
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotential antihyperlipidemic polyketones from endophytic Diaporthe sp. JC-J7 in Dendrobium nobile†
Ming Hu‡
a,
Xue-Qiong Yang‡a,
Chuen-Ping Wanb,
Bang-Yan Wanga,
Hai-Yue Yina,
Li-Jiao Shia,
Ya-Mei Wua,
Ya-Bin Yang *a,
Hao Zhoua and
Zhong-Tao Ding*a
*a,
Hao Zhoua and
Zhong-Tao Ding*a
aFunctional Molecules Analysis and Biotransformation Key Laboratory of Universities in Yunnan Province, School of Chemical Science and Technology, Yunnan University, Kunming, People's Republic of China. E-mail: ybyang@ynu.edu.cn; ztding@ynu.edu.cn
bAffiliated Hospital of Yunnan University of Traditional Chinese Medicine, Kunming 650021, PR China
First published on 14th December 2018
Abstract
Eleven new polyketones named diaporthsins A–K (1–11) were isolated from the fermentation of Diaporthe sp. JC-J7. The chemical structures of compounds (1–11) were elucidated by spectroscopic methods including HRESIMS, 2DNMR, NMR and chemical methods. Compound 11 features an unusual acyclic polyketone–phenolic polyketone hybrid structure that integrates the characteristics of different fungal metabolites (cytosporone and multiplolide). Compound 3 was the only C12-polyketone obtained in this research. These new polyketones showed inhibitory activity on triglycerides (TG) in steatosis hepatocyte L-02 cells. Among them, compound 5 and (4E)-6,7,9-trihydroxydec-4-enoic acid displayed inhibitory activities on TG in steatotic L-02 cells with inhibition ratios of 26% and 21% at concentration of 5 μg mL−1; also, inhibition ratios of 8-O-acetylmultiplolide A and phomopsisporone A at concentration of 5 μg mL−1 were calculated to be about 24% and 16%, respectively, which were equivalent to the antihyperlipidemic activity of lovastatin. The preliminary structure–activity relationship indicated that acetyl at C-8 can increase the antihyperlipidemic activity of multiplolide A and the glycol ester and hydroxyl at C-6 can also increase the corresponding activity of diaporthsin B.
Introduction
Dendrobium nobile Lindl. is a well-known edible and medicinal plant with wide distribution in southwest and south China.1 Endophytes exist in various tissues of healthy plants. Endophytes are also believed to be a rich source of valuable and novel small molecules for drug development.2,3 Diaporthe spp. living in a wide range of hosts are often regarded as plant pathogens.4 They are known to cause diseases in a wide range of plant hosts including root and fruit rot.5 Diaporthe spp. produce a variety of metabolites with different bioactivities6–8 including antioxidant,9 cytotoxic activity,10 and anti-Mycobacterium tuberculosis.11 As a part of our project for searching new bioactive compounds from endophytic fungi of medical plants in the Yunnan province in China, Diaporthe sp. was collected from the stem of Dendrobium nobile Lindl. Eleven polyketones named diaporthsins A–K (1–11) (Fig. 1) were isolated from the fermentation of Diaporthe sp. JC-J7. In this paper, we report the isolation, structure elucidation and bioactivities of these new compounds.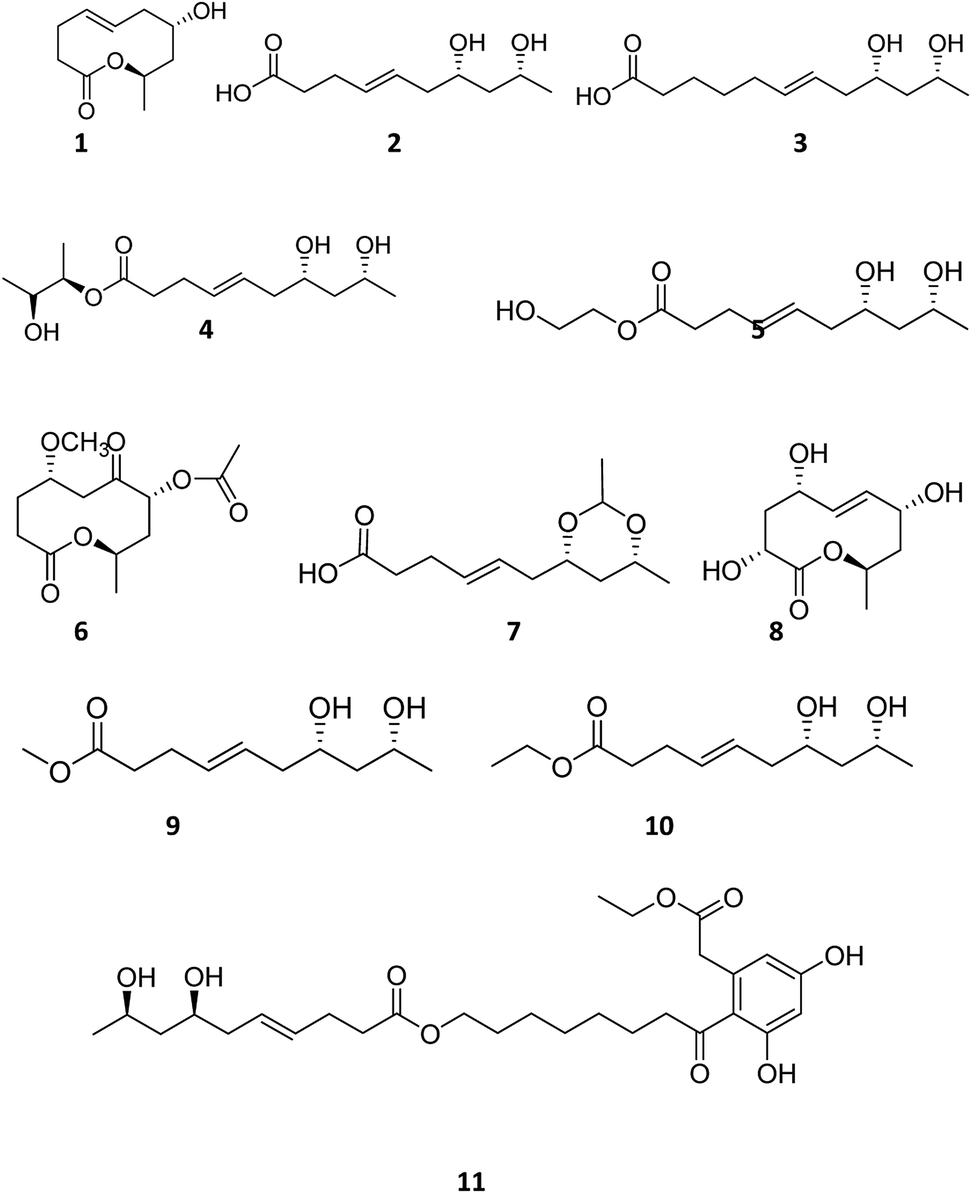 | ||
| Fig. 1 The structures of the new compounds isolated from Diaporthe sp. JC-J7. | ||
Results and discussion
