
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkylative kinetic resolution of vicinal diols under phase-transfer conditions: a chiral ammonium borinate catalysis†
Martin
Pawliczek
,
Takuya
Hashimoto
 * and
Keiji
Maruoka
*
* and
Keiji
Maruoka
*
Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo, Kyoto, 606-8502, Japan. E-mail: takuya@kuchem.kyoto-u.ac.jp; maruoka@kuchem.kyoto-u.ac.jp
First published on 12th December 2017
Abstract
Herein, we report the first alkylative kinetic resolution of vicinal alcohols realized by cooperative use of a chiral quaternary ammonium salt and an achiral borinic acid. In addition, a catalytic regioselective alkylation of a secondary alcohol in the presence of an unprotected primary one is presented, emphasizing the unique selectivity and potential of this ammonium borinate catalysis.
Introduction
Chiral phase-transfer catalysis has been extensively studied in the past three decades due to its operational simplicity and benign reaction conditions. A large body of work focusing on the development of new chiral catalysts and reactions was reported during this period.1 Despite these continuing progresses, one simple reaction, namely the asymmetric functionalization of unprotected alcohols, has remained intractable in chiral phase-transfer catalysis as a way to provide enantioenriched molecules. It is in contrast with the fact that the functionalization of alcohols has been known to be accelerated in the presence of quaternary ammonium salts long before the emergence of chiral phase-transfer catalysis.2 From our experience in this field,1a,b we surmised that an obstacle to achieve this goal is the formation of a localized, hard alkoxide anion. Compared to a delocalized and flat anion like an enolate,3 an alkoxide is structurally more flexible, rendering the recognition by a chiral cation difficult. The strong basicity of an alkoxide is also a problem since such a strong base is known to degrade the phase-transfer catalyst and is incompatible with base-sensitive substrates.Taking these facts into consideration, we planned to realize an enantioselective functionalization of alcohols by merging the chiral ammonium salt with an additional catalyst which is able to interact directly with an alcohol and modify its properties. In this regard, Taylor et al. recently reported the use of achiral borinic acids for the mono-functionalization of vicinal diols via an intermediary formed borinate (Fig. 1).4,5 In such an anionic species, the alkoxide is structurally fixed and its basicity is attenuated. Deploying a chiral quaternary ammonium salt to pair up with this anionic species is expected to create a chiral environment around the diol, facilitating asymmetric functionalization of the diol (Fig. 1).6
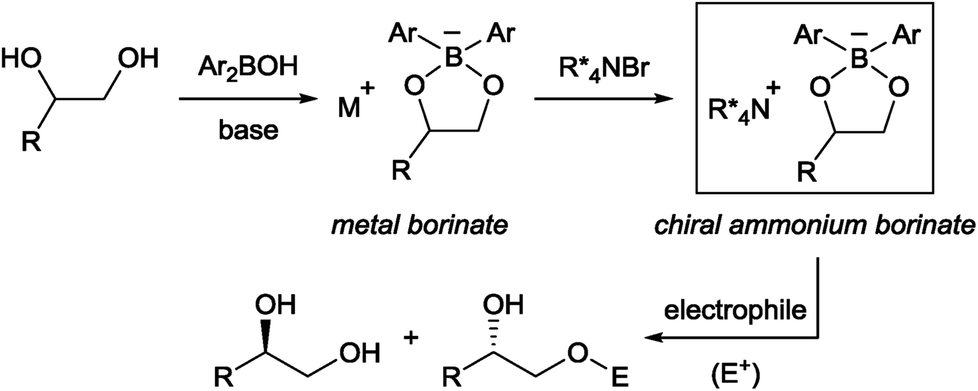 | ||
| Fig. 1 Cooperative catalysis for the asymmetric functionalization of vicinal diols. | ||
We report herein our primary investigations on this subject using an alkylative kinetic resolution of vicinal diols. Despite the fact that alkylation of alcohols directly forges ubiquitous ether moieties,7 such transformation has remained unexplored in asymmetric catalysis.8,9 High enantioselectivities were achieved in the kinetic resolution of secondary as well as tertiary diols by exploiting this chiral ammonium borinate catalysis. The availability of the optimal chiral quaternary ammonium salt, a persistent bottleneck in the binaphthyl-based chiral phase-transfer catalysts,1a,b was enhanced by applying a metal-catalyzed C–H arylation. To further highlight the unique reactivity and selectivity of this catalysis, we also demonstrate a catalyst-controlled, regioselective alkylation of an innately less reactive secondary alcohol of a terminal vicinal diol.
