
The interplay between ceria particle size, reducibility, and ethanol oxidation activity of ceria-supported gold catalysts†
Gregory M.
Mullen
a,
Edward J.
Evans
Jr.
b,
Benjamin C.
Siegert
a,
Nathan R.
Miller
c,
Benjamin K.
Rosselet
a,
Iliya
Sabzevari
a,
Adrianna
Brush
a,
Zhiyao
Duan
b and
C.
Buddie Mullins
 *abd
*abd
aMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: mullins@che.utexas.edu
bTexas Materials Institute, The University of Texas at Austin, Austin, Texas 78712, USA
cDepartment of Geosciences, Jackson School of Geosciences, University of Texas at Austin, Austin, TX 78712, USA
dDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA
First published on 5th January 2018
Abstract
The structure of a support material can have profound impacts on the behavior of a catalyst, altering the activity and selectivity of chemical reactions. In this article, we investigate the influence of the support material's structure on the activity of Au/CeO2 catalysts for selective oxidation of ethanol in a fixed-bed flow reactor. By doping the ceria support with Al, La, and Zr during synthesis and by altering the temperature of pretreatment in air after synthesis, ceria particles varying in size between 3 nm and 22 nm were prepared. The smaller ceria particles exhibited higher oxygen storage capacities as determined by temperature programmed reduction testing and resulted in more active catalysts for ethanol oxidation. We note a linear correlation between oxygen storage capacity and catalytic activity for ethanol oxidation.
Introduction
Gold, chemically inert as a bulk material, demonstrates exceptional catalytic activity for a number of reactions when stabilized in nanoparticle form.1,2 Insights into the catalytic behavior of gold have been made since the seminal studies of Haruta et al.,3,4 but many questions regarding the nature of gold as a catalytic material remain unanswered. One of the most confounding aspects of gold catalysis regards the role that the support material plays in generating an active catalyst.Support materials can influence the behavior of catalyst systems in many ways. In some cases, interactions between catalyst particles and support materials inhibit chemisorption of reactant species, harming catalytic activity.5,6 However, metal–support interactions can also promote catalytic activity, and the support material even takes part in some catalytic reactions.7 Variations in the activity of gold catalysts upon changing the supported material were noted in previous studies.8–11 By altering the properties of the support, the activity of the resulting catalyst can be influenced; therefore, interactions between the support material and the active phase may be leveraged to tune catalytic behavior.
Introducing changes to the nanoscale structure of a support material can have significant impacts on the behavior of a catalyst.12–16 This phenomenon has been well-documented for gold catalysts – especially with systems employing ceria as the support material. Carrettin et al. showed that the use of nanocrystalline ceria as a support material for gold generated a far more active catalyst for CO oxidation than microcrystalline ceria.17 Park et al. demonstrated that a pronounced enhancement in activity for the water gas shift reaction (WGSR) occurred when gold was supported on CeOX/TiO2 surfaces.18 The resulting catalyst exhibited activity surpassing both Au/CeO2 and Au/TiO2, indicating that the interaction between ceria and titania resulted in a synergistic effect on catalytic activity. The authors suggested that this effect resulted from stabilization of ceria as nanoparticles on the titania surface. Si and Stephanopoulos showed that the activity of catalysts comprised of gold supported on nanostructured ceria particles depended significantly on the crystal planes of ceria exposed at the surface of the particles.19
The formation of oxygen vacancy sites has been correlated with the enhancement of catalytic activity for several reactions.7,20,21 Catalysts comprised of gold supported on reducible materials such as ceria and titania have exhibited some of the highest reported activity for both the WGSR8,9,22 and CO oxidation,10 two of the most well-studied reactions catalyzed by gold. The authors of each study discussed in the paragraph above suggested that the influences on catalytic activity were related to the reducibility of the nanostructured supports. By influencing the reducibility of a metal oxide support through manipulation of its nanostructure, insight into the nature of the structure–function relationships that dictate catalytic behavior can be gained.
Previous studies have investigated the influence of the support material's structure on the activity of gold catalysts for selective oxidation of alcohols as well. Wang et al. showed that gold catalysts for benzyl alcohol oxidation supported on MnO2 nanorods were more active than similar catalysts supported on commerical MnO2 powder.23 Alhumaimess et al. demonstrated that the nanoscale structure of MnO2 nanowire supports influenced the activity of gold catalysts for benzyl alcohol oxidation.24 Abad et al. showed that the alcohol oxidation activity of gold catalysts supported on nanocrystalline ceria was far higher than similar catalysts supported on microcrystalline ceria. The authors suggested that the enhanced activity resulted from the formation of Ce3+ and Au+ sites on the catalyst surface, which facilitated the rate-limiting step of the reaction, hydride transfer from the reactive intermediate (an alkoxide) to the catalyst surface.25 Studies have also shown that the shape of nanostructured ceria supports plays an important role in determining the activity of Au/CeO2 catalysts for oxidation of alcohols. Gold supported on ceria nanorods resulted in more active catalysts for benzyl alcohol oxidation than gold supported on ceria nanocubes or nanopolyhedra,26,27 an effect that Wang et al. attributed to the exposure of different crystal planes at the surface the nanostructured supports.26
In this study, we have investigated the influence of ceria particle size on the activity of Au/CeO2 catalysts for the selective oxidation of ethanol in a fixed bed flow reactor. Ceria nanoparticles varying in size between 3 nm and 22 nm were prepared by doping the ceria with Al, La, and Zr during synthesis and by pretreating the ceria in air at various temperatures after synthesis. These dopants were chosen because we determined in a previous study28 that they could be homogeneously dispersed within the mixed oxide via a facile coprecipitation synthesis technique. Au/CeO2 catalysts synthesized using these materials exhibited an inverse monotonic trend between ceria particle size and ethanol oxidation activity. A linear correlation between ethanol oxidation activity and oxygen storage capacity of the Au/CeO2 catalysts for as determined by temperature programmed reaction with H2 (H2-TPR) measurements was also observed. Our results demonstrate that oxygen storage capacity serves as an excellent metric for describing the activity of Au/CeO2 catalysts for ethanol oxidation. Furthermore, we show that doping the support material can be an effective means of enhancing the activity of gold catalysts for ethanol oxidation by altering the structure and reducibility of the resulting material.
Experimental methods
Synthesis of ceria supports and Au/CeO2 catalysts
The ceria support materials were prepared via urea decomposition coprecipitation synthesis. For each synthesis, a solution of 0.11 M ammonium cerium nitrate and 2.0 M urea in deionized water was prepared. For the synthesis of doped ceria supports, the dopant precursor, a hydrated nitrate salt in each case, was incorporated into the solution at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 (dopant:Ce). The solution was then transferred to a round bottom flask and heated to 100 °C under vigorous stirring. A precipitate was formed after ∼1 h, at which point the solution was diluted by ∼50% with deionized water. The resulting mixture was aged at 100 °C for 8 h. After cooling to room temperature, the mixture was centrifuged, and the supernatant was discarded. The remaining solid was washed three times with deionized water. The product was dried for ∼24 hours under vacuum at room temperature. Portions of the undoped ceria were heated in air for 4 h at various temperatures (400 °C, 600 °C, and 800 °C). The doped ceria supports were heated in air at 400 °C for 4 h.
:9 (dopant:Ce). The solution was then transferred to a round bottom flask and heated to 100 °C under vigorous stirring. A precipitate was formed after ∼1 h, at which point the solution was diluted by ∼50% with deionized water. The resulting mixture was aged at 100 °C for 8 h. After cooling to room temperature, the mixture was centrifuged, and the supernatant was discarded. The remaining solid was washed three times with deionized water. The product was dried for ∼24 hours under vacuum at room temperature. Portions of the undoped ceria were heated in air for 4 h at various temperatures (400 °C, 600 °C, and 800 °C). The doped ceria supports were heated in air at 400 °C for 4 h.
The Au/CeO2 catalysts employed in this study were synthesized via deposition precipitation with urea as described by Zanella et al.29 For each synthesis, a solution of 6.1 × 10−3 M HAuCl4 and 0.42 M urea in deionized water was prepared. The ceria support was suspended in this solution under magnetic stirring. The mixture was heated to 80 °C and aged for 16 h to deposit gold on the support surface. After deposition, the suspension was centrifuged, and the supernatant was discarded. The catalyst was washed three times with deionized water. The product was dried for ∼24 h under vacuum at room temperature and then heated in air at 300 °C in a box furnace for 1 h. The resulting solid material was crushed and sieved to between 200 and 500 μm.
Characterization
BET surface area analysis was performed with a Quantachrome Instruments NOVA 2200e high-speed surface area BET analyzer at a temperature of 77 K. Before N2 physisorption, the samples were degassed at 100 °C overnight in a vacuum oven. Multiple data points in the pressure range of P/P0 = 0.1 to 0.3 were used to fit a line to the BET plot for each sample. Each fit achieved a correlation coefficient greater than 0.999.X-ray diffraction (XRD) was conducted with a Rigaku MiniFlex 600 diffractometer to investigate the crystal structure of the support materials and supported gold catalysts. The instrument generated radiation from a Cu sealed tube Kα X-ray source operated at a voltage of 40 kV and a current of 15 mA. The patterns were collected with a step size of 0.01° per step. Crystallite sizes were determined by Williamson–Hall analysis.