Compound 1 is a new natural product and obtained as a colorless oil; its molecular formula was determined to be C10H16O3 by analysis of its HRESI-MS spectrum and NMR data (Table 1). The NMR spectrum of 1 indicated one methyl group, four methylenes, four methines, and one carbonyl group (Table 1). The presence of a ten-membered ring was determined from the HMBC correlations from H-10 to C-2; H-3 to C-2, C-5; H-4 to C-2, C-5, C-6; H-7 to C-5, C-6, C-9; H-8 to C-9, C-10; H-10 to C-8; H-11 to C-9, C-10 (Fig. 2). In addition, the COSY correlations of H-3/H-4/H-5/H-6/H-7/H-8/H-9/H-10/H-11 also confirmed this structure. The trans orientation at C-5 and C-6 was determined on the basis of the NOESY correlations between H-5 and H-7 as well as H-8. The relative configurations of compound 1 at H-8 and H-10 were determined to be cis by the NOESY correlations between H-8 and H-9 as well as H-9 and H-10. The absolute configuration of C-10 was determined as R by comparing the NMR results with those of the compounds 2–11 and biogenesis. 1 was reacted with (S)-MTPA and (R)-MTPA, yielding (S)- and (R)-MTPA ester derivatives, respectively, at OH-8. The values of Δδ = δS − δR were calculated according to NMR data analyses. For H-7 and H-9, negative and positive Δδ values, respectively, were observed. Therefore, the absolute configuration at C-8 was determined to be S. On the basis of these data, the structure of compound 1 was determined and named diaporthsin A.| Pos. | 1 | 2 | Pos. | 3 | |||
|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | ||
| 1 | 175.0 | 1 | 181.4 | ||||
| 2 | 173.6 | 2.26(m) | 33.8 | 2 | 2.07 (m) | 37.4 | |
| 3 | 2.22 (m), 2.35 (m) | 36.9 | 2.20 (m) | 27.8 | 3 | 1.52 (m) | 25.6 |
| 4 | 2.24 (m), 2.37 (m) | 30.6 | 5.43 (m) | 131.1 | 4 | 1.30 (m) | 29.3 |
| 5 | 5.50 (m) | 132.6 | 5.43 (m) | 127.0 | 5 | 1.95 (m) | 32.1 |
| 6 | 5.40 (m) | 127.8 | 2.06 (m) | 40.6 | 6 | 5.39 (m) | 132.9 |
| 7 | 2.09 (m), 2.26 (m) | 39.0 | 3.63 (m) | 70.0 | 7 | 5.37 (m) | 125.9 |
| 8 | 3.95 (m) | 70.2 | 1.44 (m) | 44.5 | 8 | 2.06 (m) | 40.7 |
| 9 | 1.50 (d, J = 10.6 Hz), 2.18 (m) | 41.0 | 3.83 (q, J = 6.6 Hz) | 66.2 | 9 | 3.62 (m) | 70.2 |
| 10 | 5.01 (m) | 67.2 | 1.07 (d, J = 6.0 Hz) | 22.2 | 10 | 1.48 (m) | 44.6 |
| 11 | 1.24 (d, J = 6.6 Hz) | 20.3 | 11 | 3.84 (m) | 66.3 | ||
| 12 | 1.06 (d, J = 6.5 Hz) | 22.2 | |||||
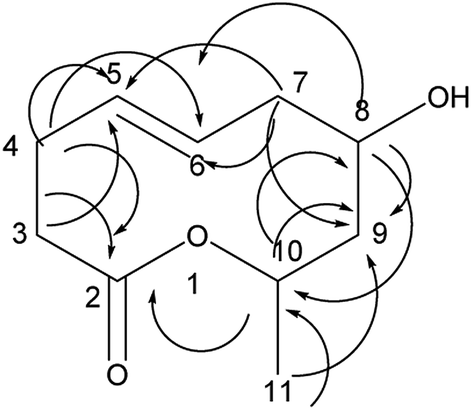 | ||
| Fig. 2 The key HMBC correlations of compound 1. | ||
Compound 2 was obtained as a colorless oil; its molecular formula was determined to be C10H18O4 by the analysis of its HRESI-MS spectrum and NMR data (Table 1). The 13C-NMR spectrum showed one methyl group, four methylenes, four methines, and one carbonyl group (Table 1). On the basis of these data, we observed that the structure of compound 2 exhibited a similar skeleton to that of (4E)-6,7,9-trihydroxydec-4-enoic acid isolated in this research12 except for the absence of OH at C-6 in 2. The structure of compound 2 was confirmed by the HMBC correlations from H-2 to C-4; H-3 to C-4, C-5; H-6 to C-4, C-5, C-7, C-8 (Fig. 3). The two OH groups connected to C-7 and C-9 were determined by the HMBC correlations from H-7 to C-5; H-9 to C-7; H-10 to C-8, C-9 (Fig. 3). The COSY correlations of H-2/H-3/H-4/H-5/H-6/H-7/H-8/H-9/H-10 also confirmed this structure. The trans orientation at C-4 and C-5 was determined by the NOESY correlations of H-4 with H-6 and H-7. The absolute configuration of 2 at C-9 was determined by comparing its NMR data with that of (4E,6R,9R)-6,9-dihydroxydec-4-enoic acid, which has a highly similar structure and is isolated from fungus Cylindrocarpon sp. and by comparing NMR data with that of 8-O-acetylmultiplolide A isolated in this research.12,13 The configuration of C-7 was determined based on its derivative (7), whose configuration was determined by the NOESY correlation between H-7 and H-9 in a cyclic ketal structure. Thus, the structure of compound 2 was determined as diaporthsin B.