Results and discussion
The first challenge to achieve our goal was the confirmation of reaction conditions to facilitate the alkylation only when both the chiral quaternary ammonium salt and the borinic acid are present. The optimization was implemented by use of (rac)-1-phenyl-1,2-ethanediol 1a and benzyl bromide as reactant in combination with 2 mol% of a simple chiral ammonium salt 4 and 10 mol% of a nitrogen-tethered borinic acid 8 as catalyst pair (Table 1).10 Initially we found out that a biphasic system consisting of CH2Cl2 and aqueous base suppresses independent catalyst activities and ensures enantioselective product formation through a cooperative catalytic pathway (entries 1–3). Screening of a collection of chiral ammonium salts (see ESI†) revealed the effectiveness of catalyst 6 (entries 4 and 5),11 which was further optimized by attaching 3,5-bis(heptafluoroisopropyl)-phenyl substituents (7) (entries 6 and 7).12 A final optimization resulted in the use of 4 mol% of 7, 5 mol% of 8, a CH2Cl2–toluene co-organic solvent system, an excess amount of benzyl bromide, 2 equivalent of Cs2CO3 and NaI as additive. Under these conditions, product 2aa was obtained in 44% yield with 91% ee (entry 8). Interestingly, a considerable amount of side-product 3aa, derived from alkylation of the secondary alcohol, was also observed with the preferential uptake of (R)-1a, indicating the divergent nature of this catalysis.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | PTC | 2aa (% yield, % ee) | 1a (% yield, % ee) | 3aa (% yield, % ee) | |||
| a Performed with (rac)-1a (0.10 mmol), benzyl bromide (0.06 mmol), phase-transfer catalyst (2 mol%) and 8 (10 mol%) in CH2Cl2/H2O (0.35 mL/0.65 mL) at rt. b NMR yield. c ee determined by chiral HPLC. d Performed without 8. e Performed at 0 °C for 48 h. f 7 (4 mol%), 8 (5 mol%), Cs2CO3 (2 eq.) and NaI (5 eq.) in CH2Cl2/toluene/H2O (0.15 mL/0.20 mL/0.65 mL) at 0 °C for 96 h. g Isolated yield. | |||||||
| 1 | 4 | 17 | 21 | 72 | 4 | — | — |
| 2d | 4 | — | — | 97 | 0 | — | — |
