Wang and B. Hu, J. Chromatogr. A, 2017, 1525, 32–41 CrossRef CAS PubMed.
- X. M. Liu, Z. W. Sun, G. Chen, W. W. Zhang, Y. P. Cai, R. M. Kong, X. Y. Wang, Y. R. Suo and J. M. You, J. Chromatogr. A, 2015, 1409, 46–52 CrossRef CAS PubMed.
- D. M. Xu, X. J. Deng, E. H. Fang, X. H Zheng, Y. Zhou, L. Y. Lin, L. P. Chen, M. Wu and Z. Q. Huang, J. Chromatogr. A, 2014, 1324, 49–56 CrossRef CAS PubMed.
- V. Fernández-González, C. Moscoso-Pérez, S. Muniategui-Lorenzo, P. López-Mahía and D. Prada-Rodríguez, Talanta, 2017, 162, 648–653 CrossRef PubMed.
- M. Behzadi, E. Noroozian and M. Mirzaeia, RSC Adv., 2014, 4, 50426–50434 RSC.
- V. L. Turco, G. Di Bella, A. G. Potortì, M. R. Fede and G. Dugo, Eur. Food Res. Technol., 2015, 240, 451–458 CrossRef.
- Q. Xu, X. Y. Yin, S. Y. Wu, M. Wang, Z. Y. Wen and Z. Z. Gu, Microchim. Acta, 2010, 168, 267–275 CrossRef CAS.
- C. Sablayrolles, M. Montréjaud-Vignoles, D. Benanou, L. Patria and M. Treilhou, J. Chromatogr. A, 2005, 1072, 233–242 CrossRef CAS PubMed.
- H. Amanzadeha, Y. Yamini, M. Moradi and Y. A. Asl, J. Chromatogr. A, 2016, 1465, 38–46 CrossRef PubMed.
- S. Lirio, C. W. Fu, J. Y. Lin, M. J. Hsu and H. Y. Huang, Anal. Chim. Acta, 2016, 927, 55–63 CrossRef CAS PubMed.
- X. L. Song, Y. Chen, J. P. Yuan, Y. J. Qin, R. S. Zhao and X. Wang, J. Chromatogr. A, 2016, 1468, 17–22 CrossRef CAS PubMed.
- M. Xu, M. H. Liu, M. R. Sun, K. Chen, X. J. Cao and Y. M. Hu, Talanta, 2016, 150, 125–134 CrossRef CAS PubMed.
- X. F. Chen, N. Ding, H. Zang, H. Yeung, R. S. Zhao, C. G. Cheng, J. H. Liu and T. W. Dominic Chan, J. Chromatogr. A, 2013, 1304, 241–245 CrossRef CAS PubMed.
- A. Mehdinia, N. Khodaee and A. Jabbari, Anal. Chim. Acta, 2015, 868, 1–9 CrossRef CAS PubMed.
- E. Tahmasebi, Y. Yamini, M. Moradi and A. Esrafili, Anal. Chim. Acta, 2013, 770, 68–74 CrossRef CAS PubMed.
- X. L. Cao, G. Y. Liu, Y. X. She, Z. J. Jiang, F. Jin, M. J. Jin, P. F. Du, F. N. Zhao, Y. X. Zhang and J. Wang, RSC Adv., 2016, 6, 113144–113151 RSC.
- M. E. S. Synaridou, V. A. Sakkas, C. D. Stalikas and T. A. Albanis, J. Chromatogr. A, 2014, 1348, 71–79 CrossRef CAS PubMed.
- S. X. Zhang, H. Y. Niu, Y. Q. Cai and Y. L. Shi, Anal. Chim. Acta, 2010, 665, 167–175 CrossRef CAS PubMed.
- Q. X. Zhou, Z. W. Zheng, J. P. Xiao, H. L. Fan and X. Y. Yan, Anal. Bioanal. Chem., 2016, 408, 5211–5220 CrossRef CAS PubMed.
- H. Li, Z. J. Jiang, X. L. Cao, H. Su, H. Shao, F. Jin, L. F. Zheng, A. M. Abd El-Aty and J. Wang, Chromatographia, 2018, 81, 359–364 CrossRef CAS.
- Analytical Detection Limit Guidance & Laboratory Guide for Determining Method Detection Limits, Wisconsin Department of Natural Resources Laboratory Certification Program, 1996, http://dnr.wi.gov/regulations/labcert/documents/guidance/-lodguide.pdf.
- D. N. Huang, X. Y. Wang, C. H. Deng, G. X. Song, H. F. Cheng and X. M. Zhang, J. Chromatogr. A, 2014, 1325, 65–71 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08643e |
| This journal is © The Royal Society of Chemistry 2018 |