XPS was conducted to probe the chemical states of Ce and Au in the Au/CeO2 catalysts. Spectra were collected with a Kratos AXIS Ultra XPS spectrometer employing an Al Kα X-ray source operated at a voltage of 13 kV and a current of 10 mA. A low energy electron flood source was used to minimize static charge buildup on the samples while collecting the XPS spectra. Analysis of XPS spectra was conducted with the CasaXPS software. The binding energies of the resulting spectra were adjusted by shifting the C 1s level for adventitious carbon to 284.8 eV.
The wt% of gold incorporated into the catalyst samples for all materials and the mol% of additive incorporated into the samples made with doped cerium oxides were determined by inductively coupled plasma-mass spectrometry (ICP-MS) analysis. A 10 mg aliquot of each material was digested in 10 mL of conc. HCl/30% H2O2 solution prepared in a 2/1 ratio. The digested samples were each diluted by a factor of 1000 with an aqueous solution containing 2% HCl (v/v) and 1% thiourea (w/v) prior to analysis. Cation concentrations in the diluted samples (Al, Zr, La, Ce, Au) were determined using an Agilent 7500ce ICP-MS. The instrument was optimized for sensitivity across the AMU range, while minimizing oxide production (CeO/Ce < 1.2%). The analytical method employed an octopole reaction system, operated in hydrogen (reaction-mode) and in no gas mode. Internal standards, mixed into unknowns during analysis, were used to compensate analyte intensities for instrumental drift. The matrix for all standards and unknowns was 2% HCl (v/v) plus 1% thiourea (w/v), which has been shown to reduce gold memory effects.38,39 Limits of detection, based upon the population of matrix blank analyses interspersed throughout the analytical sequence were typically better than 0.1 ppb (median = 0.015 ppb). Analyte recoveries obtained for replicates (n = 4) of a mid-calibration range quality control standard were typically within 5% of certified values.
A Hitachi S-5500 STEM operated at 30 kV was used to image the ceria support materials prior to deposition of gold. Samples were prepared by drop casting a suspension made by sonicating a small amount of the support material in ethanol onto lacey carbon coated copper TEM grids. The grids were then heated to 100 °C in a vacuum oven overnight to remove excess solvent prior to imaging.
H2-TPR testing was conducted in an ambient pressure fixed-bed flow reactor system to probe the reduction behavior of the materials. For each test, approximately 100 mg of the sample material was supported on a quartz wool plug in a quartz tube and held in an Applied Test Systems Model 3210 tube furnace. MKS Type M100B mass flow controllers were used to deliver H2 and Ar to the sample bed. The system was purged with argon at 50 standard cm3 per min (sccm) for 30 min before equilibrating the sample in a stream of 5% H2/Ar at 50 sccm for an additional 30 min. No measurable consumption of H2 occurred for any of the materials during this period, indicating that all of the reduction occurred during the temperature ramp. The sample was then heated from room temperature to 600 °C at a rate of 10 °C min−1. The reactor effluent composition was measured throughout this process using a custom-built gas analysis system consisting of an ExTorr XT100 residual gas analyzer operating in a stainless steel chamber maintained at a base pressure of ∼1 × 10−9 mbar. Gas from the reactor effluent stream was introduced to this chamber at a pressure of 1.0 × 10−6 mbar via a temperature-controlled leak valve sampling system. The ratio of H2 to Ar in the effluent stream was calculated by tracking the ratio of the RGA signal for m/z = 2 (H2+) to the signal for m/z = 40 (Ar+). We confirmed that this signal ratio accurately reflected the ratio of H2 to Ar in the effluent stream by calibrating the instrument across a wide range of known gas compositions. Linearly fitting a calibration parity plot of the ratio of m/z = 2 to m/z = 40 versus the ratio of H2 to Ar within the composition range used for the H2-TPR tests resulted in a correlation coefficient of <0.9999. Hydrogen consumption rates were calculated by subtracting the calculated flow rate of H2 in the effluent stream from the inlet flow rate of H2 to the reactor. H2-TPR was always conducted immediately after heating the catalyst samples in air to minimize the formation of carbonates on the surface of the materials caused by exposure to air at low temperatures.
Ethanol oxidation activity testing
The catalytic behavior of the Au/CeO2 materials for ethanol oxidation was studied in the same reactor system used to conduct H2-TPR. The reaction bed, 90 mg of catalyst diluted in 900 mg of SiC, was held between two quartz wool plugs in a quartz tube with a 7 mm ID. Ethanol was introduced to the system by bubbling the reactant feed stream through a temperature-controlled liquid–vapor saturator. All gas lines downstream from the saturator were heated to prevent ethanol from condensing on the tubing of the system.The reactant stream, comprised of 1.5 kPa (15000 ppm) ethanol in high purity dry air (<6 ppm H2O, MATHESON Gas), flowed across the catalyst bed at a space velocity of 32000 h−1. These conditions were chosen such that ethanol conversions were <10% for all tests (percent conversion of ethanol for each catalyst is shown in the ESI†) in order to ensure that the system was operating in a differential conversion regime. Each catalyst was held for several hours under these conditions to ensure that a steady state was reached. Quantitative analysis of the reactor effluent gas composition was conducted with a HP 5890 Series II gas chromatograph (GC) equipped with a stainless steel column packed with Chromosorb 101 as the stationary phase. Both a flame ionization detector and a thermal conductivity detector were used to analyze the column effluent. An additional liquid–vapor saturator was employed to deliver propanol, which was used as an internal standard, to the reactor effluent prior to injection into the GC. Carbon balances were calculated to be within 5% of unity for all experiments discussed in this study. To ensure that the reactions were not influenced by external mass and heat transfer limitations, we calculated the Mears' criterion and the Weisz–Prater criterion for each catalyst under the steady state reaction conditions (see ESI†).
Results and discussion
Characterization
Fig. 1 displays the XRD patterns for gold catalysts supported on ceria that was heated in air at 400 °C, 600 °C, and 800 °C (denoted Au/CeO2-400C, Au/CeO2-600C, and Au/CeO2-800C, respectively) and gold supported on ceria doped with Al, La, and Zr that was heated in air at 400 °C (denoted Au/Al–CeO2, Au/La–CeO2, and Au/Zr–CeO2, respectively). All features appearing in these patterns correspond to the cubic fluorite phase of CeO2 (JCPDS 34-0394). None of the patterns displayed peaks corresponding to gold, suggesting that the dispersion of gold was high for all samples. Ceria crystallite sizes determined via Williamson–Hall analyses are shown in Table 1. XRD patterns of the support materials prior to deposition of gold are included in the ESI.† The patterns taken before and after gold deposition did not exhibit any notable differences.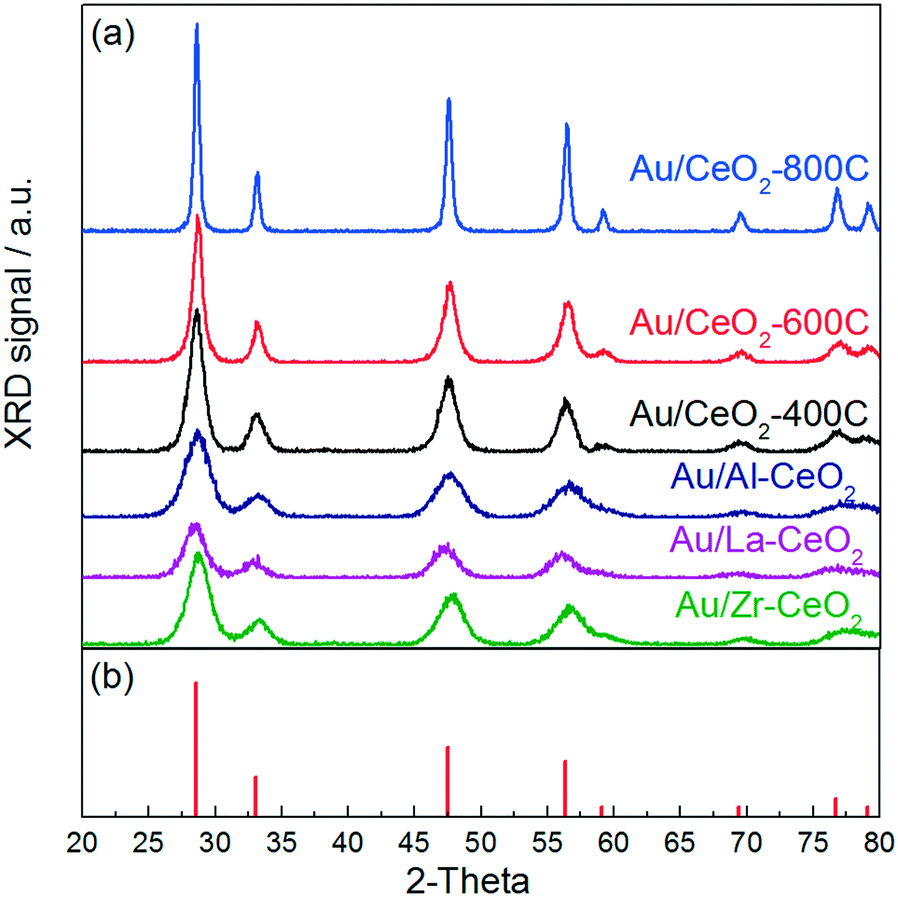 | ||
| Fig. 1 (a) XRD patterns for the Au/CeO2 catalysts. (b) Reference peaks for the cubic fluorite phase of CeO2 from JCPDS 34-0394. | ||
| Sample | CeO2 crystallite sizea (nm) | CeO2 lattice constant (nm) | CeO2 crystallite straina (%) |
|---|---|---|---|
| a Determined via Williamson–Hall analysis of XRD spectra. | |||
| Au/CeO2-800C | 24.0 | 0.5408 | 0.10 |
| Au/CeO2-600C | 8.5 | 0.5425 | 0.12 |
| Au/CeO2-400C | 6.8 | 0.5423 | 0.15 |
| Au/Al–CeO2 | 5.0 | 0.5424 | 0.89 |
| Au/La–CeO2 | 5.1 | 0.5448 | 0.41 |
| Au/Zr–CeO2 | 5.1 | 0.5411 | 0.62 |
Table 2 displays the average ceria particle size for each support material as determined by analysis of STEM images. We measured approximately 100 unique particles to calculate each average size. Representative STEM images and histograms displaying particle size distributions are included in the ESI.† The crystallite sizes determined via Williamson–Hall analysis of XRD patterns and the average particle sizes determined via analysis of STEM images correlated well with one another.