| 3 | — | — | — | 97 | 0 | — | — |
| 4 | 5 | 14 | 3 | 78 | 1 | — | — |
| 5 | 6 | 15 | 45 | 75 | 8 | — | — |
| 6e | 6 | 4 | 54 | 90 | 2 | — | — |
| 7e | 7 | 7 | 87 | 86 | 4 | — | — |
| 8f,g | 7 | 44 | 91 | 49 | 74 | 6 | 22 |
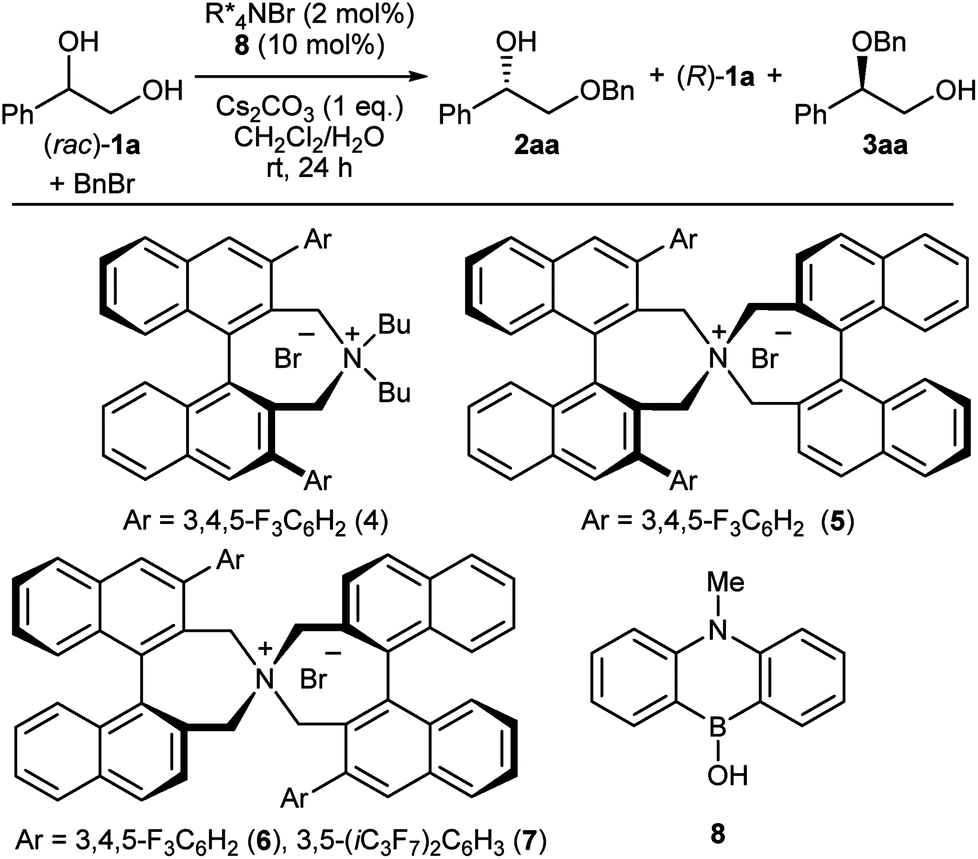
At this stage, we decided to streamline the synthesis of the structurally complex phase-transfer catalyst 7 with the help of the state-of-the-art C–H arylation.13 Starting from readily available 1,1′-binaphthyl-2,2′-dicarboxylic acid,14 direct mono C–H arylation of the 3-position was investigated using several methods. It was revealed that the ruthenium-catalyzed arylation of mono ester 9 with 3,5-(iC3F7)2C6H3I is a robust and scalable procedure (Scheme 1).15,16 Arylated compound 10 was further transformed into dibromide 11 by conventional synthetic methods, which upon treatment with aqueous ammonia yielded the desired catalyst 7.
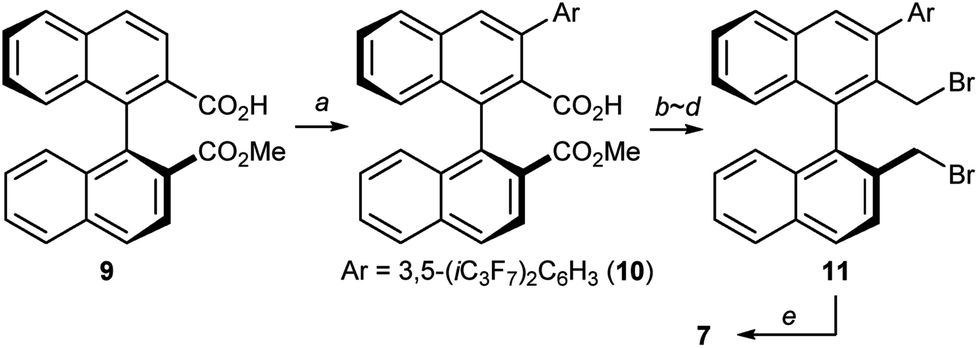 | ||
| Scheme 1 Preparation of catalyst 7. Conditions: (a) [(p-cym)RuCl2]2, Bu3P, 3,5-(iC3F7)2C6H3I, K2CO3, NMP, 110 °C, 78% yield; (b) LiOH aq., THF, reflux; (c) BH3·Me2S, THF, reflux, 86% yield (2 steps); (d) PBr3, THF, rt, 88% yield; (e) NH3 aq., CH3CN, 80 °C, 69% yield. | ||
With the optimized reaction conditions and the secure supply of the catalyst in hand, we investigated the substrate scope with various alkyl halides (Table 2, entries 1–13). As for benzylic bromides, regardless of the substituent pattern on the aromatic ring, the alkylation proceeded with good enantioselectivities at nearly half conversion of the diol (2ab–2aj). The reaction could be scaled up to 1.0 mmol without difficulty as shown in the preparation of 2ad (entry 3). Efficient kinetic resolution took place with prenyl and cinnamyl bromides to afford products 2ak and 2al with high enantioselectivities (entries 10 and 11). The mild reaction conditions allowed the use of base-sensitive methyl 4-bromocrotonate, to give 2am bearing an unsaturated ester (entry 12). The reaction using phenylpropargyl bromide was found to be slow, giving 2an in 36% yield with 90% ee after 6 d (entry 13). In these experiments, we isolated some amount of side-products derived from the alkylation of the secondary alcohol (e.g.3aa in Table 1) in a range from 10% to 30% yield in addition to the recovered (R)-diols 1 (see ESI† for detail).