
Lipase immobilised on silica monoliths as continuous-flow microreactors for triglyceride transesterification†
Mohammed
Alotaibi
ab,
Jinesh C.
Manayil
 b,
Gillian M.
Greenway
a,
Stephen J.
Haswell
a,
Stephen M.
Kelly
a,
Adam F.
Lee
*d,
Karen
Wilson
d and
Georgios
Kyriakou
*abc
b,
Gillian M.
Greenway
a,
Stephen J.
Haswell
a,
Stephen M.
Kelly
a,
Adam F.
Lee
*d,
Karen
Wilson
d and
Georgios
Kyriakou
*abc
aDepartment of Chemistry, The University of Hull, Cottingham Rd., Hull HU6 7RX, UK
bEuropean Bioenergy Research Institute, Aston University, Aston Triangle, Birmingham B4 7ET, UK. E-mail: g.kyriakou@aston.ac.uk
cChemical Engineering and Applied Chemistry, Aston University, Aston Triangle, Birmingham B4 7ET, UK
dSchool of Science, RMIT University, Melbourne, VIC 3000, Australia. E-mail: adam.lee2@rmit.edu.au
First published on 18th December 2017
Abstract
Lipase immobilised on silica monoliths has been prepared and applied as biocatalytic continuous-flow microreactors for the transesterification of tributyrin as a model bio-oil component. Candida antarctica lipase was trapped within the pores of silica monoliths, and its successful immobilisation was demonstrated by the hydrolysis of 4-nitrophenyl butyrate to 4-nitrophenol. Lipase immobilised on silica monoliths was active for the transesterification of tributyrin at ambient temperature, with reactivity as a function of the methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :tributyrin ratio, flow rate, temperature, and textural properties. Monoliths with a high surface area and large meso- and macropore channels enhanced the transesterification activity through improved molecule diffusion. The optimum immobilised lipase microreactor exhibited almost quantitative ester production for >100 h at 30 °C without deactivation.
:tributyrin ratio, flow rate, temperature, and textural properties. Monoliths with a high surface area and large meso- and macropore channels enhanced the transesterification activity through improved molecule diffusion. The optimum immobilised lipase microreactor exhibited almost quantitative ester production for >100 h at 30 °C without deactivation.
1. Introduction
Global energy consumption is predicted to increase by 61% by 2050 compared to the energy consumption in 2010 due to population growth and urbanisation.1–3 Fossil fuels currently provide 80% of the worldwide energy demand, but also account for the overwhelming majority of anthropogenic greenhouse gas (GHG) emissions.4 Despite significant recent growth in known fossil fuel reserves, concern over climate change arising from GHG emissions is driving the urgent quest for alternative energy sources.5,6 Renewable energy sources, geothermal, hydroelectric, ocean, solar, wind and bioenergy, are essential to deliver sustainable socio-economic development,7 with biodiesel as the only immediate low-cost solution for a drop-in liquid transportation fuel.3,7,8 Second-generation biodiesel comprises fatty acid methyl esters (FAME) obtained via the esterification and transesterification of free fatty acids (FFAs) and triacylglycerides (TAGs), respectively, from non-edible plant, algae or waste oil sources.9–13Biodiesel is typically produced via TAG transesterification with short-chain alcohols, notably methanol,14 catalysed by enzymes (lipases),15,16 ionic liquids,17,18 and acid19,20 or base21–24 catalysts, with glycerol as the by-product.11,20,25 Although significant research has focused on the development of solid acid (e.g. sulphonic acids, heteropolyacids and sulphated metal oxides) and base (e.g. metal oxides and layered double hydroxides) catalysts for biodiesel production, immobilised biocatalysts such as lipase have received far less attention.26,27 Lipases are carboxylesterase enzymes that hydrolyse fats28,29 and hence are amenable to the transesterification of TAGs to produce biodiesel.14,15,27Candida antarctica is a commonly used lipase catalyst for the production of biodiesel,30,31 but is prone to deactivation when the transesterification of plant oils is conducted with short-chain alcohols due to poor miscibility and substrate inhibition.32–34 This has necessitated lipase-catalysed transesterification employing low methanol:oil molar ratios.8,33 Other disadvantages of enzymes (lipases) include their comparatively high costs versus inorganic catalysts and low reaction rates. Enzyme immobilisation on a solid support facilitates continuous operation and catalyst recovery and re-use.8,30,35 Lipase can be immobilised by employing different approaches including covalent bonding, entrapping, physisorption and cross-linking.35 Commercial approaches predominantly focus on lipase adsorption onto hydrophobic polymers such as alkyl-agarose, polypropylene, and polystyrene.36,37