| Sample | Aua wt% | Dopanta mol% | CeO2 sizeb (nm) | BET surf. area (m2 g−1) | Au surf. conc. (μmol-Au m−2) |
|---|---|---|---|---|---|
| a Calculated from ICP-MS analysis. b Number averages determined from analysis of STEM images taken for the support materials prior to gold deposition. | |||||
| Au/CeO2-800C | 0.8 | — | 21.6 | 16 | 2.5 |
| Au/CeO2-600C | 1.0 | — | 7.0 | 84 | 0.60 |
| Au/CeO2-400C | 1.0 | — | 6.2 | 89 | 0.57 |
| Au/Al–CeO2 | 0.8 | 10 | 3.0 | 172 | 0.24 |
| Au/La–CeO2 | 1.0 | 11 | 3.6 | 146 | 0.35 |
| Au/Zr–CeO2 | 1.1 | 8 | 4.3 | 139 | 0.40 |
Both doping and varying the pretreatment temperature of the ceria influenced the particle size of the resulting materials. A monotonic increase in particle size occurred upon increasing the pretreatment temperature (i.e., higher pretreatment temperatures resulted in larger particles). Furthermore, each doped ceria sample exhibited a smaller average particle size than the CeO2-400C sample (which was preheated at the same temperature as the doped samples). The smaller sizes of the doped ceria supports suggest that the thermal stability of ceria was enhanced by the presence of the dopants, an effect documented previously for ceria doped with Al, La, and Zr.30
Table 1 also displays the lattice constant for CeO2 in each material, which we measured with Bragg's law using the CeO2(111) reflection that appears at a 2θ of ∼28°. The lattice parameter of bulk CeO2 is 0.5411 nm,31 but previous studies have shown that a steep expansion of the lattice parameter occurs as the size of a ceria crystallite decreases.32–34 The lattice parameters of CeO2 shown in Table 1 for the catalysts made with undoped ceria generally exhibited this phenomenon. The catalyst with the largest ceria particles Au/CeO2-800C exhibited a lattice parameter very close to that of bulk CeO2, while the catalysts made with smaller ceria particles exhibited larger lattice parameters consistent with expansion.
The CeO2 lattice parameter for Au/La–CeO2 and Au/Al–CeO2 were also consistent with expansion. These materials had very small crystallite sizes. Deconvolution of the influence of crystallite size and the influence of dopant incorporation was difficult for these materials. Qualitatively, the influences of particle size and doping could be assessed for Au/Zr–CeO2. The lattice parameter of CeO2 for Au/Zr–CeO2 was similar to that of bulk CeO2 despite the small size of the CeO2 crystallites in the doped material, suggesting that some degree of lattice contraction occurred due to incorporation of Zr within the crystallites.
The lattice strain in the CeO2 crystallites is also shown in Table 1. The strain increased slightly as the size of the ceria particles decreased for the catalysts made with undoped ceria. The strain was higher for each of the doped samples, suggesting that the dopants induced strain within the ceria crystal lattice.
Fig. 2 displays XPS spectra for the Au/CeO2 catalysts in the Ce 3d and Au 4f regions. The Ce 3d region of each spectrum exhibited a variety of peaks that have been previously attributed to Ce4+ and Ce3+ appearing at binding energies between 880 and 920 eV.35 It is widely accepted that Ce4+ and Ce3+ display three sets (u/v, u′′/v′′, u′′′/v′′′) and two sets (u0/v0, u′/v′) of spin–orbital coupled peaks in the 3d region, respectively. The approximate locations of these ten peaks are marked by the dotted lines in Fig. 2a.
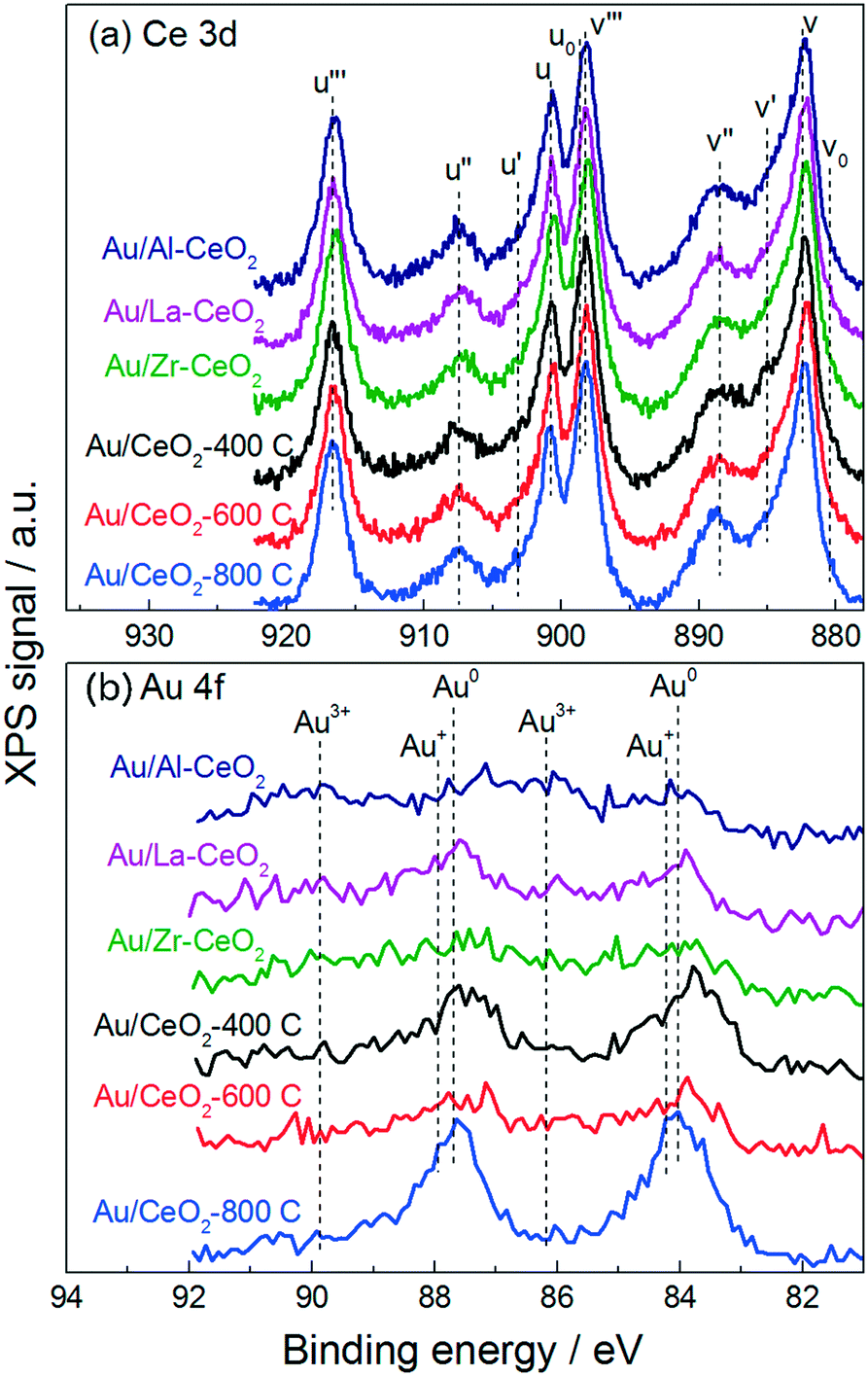 | ||
| Fig. 2 XPS spectra for the Au/CeO2 catalysts in (a) the Ce 3d region and (b) the Au 4f region. | ||
Previous studies have claimed that the concentration of Ce3+ in CeO2 particles increases as the particle size decreases – with pronounced increases for particles <10 nm in size.32,34,36 These studies measured the relative amounts of Ce4+ and Ce3+ present in ceria samples by fitting peaks in the Ce 3d region. However, some recent investigations have provided evidence suggesting that the population of Ce4+ and Ce3+ does not vary with the size of the particles. By fitting the Ce 3d region of the XPS spectra for ceria particles varying in size between 4 nm and 10 nm, Xu et al. measured a nearly identical amount of Ce3+ in each material.37 Paun et al. showed via X-ray absorption near-edge structure spectroscopy at the Ce K-edge that the amount of Ce3+ present in a series of ceria nanoparticles of varying size was low (<5%) and roughly constant, displaying no correlation with particle size.38 Cafun et al. probed the oxidation state of cerium ions using hard X-ray absorption spectroscopy during synthesis of ceria nanoparticles via coprecipitation and showed that all of the Ce3+ ions from the precursor solution were converted to Ce4+ in the precipitated product.39