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | R′ | % Yield | % ee | |
| a Performed with 7 (4 mol%), 8 (5 mol%), Cs2CO3 (2 eq.) and NaI (5 eq.). b Isolated yield. c ee determined by chiral HPLC. d Performed with 2 mol% of 7. e Yield and ee in parentheses are the results of 1 mmol scale experiment. | |||||
| 1d | Ph | 2-MeC6H4 | 51 | 87 | 2ab |
| 2 | Ph | 3-MeC6H4 | 46 | 82 | 2ac |
| 3d,e | Ph | 4-MeC6H4 | 48 (48) | 87 (86) | 2ad |
| 4 | Ph | 2-FCC6H4 | 47 | 90 | 2ae |
| 5 | Ph | 2-BrC6H4 | 45 | 85 | 2af |
| 6 | Ph | 2-CNC6H4 | 49 | 87 | 2ag |
| 7 | Ph | 4-PhC6H4 | 46 | 91 | 2ah |
| 8 | Ph | 3,5-(CF3)2C6H4 | 36 | 89 | 2ai |
| 9 | Ph | 2-Naphthyl | 36 | 91 | 2aj |
| 10 | Ph | (Me)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH CH |
40 | 89 | 2ak |
| 11 | Ph | (E)-PhCHCH |
39 | 84 | 2al |
| 12 | Ph | (E)-MeO2CHCH |
39 | 80 | 2am |
| 13 | Ph | PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C C |
36 | 90 | 2an |
| 14 | 2-MeC6H4 | Ph | 45 | 83 | 2ba |
| 15 | 3-MeC6H4 | Ph | 40 | 87 | 2ca |
| 16 | 4-MeC6H4 | Ph | 34 | 97 | 2da |
| 17 | 2-FC6H4 | Ph | 42 | 87 | 2ea |
| 18 | 3-FC6H4 | Ph | 40 | 87 | 2fa |
| 19 | 4-FC6H4 | Ph | 44 | 91 | 2ga |
| 20 | 4-BrC6H4 | Ph | 33 | 82 | 2ha |
| 21 | 4-MeOC6H4 | Ph | 46 | 88 | 2ia |
| 22 | Cy | Ph | 38 | 48 | 2ja |
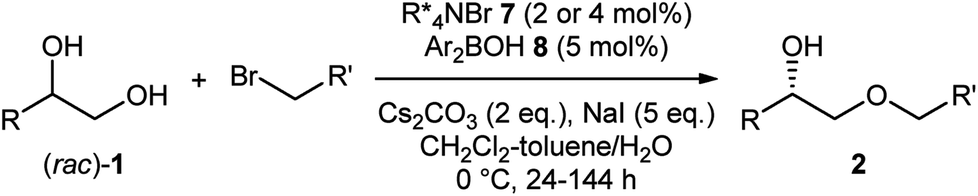
We then turned our attention to the use of various terminal vicinal diols as nucleophile (Table 2, entries 14–22). A variety of aromatic diols were found to be applicable irrespective of the substituent pattern and the electronic property (2ba–2ia). The use of an aliphatic diol gave the benzylated product 2ja with lower enantioselectivity, implying the necessity of catalyst re-optimizations for this class of substrates.