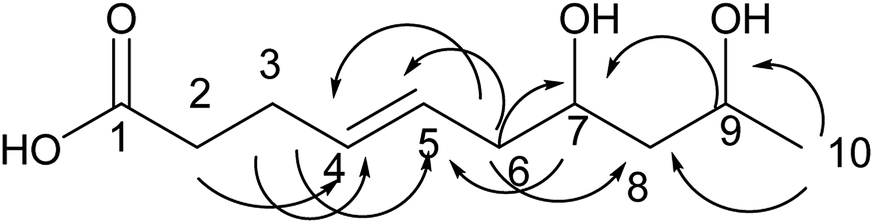 | ||
| Fig. 3 The key HMBC correlations of compound 2. | ||
Compound 3 was obtained as a colorless oil; its molecular formula was determined to be C12H22O4 by the analysis of its HRESI-MS spectrum and NMR data (Table 1). A detailed analysis of its NMR data revealed that its structure was highly similar to that of compound 2 isolated from this strain except that two more CH2 groups were observed in the NMR spectrum of 3 than that in the NMR spectrum of 2. The structure of compound 3 was determined from the HMBC correlations from H-2 to C-3, C-4; H-2, H-3 to C-1; H-5 to C-3, C-4, C-6, C-7; H-8 to C-6, C-7, C-9, C-10; H-11 to C-9; H-12 to C-10, C-11 (Fig. 4). The COSY correlations of H-2/H-3/H-4/H-5/H-6/H-7/H-8/H-9/H-10/H-11/H-12 also confirmed this structure. The configurations of 3 at C-9 and C-11 and the trans orientation at C-6 and C-7 were determined by comparing the NMR data with that of 2. On the basis of these data, the structure of compound 3 was determined as diaporthsin C. Diaporthsin C was the only C12-polyketone obtained in this research.
 | ||
| Fig. 4 The key HMBC correlations of compound 3. | ||
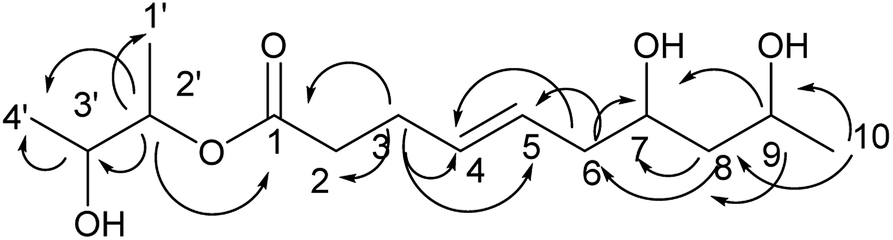
Compound 4 was obtained as a colorless oil; its molecular formula was determined to be C14H26O5 by the analysis of its HRESI-MS spectrum and NMR data (Table 2). Detailed analysis of its NMR data revealed that its structure was highly similar to that of compound 2 isolated from this strain except for one more 2,3-butanediol group in compound 4. The 2,3-butanediol group was elucidated by the COSY correlations of H-1′/H-2′/H-3′/H-4′, and the connection to C-1 was determined by the HMBC correlation from H-2′ to C-1 (Fig. 5). The structure of compound 4 was determined by the HMBC correlations from H-3 to C-1, C-2, C-4, C-5; H-6 to C-4, C-5, C-7; H-8 to C-6, C-7; H-9 to C-7, C-8; H-10 to C-8, C-9 (Fig. 5). The COSY correlations of H-2/H-3/H-4/H-5/H-6/H-7/H-8/H-9/H-10 also confirmed this structure. The configurations of 4 at C-7 and C-9 and the trans orientation at C-4 and C-5 were determined by comparing the NMR data with that of 2 and biogenesis. The configuration of 2,3-butanediol was determined as 2′S, 3′R by the diastereoisomers observed in the NMR data of compound 4. On the basis of these data, the structure of compound 4 was determined as diaporthsin D.
| Pos. | 4 | 5 | 6 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| 1 | 173.2 | 173.6 | 172.0 | |||
| 2 | 2.30 (m) | 33.9 | 2.32 (t, J = 7.5 Hz) | 33.6 | 2.34 (m) | 27.3 |
| 3 | 2.23 (m) | 27.7 | 2.24 (m) | 27.6 | 1.79 (m), 1.98 (m) | 22.5 |
| 4 | 5.42 (m) | 130.8 | 5.44 (m) | 130.8 | 3.80 (m) | 74.8 |
| 5 | 5.41 (m) | 127.3 | 5.41 (m) | 127.3 | 2.34 (m), 2.63 (m) | 39.2 |
| 6 | 2.06 (t, J = 5.5 Hz) | 40.5 | 2.06 (t, J = 5.5 Hz) | 40.5 | 208.1 | |
| 7 | 3.63 (m) | 70.0 | 3.63 (m) | 69.9 | 4.86 (d, J = 6.0 Hz) | 76.0 |
| 8 | 1.44 (m) | 44.5 | 1.45 (m) | 44.6 | 2.06 (m), 2.20 (m) | 39.0 |
| 9 | 3.84 (q, J = 6.0 Hz) | 66.2 | 3.83 (q, J = 6.0 Hz) | 66.1 | 5.10 (m) | 66.7 |
| 10 | 1.06 (d, J = 6.0 Hz) | 22.2 | 1.07 (d, J = 6.5 Hz) | 22.2 | 1.12 (d, J = 5.5 Hz) | 18.7 |
| 1′ | 1.08 (d, J = 6.5 Hz) | 14.7 | 4.03 (t, J = 5.0 Hz) | 65.5 | 170.7 | |
| 2′ | 4.67 (m) | 74.2 | 3.62 (t, J = 5.0 Hz) | 59.6 | 2.20 (s) | 19.3 |
| 3′ | 3.66 (m) | 68.8 | ||||
| 4′ | 1.05 (d, J = 6.0 Hz) | 17.5 | ||||
| OCH3 | 3.19 (s) | 55.2 | ||||
 | ||
| Fig. 5 The key HMBC correlations of compound 4. | ||
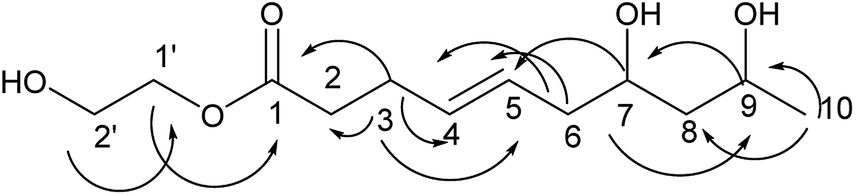
Compound 5 was obtained as a colorless oil and its molecular formula was determined to be C12H22O5 based on the analysis of its HRESI-MS spectrum and NMR data (Table 2). Detailed analysis of its NMR data revealed that its structure was highly similar to that of compound 2 isolated in this study except for one more glycol in compound 5. The two OH groups connected to C-1′ and C-2′ were determined by the HMBC correlation from H-2′ to C-1′ (Fig. 6) and the correlation of H-1′/H-2′ in the COSY spectrum, which suggested the presence of one glycol group. The glycol connected to C-1 was verified by the HMBC correlations from H-1′ to C-1 (Fig. 6). The structure of compound 5 was determined by the HMBC correlations from H-3 to C-1, C-2, C-4, C-5; H-6 to C-4, C-5; H-7 to C-5, C-9; H-9 to C-7; H-10 to C-8, C-9 (Fig. 6). The configurations of 5 at C-7 and C-9 and the trans orientation at C-4 and C-5 were determined by comparing its NMR data with that of 2. On the basis of these data, the structure of compound 5 was determined and named diaporthsin E.