Qualitatively, despite nearly an order of magnitude difference in size between the largest and smallest ceria particles, the Ce 3d regions appeared very similar for all of the materials, displaying no differences to suggest that significant changes in cerium oxidation state existed for the various materials. Analysis of the Ce 3d region is complicated by the overlapping nature of the peaks. One technique to mitigate this complication is to employ the method employed by Le Normand et al.,40 which uses the area of the well-resolved u′′′ satellite peak to estimate the area of the v′′′ peak and compares this area to the total area of the 3d 5/2 peaks (v0, v, v′, v′′, v′′′) in the Ce 3d region. The resulting ratio, which is equal to ∼0.32 for pure CeO2, decreases as the amount of Ce4+ in the material decreases. We calculated this ratio to be between 0.31 and 0.34 for all of the Au/CeO2 samples in this study, suggesting that these materials did not contain appreciable amounts of Ce3+. These calculations are discussed further in the ESI.†
Fig. 2b displays the Au 4f region of each XPS spectrum. Gold in the Au0 state exhibits one set of spin–orbital coupled peaks within this region appearing at 84.0 eV and 87.7 eV.41 The Au/CeO2-800C sample exhibited large peaks corresponding to Au0, while the Au/Al–CeO2, Au/La–CeO2, Au/Zr–CeO2, Au/CeO2-400C, and Au/CeO2-600C samples displayed broader features in the Au 4f region extending to higher binding energies. Au3+ and Au+ display coupled peaks at 86.1 eV/89.8 eV and 84.2 eV/87.9 eV, respectively.42–44 The attenuation of the Au0 peaks into broad features in these Au/CeO2 samples is consistent with the presence of oxidized gold. The increase in oxidized gold can be attributed to a higher number of defect sites on the smaller ceria particles. Previous studies have suggested that defect sites at the ceria surface interact strongly with gold, stabilizing it in the Au3+ and Au+ oxidation states,45 and smaller ceria particles contain increased concentrations of defects.33
Fig. 3 displays H2-TPR curves for the ceria supports and the Au/CeO2 catalysts. The reduction features exhibited in the TPR curves for the ceria samples prior to deposition of gold (Fig. 3a) are very similar to features exhibited in the TPR curves for ceria discussed in previous studies.46,47 Each curve exhibited a broad feature extending from low temperatures to ∼500–550 °C, consistent with the reduction of capping oxygen on the ceria surface.48 Bulk oxygen reduction during H2-TPR of CeO2 occurs at higher temperatures with a peak at ∼800 °C.48 Deposition of gold onto the ceria supports caused the reduction features to shift to lower temperatures for all of the samples. The H2-TPR curve for each of the Au/CeO2 catalysts displayed one large peak between 140 °C and 150 °C, in good agreement with TPR curves for similar materials from previous studies.42,46,49 Some of the catalysts exhibited small reduction features at higher temperatures as well.
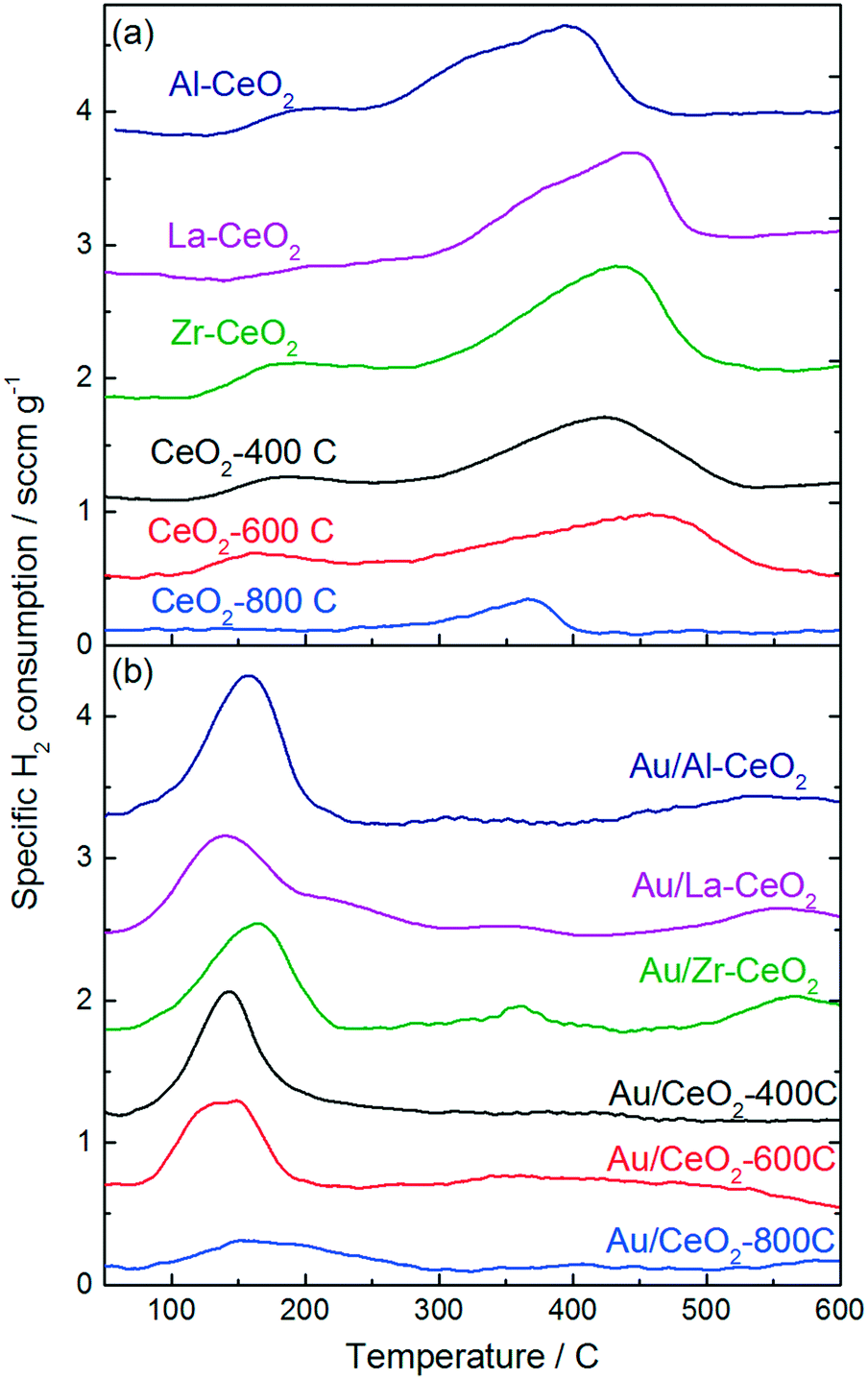 | ||
| Fig. 3 H2-TPR curves for (a) the ceria support materials prior to deposition of gold and (b) the Au/CeO2 catalysts. | ||
Studies by Sandoval et al.9 and Fu et al.50 suggested that oxygen atoms on the surface of the ceria support are the primary source of the Au/CeO2 catalyst's reducibility and that the reduction process results in the formation of oxygen vacancies generated by removal of these surface capping oxygen atoms. We calculated the amount of H2 consumed by each material, the hydrogen consumption per mole Au for the Au/CeO2 catalysts, and the percentage of surface capping oxygen that was reduced (assuming this reduction mechanism) for each material during TPR testing. The results of these calculations are shown in Table 3. H2 consumption was calculated by integrating the TPR curves for the CeO2 supports from 100 °C to 600 °C. The Au/CeO2 curves were integrated from 75 °C to 300 °C. A detailed explanation of the method employed to calculate the surface oxygen reduction percentages is included in the ESI.†
| Sample | Specific H2 consumption (μmol g-cat−1)a | Reduction of surface capping oxygen | Decrease in mol-H2 consumed per mol-Au |
|---|---|---|---|
| a The small H2 consumption features observed at higher temperatures for each Au/CeO2 catalyst was not included in these figures. b H2 consumption increased for Au/CeO2-800C. | |||
| Al–CeO2 | 475 | 21% | — |
| La–CeO2 | 394 | 21% | — |
| Zr–CeO2 | 619 | 34% | — |
| CeO2-400C | 420 | 36% | — |
| CeO2-600C | 392 | 36% | — |
| CeO2-800C | 85 | 41% | — |
| Au/Al–CeO2 | 307 | 14% | 1.3 |
| Au/La–CeO2 | 288 | 15% | 0.7 |
| Au/Zr–CeO2 | 245 | 13% | 4.5 |
| Au/CeO2-400C | 221 | 19% | 3.9 |
| Au/CeO2-600C | 186 | 17% | 2.0 |
| Au/CeO2-800C | 102 | 49% | (0.4)b |
In general, the support materials with smaller ceria particles exhibited higher specific H2 consumptions during TPR, consistent with previous studies that demonstrated an inverse correlation between ceria particle size and surface oxygen storage capacity.37,51,52 The H2 consumption correlated linearly with surface area for the undoped ceria support materials and for Zr–CeO2. The Al–CeO2 and La–CeO2 supports each consumed 40% less H2 per unit surface area than the other materials. A plot of specific surface area versus specific H2 consumption for the support materials is shown in the ESI.† The decrease in reducibility per unit surface area is consistent with the dopants stabilizing reducible sites at the ceria surface. Since Al and La are most stable in the 3+ oxidation state, substitution of a trivalent Al or La ion in the CeO2 crystal lattice in place of Ce4+ with would result in the stable formation of an acompanying vacancy site.