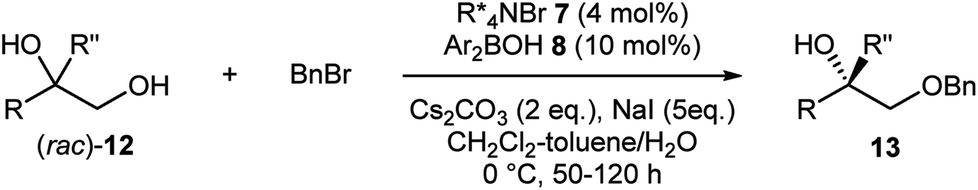
As more challenging substrates, terminal vicinal diols (rac)-12 with a tertiary alcohol moiety were examined under our reaction conditions (Table 3).9g Gratifyingly, exactly the same co-catalytic system worked for substrates having an additional methyl group (entries 1–8). At nearly half conversion, benzylated products 13a–13h were obtained with enantioselectivities ranging around 80% to 90%. Attachment of other alkyl substituents instead of the methyl group led to lower enantioselectivity as exemplified by 13i (entry 9). A general requirement for tertiary alcohols is a higher borinic acid loading and longer reaction time due to the slow turnover of the catalysis.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | R′′ | % Yield | % ee | |
| a Performed with 7 (4 mol%), 8 (10 mol%), Cs2CO3 (2 eq.) and NaI (5 eq.). b Isolated yield. c ee determined by chiral HPLC. d Performed with 15 mol% of 8. | |||||
| 1 | Ph | Me | 47 | 84 | 13a |
| 2 | 2-MeC6H4 | Me | 42 | 82 | 13b |
| 3 | 3-MeC6H4 | Me | 38 | 85 | 13c |
| 4d | 4-MeC6H4 | Me | 41 | 83 | 13d |
| 5 | 4-FC6H4 | Me | 30 | 91 | 13e |
| 6 | 4-MeOC6H4 | Me | 46 | 87 | 13f |
| 7 | 2-Naphthyl | Me | 33 | 78 | 13g |
| 8 | 3-Thienyl | Me | 44 | 72 | 13h |
| 9 | Ph | CH2CHCH2 |
25 | 69 | 13i |
We then carried out NMR experiments of the ammonium borinate prepared from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 ratio of 7, 8 and (rac)-1a, and Cs2CO3 in CDCl3 (see ESI† for detail). The analysis of the 1H and 11B NMRs confirmed the formation of two diastereomeric ammonium borinates favoring the ion pair IPS complexed with (S)-1a over IPR (Fig. 2). Addition of more of (rac)-1a to this solution shifted the ratio of ion pairs within minutes, indicating that these two diastereomeric pairs are in a rapid equilibrium relative to the reaction time frame. This observation suggested that the alkylation, not the ion pairing, is the enantio-determining step. Therefore, the enantioselectivity arguably stems from the reactivity difference of IPS and IPR for alkylation (path a > path c). As a result of deceleration of the reaction via path c, IPR is considered to react eventually at the innately less reactive secondary alcohol to give 3aa.
:1:2 ratio of 7, 8 and (rac)-1a, and Cs2CO3 in CDCl3 (see ESI† for detail). The analysis of the 1H and 11B NMRs confirmed the formation of two diastereomeric ammonium borinates favoring the ion pair IPS complexed with (S)-1a over IPR (Fig. 2). Addition of more of (rac)-1a to this solution shifted the ratio of ion pairs within minutes, indicating that these two diastereomeric pairs are in a rapid equilibrium relative to the reaction time frame. This observation suggested that the alkylation, not the ion pairing, is the enantio-determining step. Therefore, the enantioselectivity arguably stems from the reactivity difference of IPS and IPR for alkylation (path a > path c). As a result of deceleration of the reaction via path c, IPR is considered to react eventually at the innately less reactive secondary alcohol to give 3aa.