Silica monoliths are widely employed in separation science and as ‘inert’ supports in heterogeneous catalysis.38–40 Monoliths are rigid materials with interconnected pores which allow fluid transport.40 These pores may be of uniform size and of micropore to macropore diameters, or feature multiple, interconnected pore networks to offer the optimal combination of high surface area and rapid mass transport.30,38,39,41 Monolithic microreactors have shown promise in flow chemistry as a practical, economical and environmentally friendly means to formulate catalysts, offering improved control over the molecule flow rate, contact time and temperature, in addition to in situ product separation facilitating catalyst re-use.8,42,43 Porous silica monoliths are typically prepared through sol–gel syntheses in conjunction with hard/soft templating using surfactants,44 ionic liquids,45 or even ice.46 The cost and toxicity of such templates are important considerations for large-scale applications, wherein e.g. pluronic surfactants and ionic liquids remain problematic. Several monolithic microreactors have been reported for transesterification/biodiesel production utilising immobilised lipase.30,47,48 These enzymatic microreactors displayed superior performance to the free enzyme. However, it should be noted that the only monolithic study employing Candida antarctica lipase demonstrated the pre-functionalization of the silica surface with a primary amine to covalently bind the glutaraldehyde cross-linker to the enzyme.30 This approach is fundamentally flawed as it is impossible to determine whether the observed transesterification activity arose from biocatalysis by the lipase or conventional base catalysis by the organic amine since the latter is an excellent catalyst itself for biodiesel production49,50 (and essential control experiments were omitted). The commercial silica capillary employed also resulted in only a modest lipase loading of 6.34 mg.
Here, we present the first unequivocal demonstration of transesterification catalysed by silica-immobilised Candida antarctica lipase, using an amine-free silica monolith as a microreactor for the transesterification of tributyrin as a model TAG. The efficacy of lipase as a stable biocatalyst, and the role of monolith textural properties, is demonstrated.
2. Experimental
2.1 Materials and instrumentation
Candida antarctica lipase B was purchased from Sigma-Aldrich (UK) and used without further purification. Decane (99%), 4-nitrophenol (4-NP, 98%) and 4-nitrophenyl butyrate (4-NPB, 98%) were also obtained from Sigma-Aldrich. Polyethylene oxide (PEO, MW = 100000 and MW = 200000), HCl (37%), tetramethoxysilane (TMOS, 99%) and tetraethyl orthosilicate (TEOS, 99%) were purchased from Fluka. The glass capillary (0.6/0.05 mm ID and 25 mm length) was obtained from Brand GMBH. The instruments used in fabrication and detection procedures include a Baby Bee syringe pump from Bioanalytical Systems, Inc. (West Lafayette, USA), model 7971 column heater (Jones Chromatography Ltd), Chemyx Fusion 100 syringe pump from KR Analytical Ltd (Sandbach, Cheshire) and BIO Wide Pore C18 column (5 μm × 15 cm × 2.1 mm) from Phenomenex (Queens Avenue, Hurdsfield Ind. Est., United Kingdom).
2.2 Catalyst synthesis
000) was dissolved in 4 ml of 0.02 M acetic acid solution and stirred for 1 h (200 rpm) in an ice bath for complete dissolution. To this solution, 2 ml of TMOS was added and the mixture was kept under stirring for an hour. The solution was subsequently poured into a plastic mould (length = 6 cm, internal diameter = 0.45 cm) closed at both ends using PTFE thread seal tape and aged at 40 °C for 3 days. The monolithic gel was subsequently removed from the mould and thoroughly washed with deionised water. The monolithic gel was then immersed in an incubator containing 1 M aqueous NH4OH, and heated to 82 °C for 24 h to etch the silica surface and generate mesoporosity. The etched monolith was washed with deionised water until the washing reached pH 7, and dried at 40 °C for 1 day. Next, the dried monolith was calcined at 600 °C for 3 h under flowing air to remove the PEO surfactant, cooled and then cut to 4 cm length. The resulting silica monolith was placed between two pieces of borosilicate tube, and PTFE heat shrinkable tubing (Smith Scientific) was used to encapsulate all three components. Upon heating to 350 °C for 1 h, the PTFE contracted to make a leak-tight seal between the ends of the borosilicate glass capping tubes and the monolith, as shown in Fig. S1.† The resulting monolith was denoted M1. A similar protocol was adopted to prepare two additional silica monoliths using a heavier (0.305 g, MW = 200000) PEO in conjunction with either TMOS (denoted M2) or TEOS (denoted M3). In the case of the M3 monolith, 2.537 ml of 1 M nitric acid was used to hydrolyse TEOS instead of acetic acid. The bulk and surface properties of the three parent silica monoliths were characterised by N2 porosimetry using a Micromeritics TriStar porosimeter. Samples were degassed at 30 °C for 2 h prior to analysis by nitrogen adsorption at −196 °C. BET surface areas were calculated over the relative pressure range 0.01–0.2, while pore size distributions were determined by applying the Barrett–Joyner–Halenda (BJH) method to the desorption isotherm for P/P0 > 0.35. Scanning electron microscopy (SEM) images were acquired using a Zeiss EVO 60 instrument and Oxford Instruments Inca System 350 at 20 kV. Samples for SEM analysis were coated with Au–Pt (∼2 nm) using a SEMPREP 2 sputter coater (Nanotech Ltd., Sandy, UK).