The calculations shown in Table 3 are consistent with that a lower percentage of the surface capping oxygen reduction for the smaller ceria particles. The CeO2-800C support exhibited the highest percentage reduction of surface oxygen (∼40%). The CeO2-400C, CeO2-600C, and Zr–CeO2 supports each exhibited similar extents of surface reduction (∼35%), while the Al–CeO2 and La–CeO2 supports exhibited significantly lower reduction percentages (∼20%). As mentioned in the paragraph above, the decrease in surface oxygen reduction for the Al–CeO2 and La–CeO2 supports may be related to the stabilization of vacancy sites by the trivalent cations of the dopants effectively decreasing the total number of reducible sites during TPR testing. By stabilizing reduced sites on the ceria surface, less oxygen would be present on the material for reduction during TPR testing.
The specific H2 consumption and percentage reduction of surface capping oxygen were lower for all of the Au/CeO2 catalysts than their corresponding ceria support materials prior to deposition of gold (with the exception of Au/CeO2-800C), suggesting that the incorporation of gold blocked some reducible sites on the CeO2 surface. We calculated the decrease in H2 consumption per gold atom incorporated for each Au/CeO2 catalyst (shown in Table 3) and found that the extent of this blocking was significant for many of the catalysts (>4 mol-H2 per mol-Au in the case of Au/Zr–CeO2). This observation was surprising considering the significant downward shift in TPR features after incorporation of gold into the materials, indicating the gold facilitated the reduction process. In order to properly account for this behavior, it is necessary to consider the process by which H2 consumption occurred during TPR. The calculations of surface oxygen reduction percentage shown in Table 3 and discussed above assume that the reducible surface oxygen is similar in nature to lattice oxygen within the CeO2 crystallites, however, recent studies have suggested that this model may be incorrect. Xu et al. showed that the appearance of reducible oxygen present in ceria nanoparticles correlated with the observation of superoxides in electron paramagnetic resonance spectra.37 Huang and Beck also provided evidence for the formation of activated oxygen molecules on ceria nanoparticles, observing features consistent with activated O2 on the surface of CeO2 nanoparticles via FTIR spectroscopy.53 A model by Kullgren et al. based on density functional theory calculations suggested that the reduction of surface capping oxygen atoms alone cannot account for the behavior exhibited by ceria nanoparticles during TPR, and that activation of O2 provides a better description of the oxygen storage behavior demonstrated by these materials.52 These studies each attributed the activation of oxygen on ceria nanoparticles to charge transfer from reduced Ce3+ surface sites to O2 resulting in the generation of a superoxide ion. Additionally, Soria et al. demonstrated that defect sites that were active for O2 activation were generated by a mild outgassing treatment of ceria nanoparticles at temperatures as low as 100 °C.54 We note further that studies have also demonstrated that peroxides can be generated on the surface of ceria particles as well. Wu et al. probed CeO2 nanocrystals with Raman spectroscopy and demonstrated the formation of both superoxide and peroxide species.55 The type of oxide generated was dictated by the type of defect present on the CeO2 surface.
This reduction model could explain several phenomena that we observed in the TPR patterns shown in Fig. 3. The magnitude of the decrease in H2 consumption after deposition of gold was inconsistent with a reduction process involving surface oxygen atoms. If reduction occurred via surface O activation, it would be unlikely that the reduction behavior of oxygen atoms located in positions far removed from the gold atom would be influenced, therefore, we would expect that, at a maximum, each gold atom could influence the three neighboring oxygen atoms in this case. However, the decreases in H2 consumption in Table 3 are consistent with an oxygen activation mechanism occurring via reduction of superoxides generated by charge transfer from Ce3+ cations associated with vacancy sites at the surface of the ceria particles. In this case, gold atoms could block sites of superoxide formation by interacting with Ce3+ cations, which would cause a decrease in reduction of two H2 molecules per site. Lakshmanan et al. probed the nature of O2 species adsorbed to CeO2 and Au/CeO2via electron paramagnetic resonance spectroscopy and found that the Au/CeO2 material did not generate superoxides (in contrast to the behavior of CeO2 without deposited gold).56 A study by Aboukaïs et al. demonstrated similar results, suggesting that gold hindered the formation of strongly bound O2− species on Au/CeO2 surfaces.57 Furthermore, Chang and Sheu recently demonstrated that one gold atom can interact with several oxygen vacancies on a CeO2 surface, resulting in charge redistribution away from Ce3+.58 Pan et al. also demonstrated that a single gold atom can “titrate” multiple Ce3+ sites.59 Lakshmanan et al. also proposed that Au/CeO2 activated O2 in the form of peroxides rather than superoxides and that the site of this activation was associated with the interface between gold and ceria.56 Furthermore, addition of gold to ceria materials has been shown to promote the reaction of hydrogen peroxide in a manner similar to that of catalase enzymes,60,61 and this behavior could help account for the decrease in reduction temperature observed upon depositing gold onto the ceria particles. On the basis of these studies, we propose that alteration of O2 activation sites by adsorption of gold explains the significant decreases in H2 consumption of the Au/CeO2 catalysts shown in Table 3. Further analysis of Au/CeO2 catalysts via X-ray absorption techniques could help to verify that this phenomenon occurs.
The behavior of the samples with CeO2-800C was different from that of the other materials. A larger extent of reduction was noted in Table 3, and the total H2 consumption was increased after deposition of gold. Previous studies by Aneggi et al. showed that annealing ceria particles in air at 800 °C resulted in the formation of more reactive crystal planes at the surface of the particles, while the less reactive (111) planes predominated on the surface of particles that were annealed in air at 500 °C.62,63 The (110) plane has been shown to generate oxygen vacancies more readily than the (111) surface,64 which could promote a greater extent of oxygen activation via the mechanism discussed in the paragraph above. Exposure of different surface planes does not explain the increase in H2 consumption for the Au/CeO2-800C sample, however, we note that the studies mentioned in the previous paragraph by Pan et al.59 and Chang and Sheu58 which demonstrated that gold can “titrate” CeO2 oxygen vacancies both studied interactions with the CeO2(111) surface. It is possible that gold interacts differently with oxygen vacancies on different crystal planes.