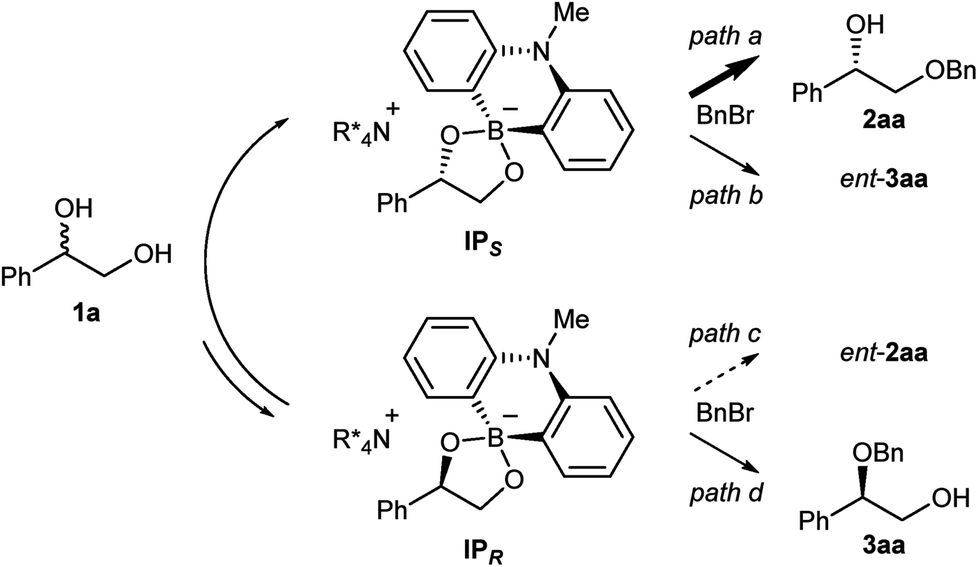 | ||
| Fig. 2 The reaction pathways of the ammonium borinate catalyzed alkylation. | ||
Intrigued by the unique regioselectivity toward secondary alcohol observed with IPR, we decided to take a closer look on this phenomenon (Scheme 2a).6b,17,18 Such a transformation is in contrast to the studies conducted by Taylor et al., in which the borinic acid exclusively functionalized the primary alcohol of a terminal diol.4b It was thus surmised that the judicious choice of an ammonium salt would reveal a novel, catalytic regioselective alkylation of the secondary alcohol of terminal vicinal diols. By analyzing the results of the previous catalyst screening (Table 1 and ESI†), we could quickly identify the optimal catalyst 14 for this purpose. Starting from commercially available (R)-1a with 87% ee, the catalyst controlled, regioselective alkylation proceeded with a ratio of 3.4:1 in favor of the secondary alcohol. A slight increase in enantiomeric excess for the desired product 3aa was observed as well.
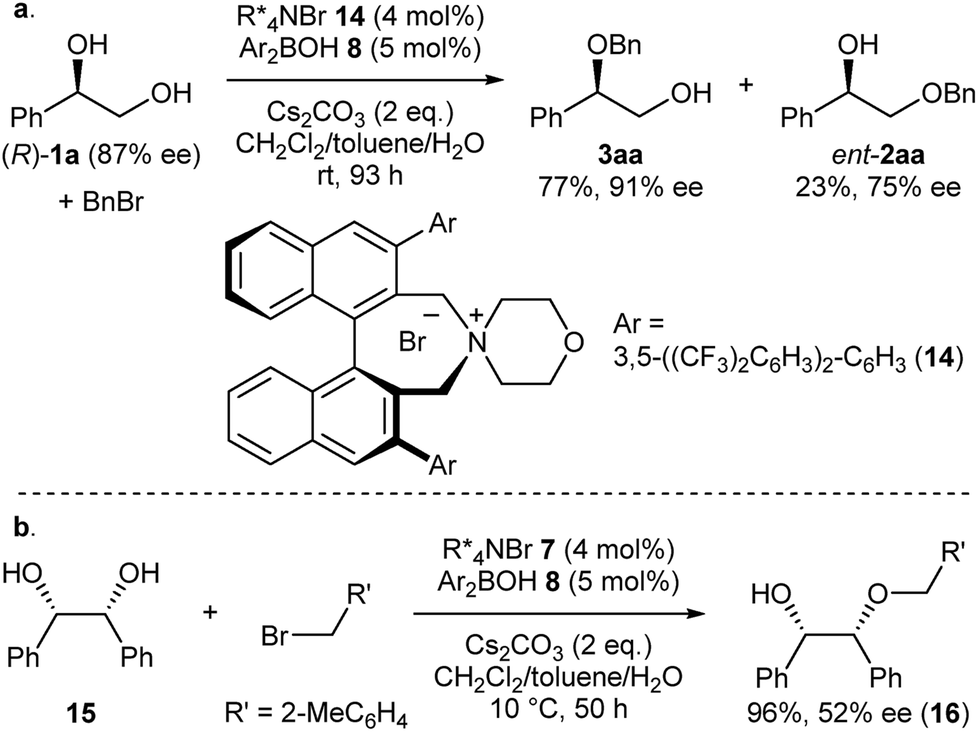 | ||
| Scheme 2 (a) Catalyst-controlled, regioselective alkylation of the secondary alcohol of (R)-1a. (b) Desymmetrization of meso-diol 12. | ||
Finally, we applied our catalyst system to the desymmetrization of a meso-diol (Scheme 2b). Treatment of meso-diol 15 with 2-methylbenzyl bromide gave the desired compound 16 in 96% yield with 52% ee paving the way for further optimizations in the future.