2.3 Determination of lipase loading
The immobilised lipase loading was quantified from the amount of unsupported lipase collected in the washing as determined from the hydrolysis of 4-nitrophenyl butyrate to 4-nitrophenol. The amount of immobilised lipase was obtained from the difference between the amount of lipase introduced into the monolith (10 mg) as described above and that observed in the residual solution collected after the immobilisation process.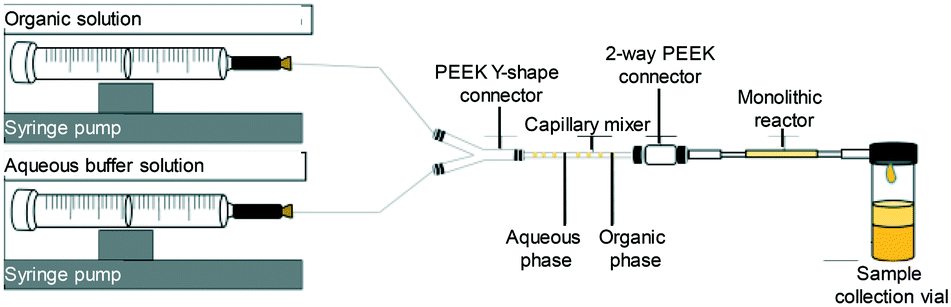 | ||
| Fig. 1 Schematic of the reactor system to assay the activity of the immobilised lipase.53 | ||
2.4 Transesterification
Transesterification of glyceryl tributyrin with methanol (Fig. 2) was performed in an isothermal flow reactor supplied by a syringe pump. A selected molar ratio of tributyrin and methanol was charged to the syringe pump and delivered into the reactor at a given temperature (Fig. 3). Samples were collected at different time intervals and analysed off-line on a Perkin Elmer Clarus HPLC with a UV detector and a BIO Wide Pore C18 column (5 μm × 15 cm × 2.1 mm), using a methanol/ultra-pure water (80:20 v/v) mobile phase, operated under isocratic conditions at 23 °C to quantify tributyrin conversion and methyl butyrate formation. Multi-point calibrations were performed for methyl butyrate to calculate the response factors. Errors in the FAME yield are the standard deviation of three experiments.
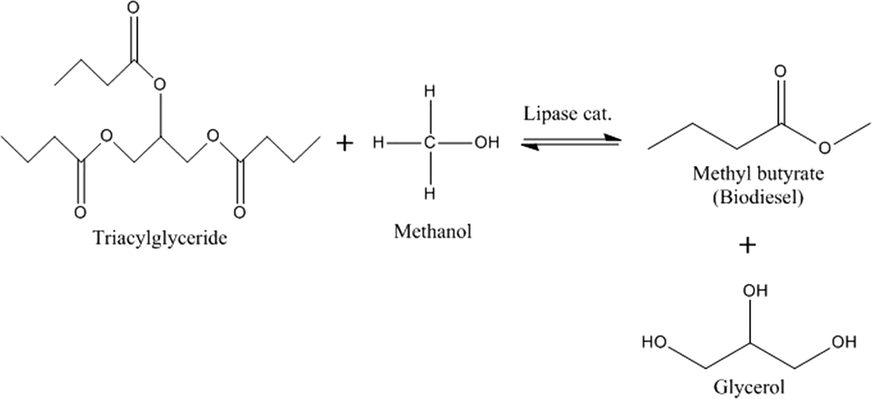 | ||
| Fig. 2 Transesterification reaction of tributyrin with methanol. | ||
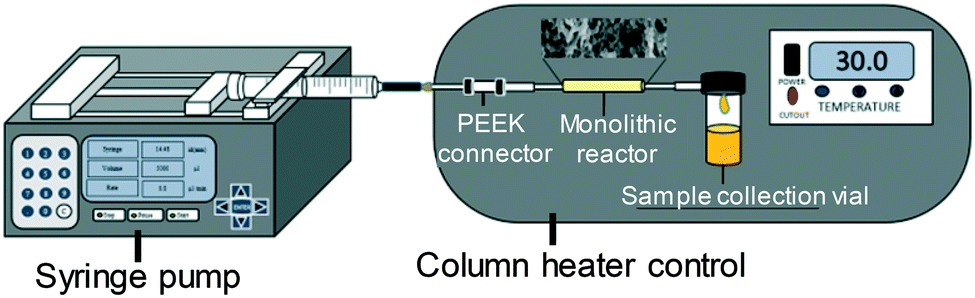 | ||
| Fig. 3 Schematic of the continuous flow reactor for testing. | ||
3. Results and discussion
3.1 Characterisation
The silica monolith morphology is expected to be strongly influenced by the synthesis protocol, notably the choice and concentration of the silane precursor, the molecular weight of the polymer surfactant, and the water:silane precursor ratio. Here, ‘coral-like’ bicontinuous architectures with interpenetrating macropores form as a result of spinodal decomposition of silica-rich and solvent-rich phases during gelation,51,53 in the presence of PEO as a surfactant-template to impart meso/macroporosity.54 Such hierarchically porous frameworks maximise the accessible internal surface area and pore volume, and minimise the pressure drop across a monolith.51,55,56 Successful monolith formation was confirmed by SEM, with micrographs in Fig. 4 evidencing coral-like porous architectures for all three samples55,57 after lipase immobilization. Macropore diameters calculated from SEM (Table 1) were similar in all cases, indicating that a common liquid volume fraction co-existed with silica sol–gel.56,58,59 However, textural differences in the architectures are apparent, with the M1 and M3 frameworks exhibiting smooth surfaces, while that of M2 is constructed from approximately 1 μm spherical beads. Table 1 summarises the textural properties of the three parent monoliths. Mesopore generation on the silica skeleton (wall) was confirmed by N2 porosimetry, revealing an increase in both mesopore and macropore diameter and total pore volume with the surfactant molecular weight (M2 versus M1) and the use of TEOS (M3) versus TMOS (M1) as