Ethanol oxidation reaction testing
All of the Au/CeO2 catalysts exhibited activity for oxidative dehydrogenation of ethanol, resulting in the production of acetaldehyde, and esterification, resulting in the production of ethyl acetate. We did not detect any other products during reaction testing, and each catalyst displayed a similar product distribution. The selectivity for acetaldehyde production varied only within the narrow range of 94–95% for the various catalysts (calculated on the basis of ethanol conversion). In a previous study, we showed that production of ethyl acetate resulted from the secondary reaction of acetaldehyde with ethanol.28,65 Acetaldehyde was first oxidized on the ceria surface to generate a surface acetate species, which reacted with ethanol to generate ethyl acetate via an esterification mechanism. Acidic surface sites catalyzed the esterification reaction. For further information about this process, we refer the reader to the studies mentioned above.28,65 The remainder of this article will only discuss the acetaldehyde production reaction.Doping and heating the ceria support materials caused significant variations to the activity of the Au/CeO2 catalysts. More than an order of magnitude difference in activity was observed between the most active (Au/Al–CeO2) and least active (Au/CeO2-800C) catalysts. Fig. 4 displays the activity of each material for acetaldehyde production plotted against the average particle size of the corresponding ceria support material. The plot exhibits an inverse monotonic trend (i.e., the activity increased as the size of the ceria particles decreased).
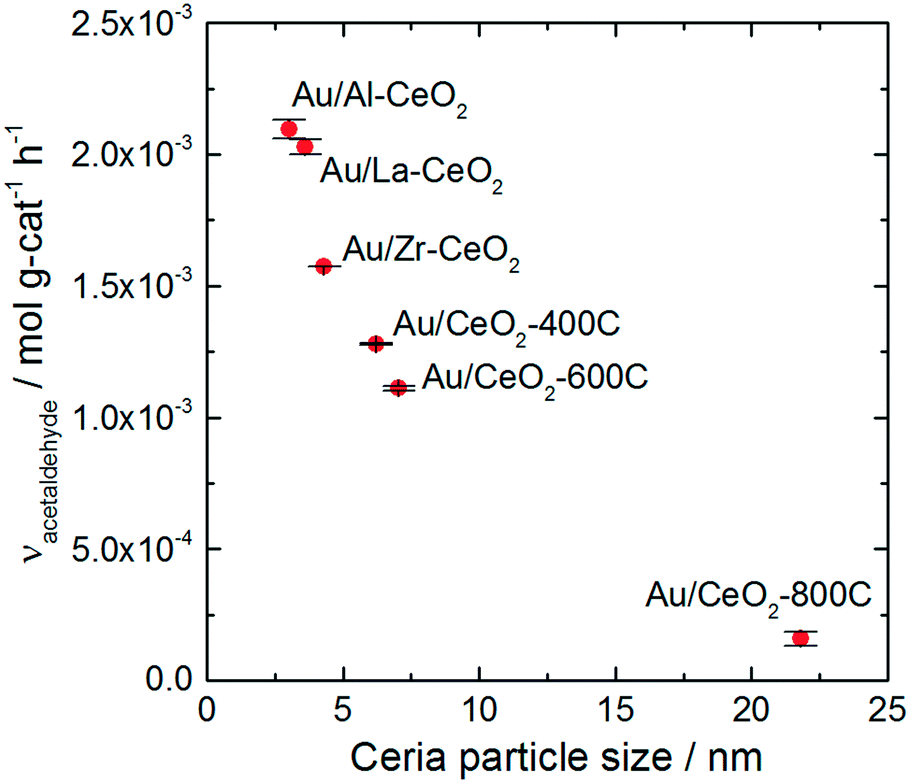 | ||
| Fig. 4 Acetaldehyde production rates for the Au/CeO2 catalysts versus average size of the corresponding ceria supports as determined by analysis of STEM images. Ethanol oxidation data were collected at a reaction temperature of 80 °C with a reactor feed consisting of 1.5 kPa (15000 ppm) ethanol in air at a space velocity of 32000 h−1. | ||
The trend in ethanol oxidation activity for the Au/CeO2 catalysts observed in Fig. 4 is similar to the trend between surface oxygen storage capacity and particle size discussed in previous studies.37,51,52 The onset of the increase in both oxygen storage capacity and in ethanol oxidation activity occurred at about the same ceria particle size (∼10 nm), suggesting that a relationship between oxygen storage capacity and ethanol oxidation activity may exist for these catalysts. To explore this relationship more directly, we plotted the specific hydrogen consumption of each Au/CeO2 catalyst during H2-TPR (Table 3) against its activity for ethanol oxidation (Fig. 4). The resulting plot, shown in Fig. 5, demonstrates that a linear correlation exists between oxygen storage capacity and acetaldehyde production.
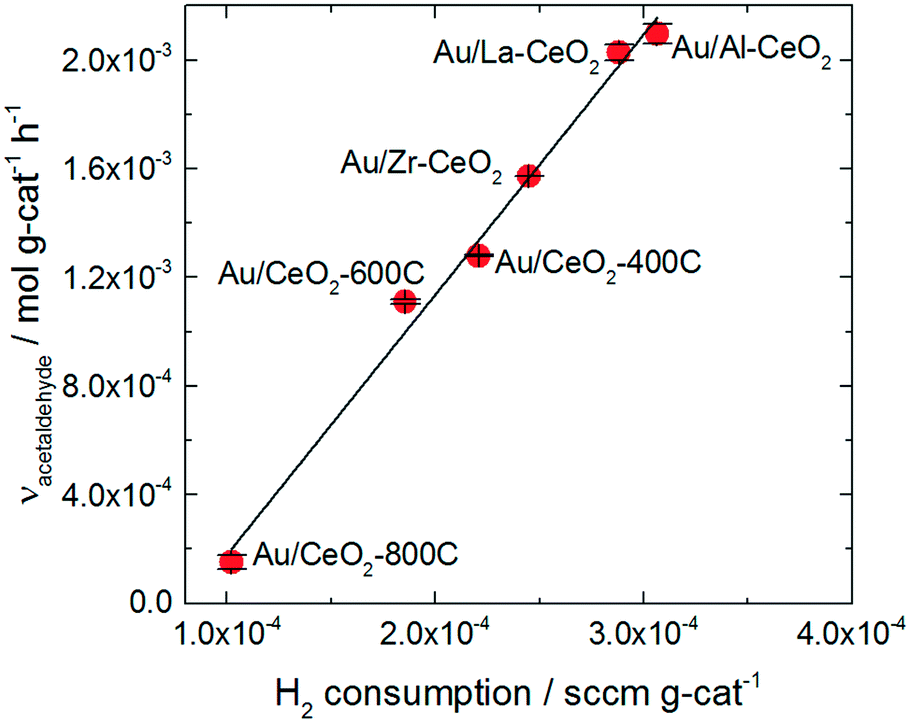 | ||
| Fig. 5 Specific H2 consumption from TPR curves for the Au/CeO2 catalysts versus acetaldehyde activity exhibited by the catalysts. | ||
H2 consumption and specific surface area are also related to one another as reduction of the ceria occurs via formation of oxygen vacancies at the surface of the particles; therefore, higher surface area materials can support greater extents of reduction. A correlation between the specific surface area of each catalyst and its activity for acetaldehyde production was also observed as shown in the ESI.† However, the H2 consumption values correlate more closely to the ethanol oxidation activity and represent a better fit of the data.
The oxygen storage capacity of each catalyst served as an excellent descriptor of activity for acetaldehyde production. Clearly, oxygen activation is an important step in the ethanol oxidation process, however, hydride transfer from an alkoxide intermediate to oxidic gold has generally been implicated as the rate-determining step for gold-catalyzed alcohol oxidation reactions.66,67 Abad et al. observed oxidic gold in XPS spectra of highly active Au/CeO2 catalyst and suggested that these species were the active sites of the alcohol oxidation reaction.25 Wang et al. also suggested that oxidic gold sites take part in alcohol oxidation reaction, assisting in hydride removal from the alkoxide intermediate.26
Gold significantly influenced the nature of oxygen reduction in H2-TPR spectra for the Au/CeO2 catalysts as shown in Fig. 3, decreasing the temperature of the reduction onset by several hundred degrees. Notably, all of the reduction of the material occurred at lower temperatures when gold was present, suggesting that the influence of gold was ubiquitous for the reduction process. The linear correlation between oxygen storage and ethanol oxidation activity suggests that H2-TPR may be used to “titrate” the active gold species. However, examination of the amount of hydrogen consumed per gold atom in the Au/CeO2 catalysts (as shown in Table 2) suggests that the oxygen reduced during H2-TPR was not likely to have been directly interacting with gold. Between 5 and 15 moles of H were consumed per mole Au for each catalyst. Furthermore, these values were calculated based on the total amount of gold in the catalyst and not on the amount of oxidic gold. The hydrogen consumption for each catalyst per mole of oxidic gold would have been even higher (as some gold was present in Au0 form). It therefore seems likely that H2 spillover occurred to some extent during TPR of the catalysts, causing oxygen not directly interacting with gold to be reduced. The correlation between oxygen storage capacity and catalytic activity suggest that this oxygen may take part in the ethanol oxidation reaction. Alternatively, it is possible that the amount of active gold is proportional to but not directly measured by the oxygen storage capacity.