Conclusions
In conclusion, we developed herein the cooperative catalytic system capable of performing asymmetric functionalization of alcohols under phase-transfer conditions as demonstrated by the first alkylative kinetic resolution of secondary and tertiary diols. In addition, this method allowed a catalyst-controlled, regioselective alkylation of the less accessible secondary alcohol in the presence of an unprotected primary one. Our ongoing research is focused on the application of this chiral ammonium borinate catalysis to other electrophiles and more complex diols like saccharides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. P. thanks Deutsche Forschungsgemeinschaft (DFG) for postdoctoral fellowship. K. M. and T. H. thank JSPS KAKENHI Grant Number JP26220803. T. H. thanks JSPS KAKENHI Grant Number JP 16H01021 in Precisely Designed Catalysts with Customized Scaffolding.Notes and references
- (a) T. Hashimoto and K. Maruoka, Chem. Rev., 2007, 107, 5656 CrossRef CAS PubMed; (b) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312 CrossRef CAS PubMed; (c) J. Tan and N. Yasuda, Org. Process Res. Dev., 2015, 19, 1731 CrossRef CAS.
- (a) H. H. Freedman and R. A. Dubois, Tetrahedron Lett., 1975, 16, 3251 CrossRef; (b) J. de la Zerda, G. Barak and Y. Sasson, Tetrahedron, 1989, 45, 1533 CrossRef CAS; (c) Y. Liu, L. Ciszewski, L. Shen and M. Prashad, Org. Process Res. Dev., 2014, 18, 1142 CrossRef CAS.
- For chiral phase-transfer catalyzed O-alkylation of enolates, see; J. D. Jolliffe, R. J. Armstrong and M. D. Smith, Nat. Chem., 2017, 9, 558 CrossRef CAS PubMed.
- (a) L. Chan and M. S. Taylor, Org. Lett., 2011, 13, 3090 CrossRef CAS PubMed; (b) D. Lee, C. L. Williamson, L. Chan and M. S. Taylor, J. Am. Chem. Soc., 2012, 134, 8260 CrossRef CAS PubMed; (c) E. Dimitrijevic and M. S. Taylor, Chem. Sci., 2013, 4, 3298 RSC; (d) K. A. D'Angelo and M. S. Taylor, J. Am. Chem. Soc., 2016, 138, 11058 CrossRef PubMed; (e) K. A. D'Angelo and M. S. Taylor, Chem. Commun., 2017, 53, 5978 RSC.
- S. O. Bajaj, E. U. Sharif, N. G. Akhmedov and G. A. O'Doherty, Chem. Sci., 2014, 5, 2230 RSC.
- The use of a borinic acid toward catalytically-activated prochiral electrophiles was reported during the course of our study, see; (a) R. Z. Li, H. Tang, K. R. Yang, L. Q. Wan, X. Zhang, J. Liu, Z. Fu and D. Niu, Angew. Chem., Int. Ed., 2017, 56, 7213 CrossRef CAS PubMed; (b) R.-Z. Li, H. Tang, L. Wan, X. Zhang, Z. Fu, J. Liu, S. Yang, D. Jia and D. Niu, Chem, 2017, 3, 834 CrossRef CAS.
- For arylation, see; N. Hamaguchi, M. Kuriyama and O. Onomura, Tetrahedron: Asymmetry, 2016, 27, 177 CrossRef CAS.