the silica source. These changes are accompanied by an inverse trend in BET surface area (representative isotherm shown for M1 in Fig. S2†), which decreased with surfactant molecular weight and the use of TEOS, reflecting the enhanced porosity. The observation that TMOS created a higher surface area monolith than TEOS is consistent with the previous literature.60 The increase in mesopore diameter for TEOS is attributed to the faster rate of hydrolysis for TMOS, which results in the formation of highly branched silica clusters for the latter, which link to form a monolith dominated by small pore diameter. Similar observations were reported by Liu et al., who observed an increase in mesopore size from 9 to 16 nm on switching from TMOS to TEOS.61 Macroporosity is essential for the immobilisation of Candida antarctica lipase B within the monolith architecture due to its high molecular weight (33273 Da) and size (120.43 nm2).62
 | ||
| Fig. 4 SEM images of silica monolith samples M1, M2, and M3. | ||
The amount of enzyme immobilised onto each monolith was calculated from the hydrolysis of 4-NPB to 4-NP (Table 1), and revealed a common loading of around 8.5 mg lipase. This represents immobilisation of 80–90% of the lipase introduced to the monolith, similar to that previously reported for an amine-functionalised monolith.30
3.2 Transesterification
Candida antarctica lipase is prone to deactivation by high concentrations of short-chain alcohols34 and hence optimisation of the methanol:oil molar ratio is critical for its application in transesterification.32–34 The impact of the methanol:tributyrin molar ratio was therefore first explored over the M2 catalyst to identify the optimum reactant stoichiometry. Fig. 5 shows a strong dependence of methyl butyrate yield on reactant stoichiometry, with a maximum of 66% obtained for 2:1 MeOH:tributyrin; this corresponds to the maximum theoretical TAG conversion to FAME for such a stoichiometry. Further increases in methanol, which usually promote transesterification over conventional solid acid/base catalysts, resulted in a significant loss in lipase activity. This difference between heterogeneous catalysts and biocatalysts is ascribed to enzyme denaturation due to methanol toxicity,63 although other reports suggest that lipase remains active for rapeseed oil methanolysis even for a MeOH:oil ratio of 6:1.64 Staged methanol introduction is proposed as a solution to avoid lipase deactivation and achieve complete oil conversion either in batch or in flow.63
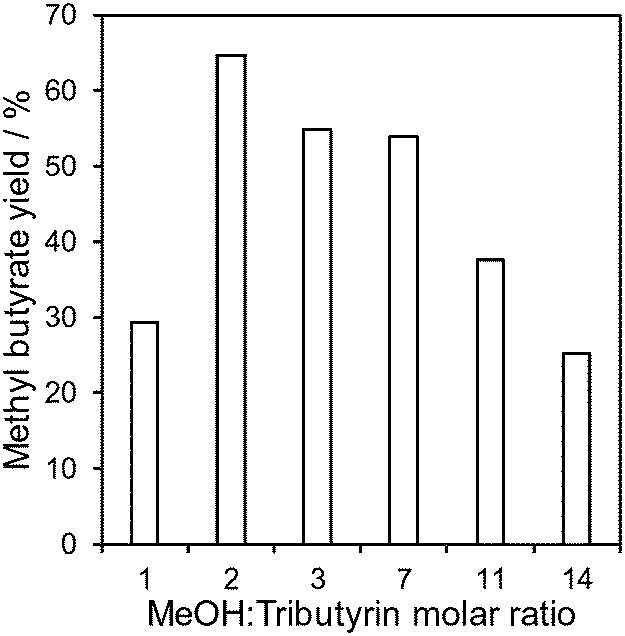 | ||
| Fig. 5 Methyl butyrate yield as a function of methanol:tributyrin mole ratio over the M2 catalyst. Reaction conditions: 0.8 μL min−1, 30 °C and 20 h on-stream. | ||
The impact of the liquid flow rate was subsequently explored over M2 (Fig. S3†), and revealed a monotonic decrease with increasing flow rate from 66% at 0.8 μL min−1 to 12% at 12.8 μL min−1. These flow rates are similar to literature ranges, when relative enzyme loadings are accounted for.47,48,65 This decrease simply reflects the corresponding fall in residence time (from 400 to 25 min) within the catalyst bed. Catalyst stability at the longest residence time (and hence the highest conversion) was also