 | ||
| Fig. 6 The key HMBC correlations of compound 5. | ||
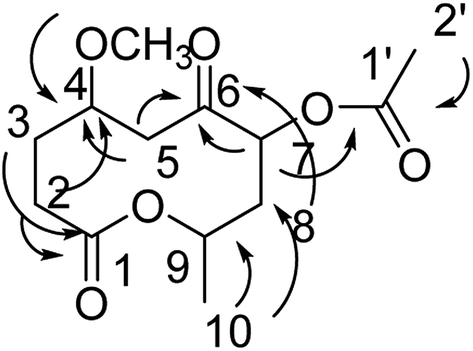
Compound 6 was obtained as a colorless oil; its molecular formula was determined to be C13H20O6 by the analysis of its HRESI-MS spectrum and NMR data (Table 2). The NMR spectra of 6 indicated two methyls, four methylenes, and three methines as well as three carbonyl groups and one methoxyl group (Table 2). The presence of acetyl signal was determined by the HMBC correlations from H-2′ to C-1′; the acetyl connected to C-7 was determined by the HMBC correlations from H-7 to C-1′ (Fig. 7). The methoxyl group connected to C-4 was determined by the HMBC correlations from OCH3 to C-4 (Fig. 7) and the COSY correlations of H-2/H-3/H-4/H-5. Furthermore, according to the HMBC correlations from H-5, H-7, H-8 to C-6; H-2 to C-4; H-2, H-3 to C-1; H-10 to C-8, C-9, these data show the positions of ketone carbonyl and two hydroxyls at C-6, C-7, and C-9 (Fig. 7). The connection of C-1 and C-9 was confirmed by 1H-NMR of H-9 at δ 5.10. The configurations of 6 at C-7 and C-9 were determined by comparing the NMR data with those of compounds 2–5 and biogenesis. The NOESY correlations of H-7/H-9 also confirmed this elucidation. The configuration of C-4 of 6 was determined as S by comparing NMR and optical rotation with those of its demethoxy product 8α-acetoxy-5α-hydroxy-7-oxodecan-10-olide isolated from Phomopsis sp.14 On the basis of these data, the structure of compound 6 was determined and termed diaporthsin F.
 | ||
| Fig. 7 The key HMBC correlations of compound 6. | ||
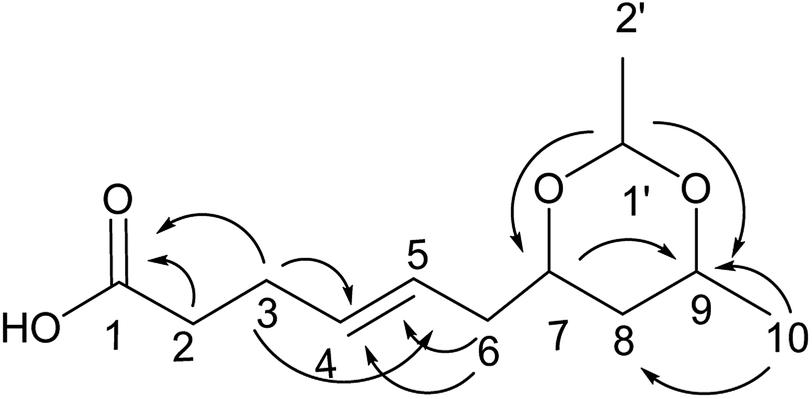
Compound 7 was obtained as a colorless oil; its molecular formula was determined to be C12H20O4 by the analysis of its HRESI-MS spectrum and NMR data (Table 3). A detailed analysis of its NMR data revealed that its structure was highly similar to that of compound 2 except for a cyclic ketal structure in compound 7. The cyclic ketal structure was determined by the COSY correlations of H-1′/H-2′ and the HMBC correlations from H-1′ to C-7 and C-9 (Fig. 8). The structure of compound 7 was confirmed by the HMBC correlations from H-3 to C-1, C-4, C-5; H-6 to C-4, C-5; H-7 to C-9; H-10 to C-8, C-9 (Fig. 8) and the COSY correlations of H-9/H-10; H-2/H-3/H-4/H-5/H-6. The relative configurations of H-7 and H-9 were determined by the NOESY correlation between H-7 and H-9. The absolute configuration of 7 and the trans orientation at C-4 and C-5 were determined by comparing its NMR data with those of compounds 2–6 and the biogenesis. On the basis of these data, the structure of compound 7 was determined as diaporthsin G.
| Pos. | 7 | 8 | Pos. | 9 | |||
|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | ||
| 1 | 173.6 | 172.2 | 1 | 173.6 | |||
| 2 | 2.33 (m) | 33.4 | 4.52 (m) | 68.1 | 2 | 2.31 (m) | 34.0 |
| 3 | 2.27 (m) | 27.8 | 2.22 (m), 2.84 (m) | 45.6 | 3 | 2.26 (m) | 28.0 |
| 4 | 5.53 (m) | 131.1 | 4.52 (m) | 70.3 | 4 | 5.46 (m) | 132.4 |
| 5 | 5.51 (m) | 126.5 | 5.60 (m) | 129.7 | 5 | 5.40 (m) | 127.0 |
| 6 | 2.06 (m), 2.18 (m) | 39.0 | 5.50 (m) | 134.7 | 6 | 2.06 (m), 2.12 (m) | 41.3 |
| 7 | 3.56 (m) | 75.7 | 3.78 (m) | 73.8 | 7 | 3.76 (m) | 72.0 |
| 8 | 1.06 (m), 1.54 (m) | 37.9 | 1.03 (m), 2.43 (m) | 34.9 | 8 | 1.42 (m), 1.49(m) | 44.3 |
| 9 | 3.70 (m) | 71.8 | 4.95 (m) | 70.1 | 9 | 3.96 (m) | 68.8 |
| 10 | 1.13 (d, J = 6.5 Hz) | 21.0 | 1.20 (d, J = 6.8 Hz) | 17.5 | 10 | 1.13 (d, J = 6.2 Hz) | 24.0 |
| 1′ | 4.66 (q, J = 5.0 Hz) | 98.0 | OCH3 | 3.60 (s) | 51.6 | ||
| 2′ | 1.20 (d, J = 5.0 Hz) | 20.6 | |||||
 | ||
| Fig. 8 The key HMBC correlations of compound 7. | ||
Compound 8 was obtained as white crystals; its molecular formula was determined to be C10H16O5 by the analysis of its HRESI-MS spectrum and NMR data (Table 3). The NMR spectra of 8 indicated one methyl group, four hydroxylated methines, two methylenes, two methines and one carbonyl group (Table 3). According to the HMBC correlation from H-2, H-3 to C-1; H-3 to C-4; H-4 to C-5, C-6; H-8 to C-7; H-10 to C-8, C-9 (Fig. 9) and COSY correlations of H-2/H-3/H-4/H-5/H-6/H-7/H-8/H-9/H-10, these data suggest the four OH groups connected to C-2, C-4, C-7, and C-9. The connection between C-1 and C-9 was confirmed by 1H-NMR of H-9 at δ 4.95. The absolute configurations of C-7 and C-9 were determined by comparing the NMR data with those of compounds 2–7 and biogenesis. The trans orientation at C-5 and C-6 was determined by NOESY correlation between H-5 and H-7. The configurations of C-2, C-4, C-7 and C-9 were determined by comparing the NMR data with those of compounds 2–7 and the biogenesis; the NOESY correlations of H-2 and H-4; H-7 and H-10; H-9 and H-10 also confirmed this elucidation. Thus, the structure of compound 8 was determined as diaporthsin H.