- For reviews, see; (a) M. D. Díaz-de-Villegas, J. A. Gálvez, R. Badorrey and M. P. López-Ram-de-Víu, Chem.–Eur. J., 2012, 18, 13920 CrossRef PubMed; (b) L. W. Xu, Y. Chen and Y. Lu, Angew. Chem., Int. Ed., 2015, 54, 9456 CrossRef CAS PubMed; (c) A. Borissov, T. Q. Davies, S. R. Ellis, T. A. Fleming, M. S. W. Richardson and D. J. Dixon, Chem. Soc. Rev., 2016, 45, 5474 RSC.
- For selected examples of catalytic kinetic resolutions of vicinal diols, see; acylation (a) S. Kuwano, S. Harada, B. Kang, R. Oriez, Y. Yamaoka, K. Takasu and K. Yamada, J. Am. Chem. Soc., 2013, 135, 11485 CrossRef CAS PubMed; (b) H. Mandai, K. Fujii, H. Yasuhara, K. Abe, K. Mitsudo, T. Korenaga and S. Suga, Nat. Commun., 2016, 7, 11297 CrossRef PubMed; (c) K. Fujii, K. Mitsudo, H. Mandai and S. Suga, Adv. Synth. Catal., 2017, 359, 2778 CrossRef CAS; silylation (d) Y. Zhao, A. W. Mitra, A. H. Hoveyda and M. L. Snapper, Angew. Chem., Int. Ed., 2007, 46, 8471–8474 CrossRef CAS PubMed; (e) N. Manville, H. Alite, F. Haeffner, A. H. Hoveyda and M. L. Snapper, Nat. Chem., 2013, 5, 768 CrossRef CAS PubMed; (f) X. Sun, A. D. Worthy and K. L. Tan, J. Org. Chem., 2013, 78, 10494 CrossRef CAS PubMed; acetalization (g) J. H. Kim, I. Čorić, C. Palumbo and B. List, J. Am. Chem. Soc., 2015, 137, 1778 CrossRef CAS PubMed.
- The use of other reported borinic acids (ref. 4) gave only a trace of the product with less than 10% ee even under optimized reaction conditions.
- T. Kano, Q. Lan, X. Wang and K. Maruoka, Adv. Synth. Catal., 2007, 349, 556 CrossRef CAS.
- (a) J. Guin, C. Rabalakos and B. List, Angew. Chem., Int. Ed., 2012, 51, 8859 CrossRef CAS PubMed; (b) A. Arlt, H. Toyama, K. Takada, T. Hashimoto and K. Maruoka, Chem. Commun., 2017, 53, 4779 RSC.
- P. Nareddy, F. Jordan and M. Szostak, ACS Catal., 2017, 7, 5721 CrossRef CAS.
- (a) T. Ooi, Y. Uematsu and K. Maruoka, J. Org. Chem., 2003, 68, 4576 CrossRef CAS PubMed; (b) T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2007, 129, 10054 CrossRef CAS PubMed.
- A. Biafora, T. Krause, D. Hackenberger, F. Berlitz and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 14752 CrossRef CAS PubMed.
- T. Furuta, M. Nikaido, J. Yamamoto, T. Kuribayashi and T. Kawabata, Synthesis, 2013, 45, 1312 CrossRef CAS.
- For a review, see; N. Funken, Y. Q. Zhang and A. Gansäuer, Chem.–Eur. J., 2017, 23, 19 CrossRef CAS PubMed.
- For catalyst-controlled, regioselective functionalization of vicinal diols, see; (a) J. M. Rodrigo, Y. Zhao, A. H. Hoveyda and M. L. Snapper, Org. Lett., 2011, 13, 3778 CrossRef CAS PubMed; (b) A. D. Worthy, X. Sun and K. L. Tan, J. Am. Chem. Soc., 2012, 134, 7321 CrossRef CAS PubMed; (c) X. Sun, H. Lee, S. Lee and K. L. Tan, Nat. Chem., 2013, 5, 790 CrossRef CAS PubMed; (d) E. Mensah, N. Camasso, W. Kaplan and P. Nagorny, Angew. Chem., Int. Ed., 2013, 52, 12932 CrossRef CAS PubMed; (e) G. Xiao, G. A. Cintron-Rosado, D. A. Glazier, B.-m. Xi, C. Liu, P. Liu and W. Tang, J. Am. Chem. Soc., 2017, 139, 4346 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04854h |
| This journal is © The Royal Society of Chemistry 2018 |