assessed for the optimum 2:1 MeOH:tributyrin stoichiometry identified above and at the 3:1 molar ratio necessary to achieve complete TAG conversion (Fig. S4†). While negligible deactivation was observed for the 2:1 MeOH:tributyrin stoichiometry >100 h of reaction on-stream, the higher methanol concentration resulted in 69% activity loss over the same time period confirming the lipase intolerance to methanol.63,66 The steady state productivity of M2 after 100 h on-stream under these optimal conditions was 0.7 μmol min−1 mglipase−1. This compares favourably with the literature values of 0.16 μmol min−1 mgenzyme−1 for the transesterification of crude safflower oil to ethyl linoleate catalysed by Thermomyces lanuginosus lipase immobilised on epoxy-functionalised silica monoliths,67 0.17 μmol min−1 mgenzyme−1 for the transesterification of cottonseed oil with methanol over resin-immobilised Candida antarctica B lipase (Novozym 435),65 and 0.007 μmol min−1 mgenzyme−1 for crude Jatropha oil methanolysis over an n-butyl-functionalised silica monolith-immobilised Burkholderia cepacia lipase.47 Finally, the temperature sensitivity of monolith-immobilised Candida antarctica was investigated. Fig. S5† reveals a significant increase in conversion from ∼30 to 63% as the reaction temperature was raised from 25 °C to 30 °C, followed by a steady loss in activity between 37–55 °C due to enzyme denaturation.68 Note that a negligible reaction occurred in any experiments in the absence of lipase at temperatures <55 °C. The optimum reaction conditions were thus 2:1 methanol:tributyrin at 30 °C and a reactant flow rate of 0.8 μL min−1.
The performance of the three immobilised lipase monolithic reactors was subsequently compared under these optimised conditions, as shown in Fig. 6. It is evident that M2 exhibited the highest FAME production and that the activity was strongly correlated with the textural properties (since all monoliths possessed a similar amount of lipase). M1 and M2 possess similar high surface areas, however the higher MW PEO-templated M2 possesses wider mesopores, which may offer superior mass transport to/from the active site, as previously reported for mesoporous69 and hierarchically porous solid acid70 and base22 catalysts for tributyrin transesterification. M2 was also superior to M3, despite the latter possessing the largest mesopores, presumably a consequence of the far lower surface area of the (TEOS-derived) M3 monolith. The superior activity of M2 may also reflect its unique textural properties, which may result in a different surface roughness and/or hydrophobicity and hence affinity for methanol and oil. Kinetic modelling of heterogeneous base-catalysed transesterification indicates that the reaction follows a Langmuir–Hinshelwood–Hougen–Watson (LHHW) mechanism, with adsorption of polar methanol being rate-limiting.71
 | ||
| Fig. 6 Methyl butyrate yield using different immobilised lipase microreactors. Reaction conditions: 0.8 μL min−1, methanol:tributyrin molar ratio = 2:1, 30 °C and 20 h on-stream. | ||
A balance of high surface area, wide meso- and macropores, and an appropriate surface texture/functionality is required to provide a high density of accessible (enzyme) active sites.