We note that although oxidic gold has been previously suggested to be associated with the active site,25,66,67 as mentioned in the paragraph above, the measurement of oxidic gold in previous studies was conducted by interpreting ex situ XPS spectra. It is possible that this oxidic gold was converted into metallic gold when exposed to the environment of the reaction. Wang et al. showed via X-ray absorption spectroscopy that such a transformation occurred for ceria–gold catalysts during the WGSR, indicating that the active site was associated with metallic rather than oxidic gold.68 This observation suggested that the correlation between oxidic gold as measured by ex situ XPS analysis and activity for Au/CeO2 catalysts described notably by Fu et al.,22 could not be used to indicate that the active site consisted of oxidic gold. It would be of interest to conduct similar studies for gold-catalyzed alcohol oxidation reactions.
To assess the behavior of the catalysts further, we carried out ethanol oxidation at various temperatures between 70 °C and 90 °C and constructed Arrhenius plots to calculate the apparent activation barriers for the acetaldehyde production process on catalysts that displayed high (Au/La–CeO2), low (Au/CeO2-800C), and intermediate (Au/CeO2-400C) activity. These Arrhenius plots are shown in Fig. 6.
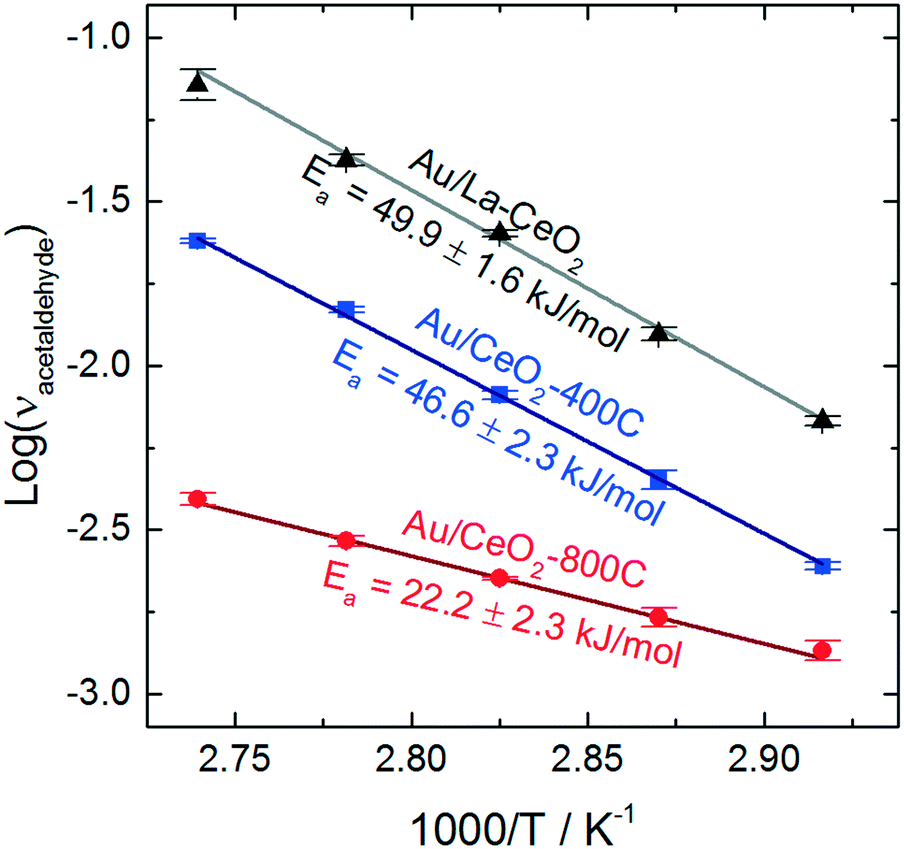 | ||
| Fig. 6 Arrhenius plots for acetaldehyde production on Au/La–CeO2, Au/CeO2-400C, and Au/CeO2-800C. | ||
Au/La–CeO2 and Au/CeO2-400C effectively the same activation barrier (49.9 ± 1.6 kJ mol−1 and 46.6 ± 2.3 kJ mol−1, respectively), suggesting that the nature of the reaction was unchanged for these materials. The shift in activity was due to an increase in the number of active sites rather than a change in the underlying behavior of the reaction, itself. The Au/CeO2-800C catalyst, however, exhibited a significantly lower activation barrier of 22.2 ± 2.3 kJ mol−1, suggesting that the nature of the reaction on this material was different.
Previous studies have investigated the influence of the exposed CeO2 crystal plane on the behavior of ceria-supported catalysts. Wang et al. measured similar activation barriers for benzyl alcohol oxidation with gold catalysts supported on ceria nanorods (24.5 kJ mol−1) and on ceria nanooctahedra (42.3 kJ mol−1).26 The authors of this study suggested that the change in activation barrier was due to a change in the exposed crystal planes of ceria. An enhancement of charge transfer from gold to the CeO2(110) facet lead to stabilization of the hydride during cleavage of the alkoxide intermediate's CH bond in the rate-determining step of the reaction. Previous studies have suggested that heating ceria in air leads to reorganization of crystal planes and the emergence of the (110) facet.62,63 Formation of the CeO2(110) facet upon calcination is a potential explanation for the change in apparent activation energy of the Au/CeO2-800C catalyst. The behavior of this catalyst was similar to that of Au/CeO2 catalysts mentioned above, but we cannot confirm that this change in behavior was caused by the emergence of the CeO2(110) facet.
As the Au/CeO2-800C catalyst exhibited different behavior for the ethanol oxidation reaction, we also compared the correlation between ethanol activity and H2 consumption with the correlation between ethanol activity and surface area after removing the data corresponding to the Au/CeO2-800C sample as shown in the ESI.† These results further illustrate that H2 consumption correlates better with ethanol oxidation activity than surface area, as the difference in the correlation is even more pronounced after removing these outlying data points.
Conclusions
We have demonstrated that the structure of the ceria support material impacts the behavior of Au/CeO2 catalysts for the selective oxidation of ethanol. By heating ceria at various temperatures in air and by introducing dopants to the material during synthesis, we were able to tune the size of the ceria particles, which influenced the interaction between the support surface and gold, affecting the reducibility of the Au/CeO2 catalysts. The activity of the catalysts for selective oxidation of ethanol varied with the size of the ceria support particles, demonstrating an inverse monotonic relationship. A linear correlation between oxygen storage capacity measured from the H2-TPR curves and catalytic activity of the Au/CeO2 materials demonstrates that reducibility can be used as a metric for catalytic activity of ethanol oxidation.Our results also show that doping of metal oxide support materials can effectively enhance the activity of Au/CeO2 catalysts for ethanol oxidation. For the catalysts tested in this study, that enhancement resulted from a decrease in the particle size of the support materials. By changing the structure of the catalysts, doping effectively enhanced their catalytic behavior. Incorporation of dopants into metal oxide supports can result in changes to the support that influence the selectivity of a catalyst, however, doping can also be used as a technique to increase the activity of a catalyst while leaving its selectivity unchanged provided the proper selection of dopants is employed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the generous support of the Department of Energy (DE-SC0018116 [C. B. M.]) and the Welch Foundation (Grants F-1436 [C. B. M.]). G. M. M. and E. J. E. acknowledge the National Science Foundation for a Graduate Research Fellowships. We also acknowledge the Jackson School of Geosciences, UT Austin for supporting the ICP-MS instrument.References
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- G. C. Bond and D. T. Thompson, Catal. Rev.: Sci. Eng., 1999, 41, 319–388 CAS.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsley, Science, 1981, 211, 1121–1125 CAS.
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Pérez, Science, 2007, 318, 1757–1760 CrossRef CAS PubMed.
- M. Shekhar, J. Wang, W.-S. Lee, W. D. Williams, S. M. Kim, E. A. Stach, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Am. Chem. Soc., 2012, 134, 4700–4708 CrossRef CAS PubMed.
- A. Sandoval, A. Gómez-Cortés, R. Zanella, G. Díaz and J. M. Saniger, J. Mol. Catal. A: Chem., 2007, 278, 200–208 CrossRef CAS.
- M. Comotti, W.-C. Li, B. Spliethoff and F. Schüth, J. Am. Chem. Soc., 2006, 128, 917–924 CrossRef CAS PubMed.
- M. Haruta, Catal. Today, 1997, 861, 153–166 CrossRef.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. C. Prince, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef CAS PubMed.
- T. Bunluesin, R. J. Gorte and G. W. Graham, Appl. Catal., B, 1998, 15, 107–114 CrossRef CAS.
- Y. Li and W. Shen, Chem. Soc. Rev., 2014, 43, 1543–1574 RSC.
- D. Zhang, X. Du, L. Shi and R. Gao, Dalton Trans., 2012, 41, 14455 RSC.
- F. Zaera, Chem. Soc. Rev., 2012, 42, 2746–2762 RSC.
- S. Carrettin, P. Concepción, A. Corma, J. M. López Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538–2540 CrossRef CAS PubMed.
- J. B. Park, J. Graciani, J. Evans, D. Stacchiola, S. Ma, P. Liu, A. Nambu, J. F. Sanz, J. Hrbek and J. A. Rodriguez, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4975–4980 CrossRef CAS PubMed.