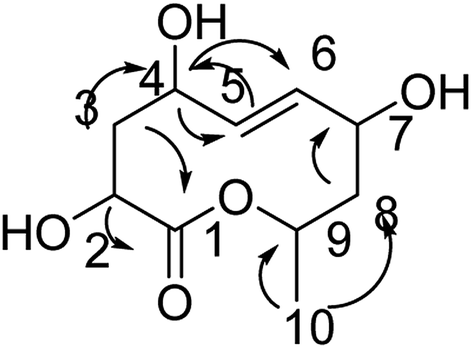 | ||
| Fig. 9 The key HMBC correlations of compound 8. | ||
Compound 9 was obtained as a colorless oil. The molecular formula of 9 was established as C11H20O4 by the HRESI-MS and NMR data (Table 3). The 1H NMR and 13C NMR data showed that 9 exhibited a similar skeleton to that of compound 2 except for one more methoxyl in compound 9. The structure of compound 9 was determined by the HMBC correlations from H-2, H-3 to C-1, H-4 to C-2, C-3; H-5 to C-3, C-4; H-9 to C-7, H-10 to C-8, C-9; OCH3 to C-1 (Fig. 10). The absolute configurations of C-7 and C-9 and the trans orientation at C-4 and C-5 were determined by comparing the NMR data with those of compounds 2–7 and biogenesis. Thus, the structure of compound 9 was determined as diaporthsin I.
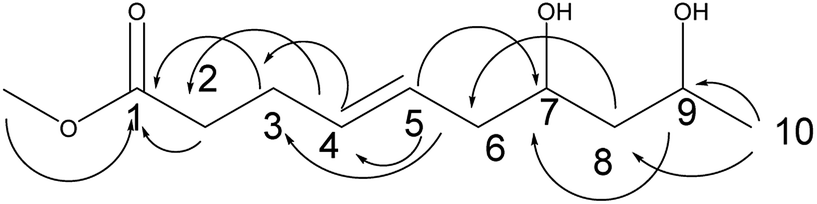 | ||
| Fig. 10 The key HMBC correlations of compound 9. | ||
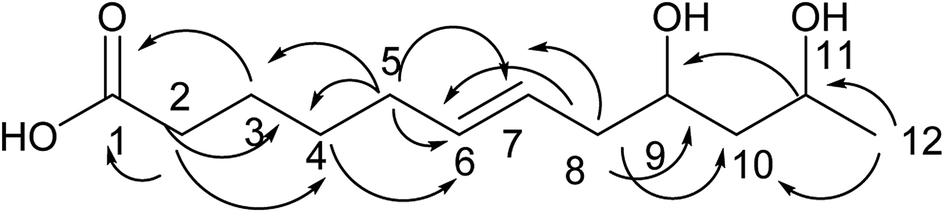
Compound 10 was obtained as a colorless oil. The molecular formula of 10 was established as C12H22O4 by the HRESI-MS and NMR data (Table 3). A detailed analysis of its NMR data revealed that its structure was highly similar to that of compound 2 isolated in this study except for one more alcohol in compound 10. The oxethyl group was elucidated by the COSY correlations of H-11/H-12 and its connection to C-1 was determined by the HMBC correlations from H-11 to C-1 (Fig. 11). The structure of compound 10 was determined by the HMBC correlations from H-3 to C-1, C-4, C-5; H-4 to C-3; H-10 to C-8, C-9 (Fig. 11) and the COSY correlations of H-2/H-3/H-4/H-5/H-6/H-7/H-8/H-9/H-10. Moreover, the absolute configurations of C-7 and C-9 and the trans orientation at C-4 and C-5 were determined by comparing the NMR data with those of new compounds. Thus, the structure of compound 10 was determined as diaporthsin J.
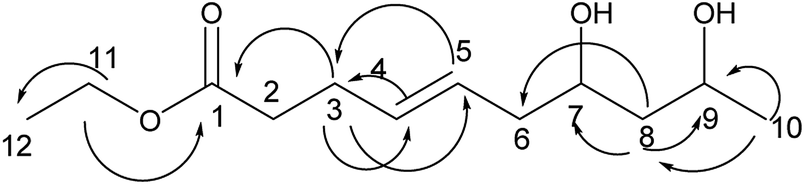 | ||
| Fig. 11 The key HMBC correlations of compound 10. | ||
Compound 11 was obtained as an orange-red oil; its molecular formula was determined to be C28H42O9 by the analysis of its HRESI-MS spectrum and NMR data (Table 4). In 1H NMR spectrum, two aromatic protons at δ 6.22 (1H, d, J = 2.0 Hz) and 6.28 (1H, d, J = 2.0 Hz) were observed; the 13C NMR spectrum showed six carbon signals at δ 101.4, 110.3, 120.0, 135.6, 158.4, and 159.9, suggesting the existence of four substituted benzene species in compound 11 (Table 4). The structure of the cytosporone fraction of this compound was confirmed by the HMBC correlations from H-27 to C-1, C-28; H-4 to C-2, C-5, and C-6; H-6 to C-5, C-7, C-8; H-2 to C-8; H-10, H-11 to C-9; H-10 to C-11, C-12; H-16 to C-14, C-15, C-17 (Fig. 12). The structure of the multiplolide fraction of this compound was confirmed by the HMBC correlations from H-18, H-19 to C-17; H-19 to C-20, C-21; H-23 to C-21, C-25; H-26 to C-24, C-25; H-25 to C-23. According to these data, the structure of compound 11 was obtained by esterification reaction of compound 2 and dothiorelone C.15 Thus, the structure of compound 11 was determined as diaporthsin K.