4. Conclusions
Lipase immobilised on silica monoliths has been successfully synthesised and screened for the transesterification of tributyrin in continuous flow at mild temperatures. Successful Candida antarctica immobilisation was demonstrated by calculating the amount of trapped enzyme via 4-nitrophenyl butyrate hydrolysis. Reaction conditions were optimised for the methanol:tributyrin molar ratio, flow rate, and reaction temperature. Although enzyme deactivation occurred at high methanol concentrations and temperatures >30 °C, excellent stability and quantitative methyl tributyrate yields were obtained for >100 h reaction for a 2:1 methanol:tributyrin feed and a flow rate of 0.8 μL min−1 at 30 °C. Although lipase immobilised on silica monoliths prepared using different silica precursors and molecular weight surfactants were all active for tributyrin transesterification, their different textural properties significantly influenced the catalytic activity. A combination of high surface area and large meso- and macropores was necessary to overcome diffusion limitations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MA thanks the Ministry of Education, Saudi Arabia, for financial support through the King's scholarship program. GK thanks the Royal Society for an Equipment Research Grant. KW and AFL acknowledge support from a Leverhulme-Royal Society Africa Award. Mr. Anthony Sinclair is acknowledged for assistance with SEM.References
- M. Hajjari, M. Tabatabaei, M. Aghbashlo and H. Ghanavati, Renewable Sustainable Energy Rev., 2017, 72, 445–464 CrossRef CAS.
- G. Köktürk and A. Tokuç, Renewable Sustainable Energy Rev., 2017, 73, 332–345 CrossRef.
- T. Taparia, M. Mvss, R. Mehrotra, P. Shukla and S. Mehrotra, Biotechnol. Appl. Biochem., 2016, 63, 715–726 CrossRef CAS PubMed.
- H. M. Mahmudul, F. Y. Hagos, R. Mamat, A. A. Adam, W. F. W. Ishak and R. Alenezi, Renewable Sustainable Energy Rev., 2017, 72, 497–509 CrossRef CAS.
- H. H. Mardhiah, H. C. Ong, H. H. Masjuki, S. Lim and H. V. Lee, Renewable Sustainable Energy Rev., 2017, 67, 1225–1236 CrossRef CAS.
- N. Mansir, Y. H. Taufiq-Yap, U. Rashid and I. M. Lokman, Energy Convers. Manage., 2017, 141, 171–182 CrossRef CAS.
- O. Edenhofer, R. Pichs-Madruga, Y. Sokona, K. Seyboth, P. Matschoss, S. Kadner, T. Zwickel, P. Eickemeier, G. Hansen, S. Schlömer and C. von Stechow, IPCC, 2011: Summary for Policymakers, in IPCC Special Report on Renewable Energy Sources and Climate Change Mitigation, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2011 Search PubMed.
- R. E. Gumba, S. Saallah, M. Misson, C. M. Ongkudon and A. Anton, Biofuel Res. J., 2016, 3, 431–447 CrossRef.
- D. Y. C. Leung, X. Wu and M. K. H. Leung, Appl. Energy, 2010, 87, 1083–1095 CrossRef CAS.
- A. Demirbas, Biodiesel, Springer, 2008 Search PubMed.
- A. F. Lee, J. A. Bennett, J. C. Manayil and K. Wilson, Chem. Soc. Rev., 2014, 43, 7887–7916 RSC.
- K. Wilson and A. F. Lee, Catal. Sci. Technol., 2012, 2, 884–897 CAS.
- B. K. Barnwal and M. P. Sharma, Renewable Sustainable Energy Rev., 2005, 9, 363–378 CrossRef.
- A. Sivasamy, K. Y. Cheah, P. Fornasiero, F. Kemausuor, S. Zinoviev and S. Miertus, ChemSusChem, 2009, 2, 278–300 CrossRef CAS PubMed.
- H. Fukuda, A. Kondo and H. Noda, J. Biosci. Bioeng., 2001, 92, 405–416 CrossRef CAS PubMed.
- Y. Shimada, Y. Watanabe, A. Sugihara and Y. Tominaga, J. Mol. Catal. B: Enzym., 2002, 17, 133–142 CrossRef CAS.
- N. Muhammad, Y. A. Elsheikh, M. I. A. Mutalib, A. A. Bazmi, R. A. Khan, H. Khan, S. Rafiq, Z. Man and I. Khan, J. Ind. Eng. Chem., 2015, 21, 1–10 CrossRef CAS.
- F. Liu, L. Wang, Q. Sun, L. Zhu, X. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2012, 134, 16948–16950 CrossRef CAS PubMed.
- C. Pirez, A. F. Lee, J. C. Manayil, C. M. A. Parlett and K. Wilson, Green Chem., 2014, 16, 4506–4509 RSC.
- E. Lotero, Y. Liu, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin, Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef CAS.
- J. J. Creasey, C. M. A. Parlett, J. C. Manayil, M. A. Isaacs, K. Wilson and A. F. Lee, Green Chem., 2015, 17, 2398–2405 RSC.
- J. J. Woodford, J.-P. Dacquin, K. Wilson and A. F. Lee, Energy Environ. Sci., 2012, 5, 6145–6150 CAS.
- Z. Wen, X. Yu, S.-T. Tu, J. Yan and E. Dahlquist, Appl. Energy, 2010, 87, 743–748 CrossRef CAS.
- Z. Wen, X. Yu, S.-T. Tu, J. Yan and E. Dahlquist, Bioresour. Technol., 2010, 101, 9570–9576 CrossRef CAS PubMed.
- F. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS.
- R. A. Sheldon, R. M. Lau, M. J. Sorgedrager, F. van Rantwijk and K. R. Seddon, Green Chem., 2002, 4, 147–151 RSC.
- A. Bajaj, P. Lohan, P. N. Jha and R. Mehrotra, J. Mol. Catal. B: Enzym., 2010, 62, 9–14 CrossRef CAS.
- R. Sharma, Y. Chisti and U. C. Banerjee, Biotechnol. Adv., 2001, 19, 627–662 CrossRef CAS PubMed.
- K.-E. Jaeger and T. Eggert, Curr. Opin. Biotechnol., 2002, 13, 390–397 CrossRef CAS PubMed.
- S. T. Anuar, Y.-Y. Zhao, S. M. Mugo and J. M. Curtis, J. Mol. Catal. B: Enzym., 2013, 92, 62–70 CrossRef CAS.
- R. Irimescu, Y. Iwasaki and C. T. Hou, J. Am. Oil Chem. Soc., 2002, 79, 879–883 CrossRef CAS.
- Y. Watanabe, Y. Shimada, A. Sugihara and Y. Tominaga, J. Mol. Catal. B: Enzym., 2002, 17, 151–155 CrossRef CAS.
- T. Samukawa, M. Kaieda, T. Matsumoto, K. Ban, A. Kondo, Y. Shimada, H. Noda and H. Fukuda, J. Biosci. Bioeng., 2000, 90, 180–183 CrossRef CAS PubMed.
- J.-W. Chen and W.-T. Wu, J. Biosci. Bioeng., 2003, 95, 466–469 CrossRef CAS PubMed.
- M. C. Franssen, P. Steunenberg, E. L. Scott, H. Zuilhof and J. P. Sanders, Chem. Soc. Rev., 2013, 42, 6491–6533 RSC.
- F. X. Malcata, H. R. Reyes, H. S. Garcia, C. G. Hill Jr and C. H. Amundson, J. Am. Oil Chem. Soc., 1990, 67, 890–910 CrossRef CAS.
- A. Bastida, P. Sabuquillo, P. Armisen, R. Fernandez–Lafuente, J. Huguet and J. M. Guisan, Biotechnol. Bioeng., 1998, 58, 486–493 CrossRef CAS PubMed.
- T. A. Nijhuis, A. E. W. Beers, T. Vergunst, I. Hoek, F. Kapteijn and J. A. Moulijn, Catal. Rev.: Sci. Eng., 2001, 43, 345–380 CAS.
- C. M. A. Parlett, K. Wilson and A. F. Lee, Chem. Soc. Rev., 2013, 42, 3876–3893 RSC.
- G. Guiochon, J. Chromatogr. A, 2007, 1168, 101–168 CrossRef CAS PubMed.
- A. Šalić and B. Zelić, Goriva Maziva, 2011, 50, 98–110 Search PubMed.
- A. Zampieri, P. Colombo, G. T. P. Mabande, T. Selvam, W. Schwieger and F. Scheffler, Adv. Mater., 2004, 16, 819–823 CrossRef CAS.
- A. Sachse, N. Linares, P. Barbaro, F. Fajula and A. Galarneau, Dalton Trans., 2013, 42, 1378–1384 RSC.
- J. García-Aguilar, I. Miguel-García, Á. Berenguer-Murcia and D. Cazorla-Amorós, ACS Appl. Mater. Interfaces, 2014, 6, 22506–22518 Search PubMed.
- Y. Zhou, J. H. Schattka and M. Antonietti, Nano Lett., 2004, 4, 477–481 CrossRef CAS.
- H. Nishihara, S. R. Mukai, D. Yamashita and H. Tamon, Chem. Mater., 2005, 17, 683–689 CrossRef CAS.
- K. Kawakami, Y. Oda and R. Takahashi, Biotechnol. Biofuels, 2011, 4, 42 CrossRef CAS PubMed.
- K. Kawakami, Y. Sera, S. Sakai, T. Ono and H. Ijima, Ind. Eng. Chem. Res., 2005, 44, 236 CrossRef CAS.
- A. L. de Lima, A. Mbengue, R. A. S. San Gil, C. M. Ronconi and C. J. A. Mota, Catal. Today, 2014, 226, 210–216 CrossRef.
- V. Varela Guerrero and D. F. Shantz, Ind. Eng. Chem. Res., 2009, 48, 10375–10380 CrossRef.
- P. D. I. Fletcher, S. J. Haswell, P. He, S. M. Kelly and A. Mansfield, J. Porous Mater., 2011, 18, 501–508 CrossRef CAS.
- H. Gustafsson, E. M. Johansson, A. Barrabino, M. Odén and K. Holmberg, Colloids Surf., B, 2012, 100, 22–30 CrossRef CAS PubMed.
- P. He, G. Greenway and S. J. Haswell, Process Biochem., 2010, 45, 593–597 CrossRef CAS.
- J.-L. Blin, A. Léonard, Z.-Y. Yuan, L. Gigot, A. Vantomme, A. K. Cheetham and B.-L. Su, Angew. Chem., Int. Ed., 2003, 42, 2872–2875 CrossRef CAS PubMed.
- H. Minakuchi, K. Nakanishi, N. Soga, N. Ishizuka and N. Tanaka, J. Chromatogr. A, 1998, 797, 121–131 CrossRef CAS.
- K. Nakanishi, H. Minakuchi, N. Soga and N. Tanaka, J. Sol-Gel Sci. Technol., 1997, 8, 547–552 CAS.
- O. Núñez, K. Nakanishi and N. Tanaka, J. Chromatogr. A, 2008, 1191, 231–252 CrossRef PubMed.
- K. Nakanishi, J. Porous Mater., 1997, 4, 67–112 CrossRef CAS.
- K. Nakanishi, H. Minakuchi, N. Soga and N. Tanaka, J. Sol-Gel Sci. Technol., 1998, 13, 163–169 CrossRef CAS.
- A. Soleimani Dorcheh and M. H. Abbasi, J. Mater. Process. Technol., 2008, 199, 10–26 CrossRef CAS.
- J. Liu, L. Zhang, Q. Yang and C. Li, Microporous Mesoporous Mater., 2008, 116, 330–338 CrossRef CAS.
- P. Trodler and J. Pleiss, BMC Struct. Biol., 2008, 8, 9 CrossRef PubMed.
- K. Nie, F. Xie, F. Wang and T. Tan, J. Mol. Catal. B: Enzym., 2006, 43, 142–147 CrossRef CAS.
- W. Du, D. Liu, L. Li and L. Dai, Biotechnol. Prog., 2007, 23, 1087–1090 CrossRef CAS PubMed.
- D. Royon, M. Daz, G. Ellenrieder and S. Locatelli, Bioresour. Technol., 2007, 98, 648–653 CrossRef CAS PubMed.
- A. E. M. Janssen, A. M. Vaidya and P. J. Halling, Enzyme Microb. Technol., 1996, 18, 340–346 CrossRef CAS PubMed.
- N. Brun, A. Babeau-Garcia, M.-F. Achard, C. Sanchez, F. Durand, G. Laurent, M. Birot, H. Deleuze and R. Backov, Energy Environ. Sci., 2011, 4, 2840–2844 CAS.
- R. K. Scopes, Clin. Chim. Acta, 1995, 237, 17–23 CrossRef CAS.
- C. Pirez, J.-M. Caderon, J.-P. Dacquin, A. F. Lee and K. Wilson, ACS Catal., 2012, 2, 1607–1614 CrossRef CAS.
- J. Dhainaut, J.-P. Dacquin, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 296–303 RSC.
- A. Kapil, K. Wilson, A. F. Lee and J. Sadhukhan, Ind. Eng. Chem. Res., 2011, 50, 4818–4830 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Effect of flow rate, time and temperature on transesterification of tributyrin. See DOI: 10.1039/c7re00162b |
| This journal is © The Royal Society of Chemistry 2018 |