- R. Si and M. Flytzani-Stephanopoulos, Angew. Chem., 2008, 120, 2926–2929 CrossRef.
- J. A. Rodriguez, X. Wang, P. Liu, W. Wen, J. C. Hanson, J. Hrbek, M. Pérez and J. Evans, Top. Catal., 2007, 44, 73–81 CrossRef CAS.
- J. Guzman, S. Carrettin and A. Corma, J. Am. Chem. Soc., 2005, 127, 3286–3287 CrossRef CAS PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- L.-C. Wang, Y.-M. Liu, M. Chen, Y. Cao, H.-Y. He and K.-N. Fan, J. Phys. Chem. C, 2008, 112, 6981–6987 CAS.
- M. Alhumaimess, Z. Lin, Q. He, L. Lu, N. Dimitratos, N. F. Dummer, M. Conte, S. H. Taylor, J. K. Bartley, C. J. Kiely and G. J. Hutchings, Chem. – Eur. J., 2014, 20, 1701–1710 CrossRef CAS PubMed.
- A. Abad, P. Concepción, A. Corma and H. García, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS PubMed.
- M. Wang, F. Wang, J. Ma, M. Li, Z. Zhang, Y. Wang, X. Zhang and J. Xu, Chem. Commun., 2014, 50, 292–294 RSC.
- Y. Guan, D. A. J. M. Ligthart, Ö. Pirgon-Galin, J. A. Z. Pieterse, R. A. van Santen and E. J. M. Hensen, Top. Catal., 2011, 54, 424–438 CrossRef CAS.
- G. M. Mullen, B. C. Siegert, A. Dolocan, N. R. Miller, B. K. Rosselet, I. Sabzevari and C. B. Mullins, J. Phys. Chem. C, 2017, 121, 19269–19279 CAS.
- R. Zanella, S. Giorgio, C. R. Henry and C. Louis, J. Phys. Chem. B, 2002, 106, 7634–7642 CrossRef CAS.
- M. Pijolat, M. Prin, M. Soustelle, O. Touret and P. Nortier, J. Chem. Soc., Faraday Trans., 1995, 91, 3941 RSC.
- M. Mogensen, N. M. Sammes and G. A. Tompsett, Solid State Ionics, 2000, 129, 63–94 CrossRef CAS.
- S. Deshpande, S. Patil, S. V. Kuchibhatla and S. Seal, Appl. Phys. Lett., 2005, 87, 133113 CrossRef.
- F. Zhang, S.-W. Chan, J. E. Spanier, E. Apak, Q. Jin, R. D. Robinson and I. P. Herman, Appl. Phys. Lett., 2002, 80, 127 CrossRef CAS.
- L. Wu, H. J. Wiesmann, A. R. Moodenbaugh, R. F. Klie, Y. Zhu, D. O. Welch and M. Suenaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 125415 CrossRef.
- D. R. Mullins, S. H. Overbury and D. R. Huntley, Surf. Sci., 1998, 409, 307–319 CrossRef CAS.
- L. Chen, P. Fleming, V. Morris, J. D. Holmes and M. A. Morris, J. Phys. Chem. C, 2010, 114, 12909–12919 CAS.
- J. Xu, J. Harmer, G. Li, T. Chapman, P. Collier, S. Longworth and S. C. Tsang, Chem. Commun., 2010, 46, 1887 RSC.
- C. Paun, O. V. Safonova, J. Szlachetko, P. M. Abdala, M. Nachtegaal, J. Sa, E. Kleymenov, A. Cervellino, F. Krumeich and J. A. van Bokhoven, J. Phys. Chem. C, 2012, 116, 7312–7317 CAS.
- J.-D. Cafun, K. O. Kvashnina, E. Casals, V. F. Puntes and P. Glatzel, ACS Nano, 2013, 7, 10726–10732 CrossRef CAS PubMed.
- F. Le Normand, L. Hilaire, K. Kili, G. Krill and G. Maire, J. Phys. Chem., 1988, 92, 2561–2568 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, 1992, vol. 3 Search PubMed.
- W. Deng, J. De Jesus, H. Saltsburg and M. Flytzani-Stephanopoulos, Appl. Catal., A, 2005, 291, 126–135 CrossRef CAS.
- M. Peuckert, F. P. Coenen and H. P. Bonzel, Surf. Sci., 1984, 141, 515–532 CrossRef CAS.
- T. Dickinson, A. F. Povey and P. M. A. Sherwood, J. Chem. Soc., Faraday Trans., 1975, 71, 298 RSC.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- Q. Fu, A. Weber and M. Flytzani-Stephanopoulos, Catal. Lett., 2001, 77, 87–95 CrossRef CAS.
- N. K. Renuka, N. Harsha and T. Divya, RSC Adv., 2015, 5, 38837–38841 RSC.
- F. Giordano, A. Trovarelli, C. de Leitenburg and M. Giona, J. Catal., 2000, 193, 273–282 CrossRef CAS.
- D. Andreeva, V. Idakiev, T. Tabakova, L. Ilieva, P. Falaras, A. Bourlinos and A. Travlos, Catal. Today, 2002, 72, 51–57 CrossRef CAS.
- Q. Fu, S. Kudriavtseva, H. Saltsburg and M. Flytzani-stephanopoulos, Chem. Eng. J., 2003, 93, 41–53 CrossRef CAS.
- C. Sun and D. Xue, Phys. Chem. Chem. Phys., 2013, 15, 14414 RSC.
- J. Kullgren, K. Hermansson and P. Broqvist, J. Phys. Chem. Lett., 2013, 4, 604–608 CrossRef CAS PubMed.
- X. Huang and M. J. Beck, Chem. Mater., 2015, 27, 5840–5844 CrossRef CAS.
- J. Soria, A. Martínez-Arias and J. C. Conesa, J. Chem. Soc., Faraday Trans., 1995, 91, 1669–1678 RSC.
- Z. Wu, M. Li, J. Howe, H. M. Meyer and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- P. Lakshmanan, F. Averseng, N. Bion, L. Delannoy, J.-M. Tatibouët and C. Louis, Gold Bull., 2013, 46, 233–242 CrossRef.
- A. Aboukaïs, S. Aouad, M. Skaf, S. Hany, M. Labaki, R. Cousin and E. Abi-Aad, Mater. Chem. Phys., 2016, 170, 285–293 CrossRef.
- M.-W. Chang and W.-S. Sheu, Phys. Chem. Chem. Phys., 2016, 18, 15884–15893 RSC.
- Y. Pan, N. Nilius, H.-J. Freund, J. Paier, C. Penschke and J. Sauer, Phys. Rev. Lett., 2013, 111, 206101 CrossRef PubMed.
- A. N. Eryomin, A. V. Abakshonok, V. E. Agabekov and A. A. Kvasyuk, Russ. J. Gen. Chem., 2017, 87, 2358–2368 CrossRef CAS.
- S. Bhagat, N. V. Srikanth Vallabani, V. Shutthanandan, M. Bowden, A. S. Karakoti and S. Singh, J. Colloid Interface Sci., 2018, 513, 831–842 CrossRef CAS PubMed.
- E. Aneggi, C. de Leitenburg, J. Llorca and A. Trovarelli, Catal. Today, 2012, 197, 119–126 CrossRef CAS.
- E. Aneggi, J. Llorca, M. Boaro and A. Trovarelli, J. Catal., 2005, 234, 88–95 CrossRef CAS.
- M. Nolan, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 595, 223–232 CrossRef CAS.
- G. M. Mullen, E. J. Evans, I. Sabzevari, B. E. Long, K. Alhazmi, B. D. Chandler and C. B. Mullins, ACS Catal., 2017, 7, 1216–1226 CrossRef CAS.
- B. Jørgensen, S. E. Christiansen, M. L. D. Thomsen and C. H. Christensen, J. Catal., 2007, 251, 332–337 CrossRef.
- P. Fristrup, L. B. Johansen and C. H. Christensen, Catal. Lett., 2007, 120, 184–190 CrossRef.
- X. Wang, J. A. Rodriguez, J. C. Hanson, M. Pérez and J. Evans, J. Chem. Phys., 2005, 123, 221101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD spectra for CeO2 samples prior to gold deposition, representative STEM images of ceria supports prior to gold deposition and histograms of ceria particle size from STEM image analysis, analysis of Ce 3d XPS spectra for determination of Ce3+ species by method of Le Normand et al., BET surface area versus H2 consumption from TPR for ceria support materials, calculation of the percent reduction of surface capping oxygen during H2-TPR of Au/CeO2 catalysts, ethanol conversion data, Mears' criterion and the Weisz–Prater criterion for each catalyst during ethanol oxidation, correlation between ethanol oxidation activity and surface area and between ethanol oxidation activity and H2 consumption. See DOI: 10.1039/c7re00175d |
| This journal is © The Royal Society of Chemistry 2018 |