| Pos. | 10 | 11 | Pos. | 11 | |||
|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | ||
| 1 | 172.2 | 172.1 | 16 | 4.07 (t, J = 6.5 Hz) | 64.2 | ||
| 2 | 2.33 (m) | 33.8 | 3.59 (s) | 39.1 | 17 | 173.7 | |
| 3 | 2.28 (m) | 27.8 | 135.6 | 18 | 2.41 (m), 2.36 (m) | 33.7 | |
| 4 | 5.50 (m) | 130.7 | 6.22 (d, J = 2.0 Hz) | 110.3 | 19 | 2.32 (m) | 27.7 |
| 5 | 5.52 (m) | 127.7 | 159.9 | 20 | 5.54 (m) | 130.8 | |
| 6 | 2.13 (m) | 41.2 | 6.28 (d, J = 2.0 Hz) | 101.4 | 21 | 5.51 (m) | 127.3 |
| 7 | 3.78 (m) | 71.2 | 158.4 | 22 | 2.17 (m) | 40.5 | |
| 8 | 1.42 (m), 1.54 (m) | 44.6 | 120.0 | 23 | 3.65 (m) | 70.0 | |
| 9 | 3.95 (m) | 67.3 | 207.6 | 24 | 1.51 (m) | 44.5 | |
| 10 | 1.12 (d, J = 6.2 Hz) | 23.5 | 2.92 (t, J = 7.0 Hz) | 43.7 | 25 | 3.90 (m) | 66.2 |
| 11 | 4.07 (q, J = 6.1 Hz) | 59.6 | 1.64 (m) | 24.0 | 26 | 1.15 (d, J = 6.2 Hz) | 22.3 |
| 12 | 1.20 (t, J = 6.0 Hz) | 13.7 | 1.37 (s) | 28.2 | 27 | 4.13 (q, J = 7.0 Hz) | 60.5 |
| 13 | 1.37 (s) | 28.2 | 28 | 1.29 (t, J = 7.0 Hz) | 13.1 | ||
| 14 | 1.37 (s) | 25.5 | |||||
| 15 | 1.64 (m) | 28.9 | |||||
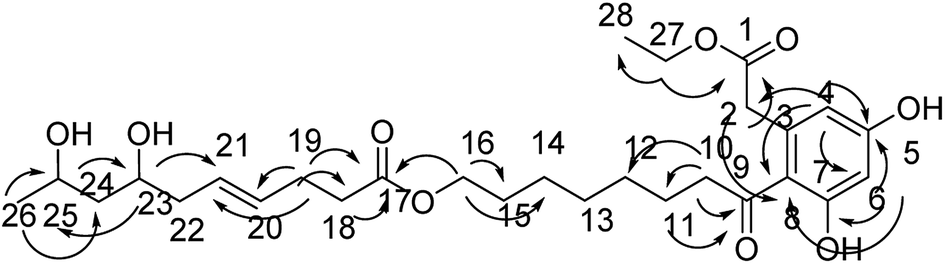 | ||
| Fig. 12 The key HMBC correlations of compound 11. | ||
Other known compounds were isolated and determined as (4E)-6,7,9-trihydroxydec-4-enoic acid,12 multiplolide A,12 8-O-acetylmultiplolide A,12 and phomopsisporone A.16 Most of the new polyketones were esterified products of 2. We tested the possibility of artificial reaction of compound 1 in solvents of methanol and ethanol at 76 °C for 10 hours, but the reaction was unsuccessful. Thus, we infer that esterification should occur in fungal biotransformation.
Cardiovascular disease (CVD), which is currently the leading cause of death in developed countries, is expected to become a predominant health problem worldwide. Atherosclerosis, a progressively evolving disease characterized by the accumulation of lipids and fibrous elements in large arteries, constitutes the single most important contributor to this growing burden of CVD.17 Currently, treatment of atherosclerosis is aimed at reducing blood cholesterol and triglyceride contents.18 Decarestrictines, which are novel 10-membered lactones, are produced by different strains of Penicillium. Some of them have inhibitory effects on the cholesterol biosynthesis.19 Our isolated polyketones belonged to the skeleton of decarestrictine; thus, the antihyperlipidemic activities of these compounds 1, 2, 3, 5, 8, 10, (4E)-6,7,9-trihydroxydec-4-enoic acid (THEA), 8-O-acetylmultiplolide A, multiplolide A, and phomopsisporone A were investigated. Compound 5, (4E)-6,7,9-trihydroxydec-4-enoic acid, 8-O-acetylmultiplolide A, and phomopsisporone A displayed lipid lowering effects on triglycerides (TG) in steatotic L-02 cells with inhibition ratios of 26%, 21%, 24%, and 16%, respectively, at a concentration of 5 μg mL−1. Lovastatin was used as a positive control with inhibition ratio of 31.80% at concentration of 5 μg mL−1 8-O-acetylmultiplolide A exhibited better lipid lowering effect than multiplolide A, whereas compound 5 and (4E)-6,7,9-trihydroxydec-4-enoic acid exhibited more activities than compound 2 in antihyperlipidemic assay (Table 5). The structural difference between 8-O-acetylmultiplolide A and multiplolide A was one more acetyl group at C-8 in 8-O-acetylmultiplolide A than that in multiplolide A; the other differences observed in this study include one more glycol ester in 5 than that in 2 and one more hydroxyl at C-6 in (4E)-6,7,9-trihydroxydec-4-enoic acid than that in 2. Compound 2 and multiplolide A showed no clear antihyperlipidemic activity with inhibition ratios of <5% at the concentration of 5 μg mL−1 Hence, the acetyl at C-8 can increase the antihyperlipidemic activity of multiplolide A, and the glycol ester and hydroxyl at C-6 can also increase the corresponding activity of diaporthsin B. These compounds were evaluated for their cytotoxicity, but no activity was observed with IC50 > 100 μM.
| Sample | TG concentration (mmol g−1) | Inhibition ratio |
|---|---|---|
| 5 | 0.307 | 26% |
| THEA | 0.326 | 21% |
| 8-O-Acetylmultiplolide A | 0.312 | 24% |
| Phomopsisporone A | 0.332 | 16% |
| Lovastatin | 0.283 | 31% |
| Other compounds | <10% | |
| Control | 0.415 | |
| Normal | 0.015 |
Conclusions
Eleven new polyketones named diaporthsins A–K (1–11) were isolated from the fermentation of Diaporthe sp. JC-J7. Compound 11 featured an unusual acyclic polyketone–phenolic polyketone hybrid structure that integrates the characteristics of different fungal metabolites (cytosporone and multiplolide). Compound 3 was the only C12-polyketone obtained in this study. These polyketones showed significant inhibitory activity on triglycerides (TG) in steatotic L-02 cells, which exhibited potential antihyperlipidemic activities in corresponding drug research.Experimental
General experimental procedures
Silica gel (200–300 mesh, Qingdao Marine Chemical Group Co., Qingdao, China), LiChroprep RP-18 (40–63 mm; Merck, Darmstadt, Germany) and Sephadex LH-20 (GE Healthcare Co., Buckinghamshire, UK) were used for column chromatography. 1D and 2D NMR spectra were obtained on a Bruker AVANCE 400, 500, 600 MHz NMR instrument (Bruker, Karlsruhe, Germany). MS spectra were recorded with Agilent G3250AA (Agilent, Santa Clara, USA) and AutoSpec Premier P776 spectrometers (Waters, Milford, USA). ORs were obtained on a Jasco P-1020 polarimeter.Fungal material and fermentation
The endophytic fungus JC-J7 was isolated using potato dextrose agar medium (PDA) (peeled and cut potato 200 g L−1, glucose 20 g L−1, agar 15 g L−1) from stems of Dendrobium nobile isolated from Honghe of Yunnan province and identified as Diaporthe sp. JC-J7 by ITS gene sequencing. The fungus has been preserved at the School of Chemical Science and Technology, Yunnan University, China. The pure strain was stored in 50% glycerol at −80 °C. Diaporthe sp. JC-J7 was maintained in PDA medium. Small agar plugs (approximately 5 mm × 5 mm) of the fungus were cultured in 0.5 L Erlenmeyer flasks containing 100 mL of potato dextrose broth (PDB, potato infusion of 200 g fresh potato, dextrose 15 g, distilled water 1.0 L, pH 7.0) at 130 rpm and 28 °C for 3 days. Each 20–25 mL of seed culture was transferred into a 1.0 L Erlenmeyer flask containing 250 mL of PDB and incubated at 130 rpm and 28 °C for 7 days.Extraction and isolation of new compounds
The production culture (50 L) was centrifuged to separate the mycelia from the supernatant. The extracts of the fermentation broth and the mycelia were combined after TLC analysis to yield a crude extract (30 g). The residue was first subjected to column chromatography (CC) (silica gel, CHCl3/MeOH 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 50:1, 30:1, 10:1 and 3:1 (v/v)) to afford fractions 1–5, respectively.
:0, 50:1, 30:1, 10:1 and 3:1 (v/v)) to afford fractions 1–5, respectively.
Fr. 1 was fractionated by silica gel chromatography eluted with petroleum ether/EtOAc (80:1, 40:1, 20:1, 10:1, 3:1, 1:1) to give six subfractions (Fr. 1.1 – Fr. 1.6). Then, Fr. 1.3 was eluted using Sephadex LH-20 (methanol) to obtain three subfractions (Fr. 1.3.1–Fr. 1.3.3). Fr. 1.3.1 was eluted through Sephadex LH-20 (methanol) to afford compound 1 (27 mg).
Fr. 3 was fractionated by silica gel CC eluted with CHCl3/MeOH (50:1, 20:1, 5:1) to give five subfractions (Fr. 3.1–Fr. 3.5). Fr. 3.3 was further subjected to CC (silica gel, petroleum ether/acetone 3:1) and Sephadex LH-20 (methanol) to afford compound 2 (2 mg) and compound 3 (4 mg). Fr. 3.2 was further separated by CC eluted with petroleum ether/EtOAc (8:1) to give compound 6 (3 mg). Fr. 3.1 was fractionated by column chromatography on Lichroprep RP-18 eluted with MeOH–H2O (20–100%) into four subfractions (Fr. 3.1.1 – Fr. 3.1.4). Fr. 3.1.1 was eluted using Sephadex LH-20 (methanol) to afford compound 4 (5 mg) and compound 5 (7 mg). Fr. 3.1.2 was further purified using CC (silica gel, petroleum ether/EtOAc 6:1) and SephadexLH-20 (methanol) to yield compounds 7 (3 mg), 9 (6.4 mg) and 10 (4 mg). Fr. 4 was further subjected to CC (silica gel, CHCl3/MeOH 20:1, 10:1, 5:1) to afford Fr. 4.1–Fr. 4.4. Compounds 8 (12 mg) and 11 (10 mg) were obtained from Fr. 4.2 by Sephadex LH-20 (methanol).
Diaporthsin A (1): colorless oil; [α]23D 8.95 (c 0.27, CHCl3); IR (KBr) νmax 3438, 2955, 2852, 1636, 1460 cm−1. 1H NMR (600 MHz) and 13C NMR (150 MHz) in CDCl3, see Table 1. HR-ESIMS m/z 185.1171 [M + H]+ (calcd for C10H17O3 185.1178).
Diaporthsin B (2): colorless oil; [α]23D 30.15 (c 0.1, MeOH); IR (KBr) νmax 3428, 2925, 1637, 1438 cm−1. 1H NMR (600 MHz) and 13C NMR (150 MHz) in MeOD, see Table 1. HR-ESIMS m/z 225.1098 [M + Na]+ (calcd for C10H18O4 Na 225.1103).
Diaporthsin C (3): colorless oil; [α]23D 341.93 (c 0.01, MeOH); IR (KBr) νmax 3431, 2963, 1624, 1414 cm−1. 1H NMR (600 MHz) and 13C NMR (150 MHz) in MeOD, see Table 1. HR-ESIMS m/z 253.1410 [M + Na]+ (calcd for C12H22O4Na 253.1416).
Diaporthsin D (4): colorless oil; [α]23D 79.95 (c 0.044, MeOH); IR (KBr) νmax 3393, 2969, 1724, 1437 cm−1. 1H NMR (500 MHz) and 13C NMR (125 MHz) in MeOD, see Table 2. HR-ESIMS m/z 297.1672 [M + Na]+ (calcd for C14H26O5Na 297.1678).
Diaporthsin E (5): colorless oil; [α]23D 305.63 (c 0.011, MeOH); IR (KBr) νmax 3423, 2923, 1724, 1636, 1384 cm−1. 1H NMR (500 MHz) and 13C NMR (125 MHz) in MeOD, see Table 2. HR-ESIMS m/z 247.1542 [M + H]+ (calcd for C12H23O5 247.1545).
Diaporthsin F (6): colorless oil; [α]23D 242.10 (c 0.01, MeOH); IR (KBr) νmax 3431, 2924, 1734, 1458 cm−1. 1H NMR (500 MHz) and 13C NMR (125 MHz) in MeOD, see Table 2. HR-ESIMS m/z 295.1161 [M + Na]+ (calcd for C13H20O6 Na 295.1158).
Diaporthsin G (7): colorless oil; [α]23D 234.69 (c 0.014, acetone); IR (KBr) νmax 3443, 2925, 1630, 1384 cm−1. 1H NMR (500 MHz) and 13C NMR (125 MHz) in acetone-d6, see Table 3. HR-ESIMS m/z 227.1289 [M − H]− (calcd for C12H19O4 227.1283).
Diaporthsin H (8): white crystal; [α]23D 73.14 (c 0.04, MeOH); IR (KBr) νmax 3420, 2926, 1721, 1362 cm−1. 1H NMR (600 MHz) and 13C NMR (150 MHz) in MeOD, see Table 3. HR-ESIMS m/z 239.0938 [M + Na]+ (calcd for C10H16O5Na 239.0895).
Diaporthsin I (9): colorless oil; [α]23D 61.11 (c 0.064, CDCl3); IR (KBr) νmax 3416, 2924, 1736, 1377 cm−1. 1H NMR (600 MHz) and 13C NMR (150 MHz) in CDCl3, see Table 3. HR-ESIMS m/z 239.1257 [M + Na]+ (calcd for C11H20O4Na 239.1259).
Diaporthsin J (10): colorless oil; [α]23D 332.90 (c 0.01, acetone); IR (KBr) νmax 3447, 2963, 1631, 1384 cm−1. 1H NMR (600 MHz) and 13C NMR (150 MHz) in acetone-d6, see Table 4. HR-ESIMS m/z 253.1409 [M + Na]+ (calcd for C12H22O4Na 253.1416).
Diaporthsin K (11): orange-red oil; [α]23D 63.20 (c 0.095, MeOH); IR (KBr) νmax 3426, 2930, 1730, 1618, 1384 cm−1. UV (MeOH) λmax (logε): 270 (3.45) nm. 1H NMR (500 MHz) and 13C NMR (125 MHz) in MeOD, see Table 4. HR-ESIMS m/z 545.2720 [M + Na]+ (calcd for C28H42O9Na 545.2727).
Antihyperlipidemic activity assay
Steatosis hepatocyte L-02 cells (3 × 105 cells per well) were seeded into 6-well microtiter plates (Corning) and divided into control, model, and treatment groups. The steatosis hepatocyte L-02 model was established by exposure to fat emulsion (5%) for 48 h. The resulting hepatocytes were treated with control or various concentrations of compounds for 24 h; after culture, the cells were washed with PBS for two times and whole cell lysates were prepared by incubating in a cell lysis buffer (1% Triton X-100) for 30 min to extract total protein. Whole cell extracts were obtained by centrifugation for 10 min. Protein concentrations were determined using the Bio-Rad microprotein assay with bovine serum albumin as the standard. The level of TG was detected by TG determination kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).Cytotoxicity assay
The cytotoxicities of these compounds against A-549 cells were determined in vitro by the MTT method. Cisplatin was used as a positive control with IC50 at 12.01 μM.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research work was financially supported by the National Natural Science Foundation of China (81560571, 81660582) and a project of Yunling Scholars of Yunnan Province.References
- J. H. Wang, Y. D. Zhang and J. P. Luo, Biotechnol. Biotechnol. Equip., 2018, 32, 744–750 CrossRef.
- A. Schueffler and T. Anke, Nat. Prod. Rep., 2014, 31, 1425–1448 RSC.
- A. Evidente, A. Kornienko, A. Cimmino, A. Andolfi, F. Lefranc, V. Mathieu and R. Kiss, Nat. Prod. Rep., 2014, 31, 617–627 RSC.
- R. R. Gomes, C. Glienke, S. I. Videira, L. Lombard, J. Z. Groenewald and P. W. Crous, Persoonia, 2013, 31, 1–41 CrossRef CAS PubMed.
- L. Sessa, E. Abreo, L. Bettucci and S. Lupo, Phytopathol. Mediterr., 2017, 56, 431–444 Search PubMed.
- Z. Liu, J. Y. Zhao, X. Liang, X. X. Lv, Y. Li, J. Qu and Y. B. Liu, Fitoterapia, 2018, 127, 7–14 CrossRef CAS PubMed.
- H. Cui, Y. N. Liu, J. Li, X. S. Huang, T. Yan, W. H. Cao, H. J. Liu, Y. H. Long and Z. G. She, J. Org. Chem., 2018, 83, 11804–11813 CrossRef CAS PubMed.
- K. I. Nakashima, J. Tomida, T. Kamiya, T. Hirai, Y. Morita, H. Hara, Y. Kawamura, T. Adachi and M. Inoue, Tetrahedron Lett., 2018, 59, 1212–1215 CrossRef CAS.
- C. Tanapichatsakul, S. Monggoot, E. Gentekaki and P. Pripdeevech, Curr. Microbiol., 2018, 75, 476–483 CrossRef CAS PubMed.
- C. J. Chen, X. X. Liu, W. J. Zhang, L. Y. Zang, G. Wang, S. W. Ng, R. X. Tan and H. M. Ge, RSC Adv., 2015, 5, 17559–17565 RSC.
- S. Dettrakul, P. Kittakoop, M. Isaka, S. Nopichai, C. Suyarnsestakorn, M. Tanticharoenb and Y. Thebtaranonth, Bioorg. Med. Chem. Lett., 2003, 13, 1253–1255 CrossRef CAS.
- Q. F. Tan, X. F. Yan, X. Lin, Y. J. Huang, Z. H. Zheng, S. Y. Song, C. H. Lu and Y. M. Shen, Helv. Chim. Acta, 2007, 90, 1811–1817 CrossRef CAS.
- S. Yoshida, T. Suzuki, H. Furuno, T. Aboshi, T. Murayama, T. Koseki and Y. Shiono, Nat. Prod. Res., 2018, 32, 60–64 CrossRef CAS.
- S. H. Wu, Y. W. Chen, S. C. Shao, L. D. Wang, Z. Y. Li, L. Y. Yang, S. L. Li and R. Huang, J. Nat. Prod., 2008, 71, 731–734 CrossRef CAS.
- Q. W. Tan, P. H. Fang, J. C. Ni, F. L. Gao and Q. J. Chen, Molecules, 2017, 22, 2073 CrossRef PubMed.
- T. Kongprapan, X. Xu, V. Rukachaisirikul, S. Phongpaichit, J. Sakayaroj, J. Chen and X. Shen, Phytochem. Lett., 2017, 22, 219–223 CrossRef CAS.
- A. N. Matralis and A. P. Kourounakis, J. Med. Chem., 2014, 57, 2568–2581 CrossRef CAS.
- A. N. Matralis, M. G. Katselou, A. Nikitakis and A. P. Kourounakis, J. Med. Chem., 2011, 54, 5583–5591 CrossRef CAS PubMed.
- S. Grabley, E. Granzer, K. Hutter, D. Ludwig, M. Mayer, R. Thiericke, G. Till, J. Wink, S. Philipps and A. Zeeck, J. Antibiot., 1992, 45, 56–65 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR, HSQC, HMBC, 1H–1H COSY, ROESY, IR, UV and HR-ESIMS spectra of compounds 1–11. See DOI: 10.1039/c8ra08822e |
| ‡ M. Hu and X. Q. Yang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |