
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceO 2-3-Aminopropyl diazeniumdiolates suppress the progression of highly metastatic triple-negative breast cancer by inhibition of microvesicle formation via nitric oxide-based epigenetic regulation†‡
Fenghua
Kang
a,
Jiayi
Zhu
a,
Jianbing
Wu
a,
Tian
Lv
a,
Hua
Xiang
 b,
Jide
Tian
c,
Yihua
Zhang
*a and
Zhangjian
Huang
*a
b,
Jide
Tian
c,
Yihua
Zhang
*a and
Zhangjian
Huang
*a
aState Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, Center of Drug Discovery, China Pharmaceutical University, Nanjing 210009, China. E-mail: zyhtgd@163.com; zhangjianhuang@cpu.edu.cn
bDepartment of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, Nanjing 210009, China
cDepartment of Molecular and Medical Pharmacology, University of California, Los Angeles, California 90095, USA
First published on 3rd August 2018
Abstract
Currently, there is no effective therapy for the treatment of highly metastatic triple-negative breast cancer (TNBC). Microvesicle (MV) formation is crucial for the metastasis of TNBC. Here we report a novel strategy to inhibit the generation of MVs for the intervention of TNBC. O2-3-Aminopropyl diazeniumdiolates 3a–f are designed and synthesized, which can be activated by lysyloxidase over-expressed in TNBC cells. The most active compound 3f is able to selectively release high levels of NO in TNBC cells, inhibit the cell proliferation, and reduce the adhesion, invasion and migration of TNBC cells in vitro. Furthermore, 3f significantly suppresses the growth and metastasis of implanted TNBC in vivo through attenuating MV formation by an epigenetic modification of miR-203/RAB22A expression in an NO-dependent manner, providing the first evidence of NO donor(s) acting as epigenetic modulators to fight highly metastatic TNBC.
Introduction
Tumor invasion and metastasis remain principal causes for the resultant mortality of patients with cancer, including triple-negative breast cancer (TNBC).1 TNBC is a subtype of breast cancer characterized by its high aggressiveness. Patients with TNBC tend to have distant metastasis and a very poor prognosis due to the lack of efficient targeted therapies.2 A recent study has shown that microvesicles (MVs) shed from the surface of cancer cells are crucial for the invasion and metastasis of cancer cells.3 The tumor-derived MVs can act as messengers to transfer bioactive lipids and proteins, including oncogene products and receptors, from the origin cells to recipient ones in distal sites during the process of metastasis.3 Indeed, the MVs derived from TNBC have been shown to promote tumor invasion and metastasis.4 Thereby, a novel strategy to inhibit or minimize the generation of MVs in TNBC may be valuable for the prevention and treatment of TNBC metastasis. However, unfortunately, there has been no application of a strategy to fight TNBC until now.Nitric oxide (NO) is a gaseous signalling molecule and can modulate the epigenetic behavior of cells.5 Several classes of NO donors inhibit the invasive activity of primary cancer cells by regulation of metalloproteinase (MMP),6N-myc downstream-regulated gene-1 (NDRG1)7 and others in cancer cells. Additionally, some NO donors, such as CAP-NO (S-nitrosocaptoprilnin), prevent hetero-adhesion of cancer cells to vascular endothelial cells by down-regulating the expression of cell adhesion molecules (CAMs) onto the endothelial cells.8 Hence, inhibition of TNBC cell-derived MVs through an NO-based epigenetic mechanism could suppress both the invasion and metastasis of TNBC.
O 2-Protected diazeniumdiolates have unique advantages because a rationally designed protecting group in diazeniumdiolates can be metabolically removed by enzyme(s) highly expressed in cancer cells, to selectively release high levels of NO,9 which can induce apoptosis, and retard the proliferation and metastatic cascades.10
Lysyloxidase (LOX) is a secreted primary amine oxidase and frequently over-expressed in cancer cells, particularly for those with a highly invasive potential, such as TNBC cells.11–13 LOX expression is associated with poor overall survival in breast cancer patients.14 A previous study has revealed that LOX is able to convert the ε-amino group of hydroxylysine and its derivatives into an aldehyde through an oxidative deamination process, accompanied by the production of hydrogen peroxide.11
Accordingly, we hypothesized that O2-(3-aminopropyl) diazeniumdiolates 3a–f could be metabolized by LOX to generate three toxic molecules, i.e. hydrogen peroxide, acrolein and diazeniumdiolate in TNBC cells via an oxidative deamination/β-elimination process (Fig. 1a), and the diazeniumdiolate would subsequently release NO in situ to attenuate the metastasis of TNBC by inhibiting MV formation, and that the NO along with hydrogen peroxide and acrolein may synergistically exert potent anticancer activity.
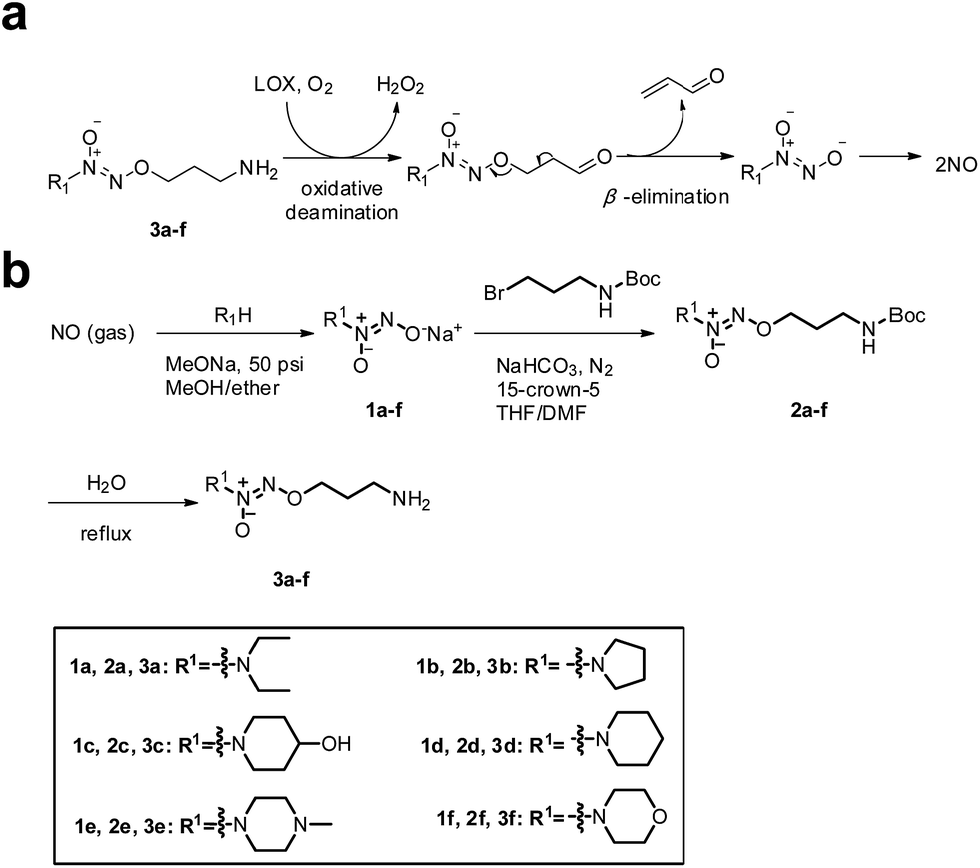 | ||
| Fig. 1 (a) Mechanism of LOX-triggered NO release from 3a–f. (b) The synthetic route to target compounds 3a–f. | ||
Results and discussion
To test the hypothesis, we firstly designed and synthesized compounds 3a–f. The synthetic route to 3a–f is depicted in Fig. 1b. Six secondary amines were allowed to react with NO gas at high pressure (50 psi) in the presence of 30% sodium methoxide in methanol at room temperature as previously reported15 to generate diazeniumdiolate sodium salts 1a–f. The condensation of 1a–f with N-Boc-3-bromopropylamine in a mixed solvent of THF and DMF in the presence of 15-crown-5 gave N-protected O2-(3-aminopropyl)diazeniumdiolates 2a–f, which were subsequently deprotected in boiling water to provide target compounds 3a–f, respectively.Next, we examined the anti-proliferative activity of 3a–f by MTT. Treatment with these compounds significantly inhibited the proliferation of TNBC MDA-MB-231, MDA-MB-436 and MDA-MB-468 cells as well as melanoma B16F10, glioma U251 and prostate PC-3 cells, which express relatively high levels of LOX. Among them, 3f was the most potent (IC50 = 0.68–0.98 μM) against these TNBC cells, especially for MDA-MB-231 cells (0.68 ± 0.10 μM), and was superior to positive control cisplatin (Tables S1 and S2, and Fig. S1–S3‡). Notably, 3f had 27–30 fold less inhibitory activity against poorly invasive breast cancer MCF-7 cells and non-tumor MCF10A cells that had lower levels of LOX expression than highly invasive/metastatic TNBC MDA-MB-231 cells (Fig. S1 and S2, and Table S2‡), suggesting that 3f may selectively inhibit the proliferation of TNBC cells.
The stability of 3f in the absence or presence of LOX was examined by HPLC and Griess assays,16 respectively. There was no detectable NO from 3f cultured in a PBS buffer (pH = 7.4) and rat plasma for 7 h (Fig. S4‡), whereas nearly 50% of the 3f was rapidly metabolized into NO upon addition of LOX for 120 min (Fig. 2a). Actually, 3f released relatively high levels of NO in MDA-MB-231 cells but a little NO in MCF-7 and MCF10A cells (Fig. 2b and S5‡). Furthermore, pretreatment with an LOX inhibitor,17 β-aminopropionitrile (BAPN) potently diminished the amount of NO produced by 3f (Fig. S6b‡). In addition, 3f generated moderate levels of acrolein and hydrogen peroxide (Fig. S8,‡2c and d) in MDA-MB-231 cells, which were mitigated by pre-treatment with BAPN. Collectively, such data indicated that 3f was metabolized by LOX to produce NO, acrolein and hydrogen peroxide in TNBC cells.
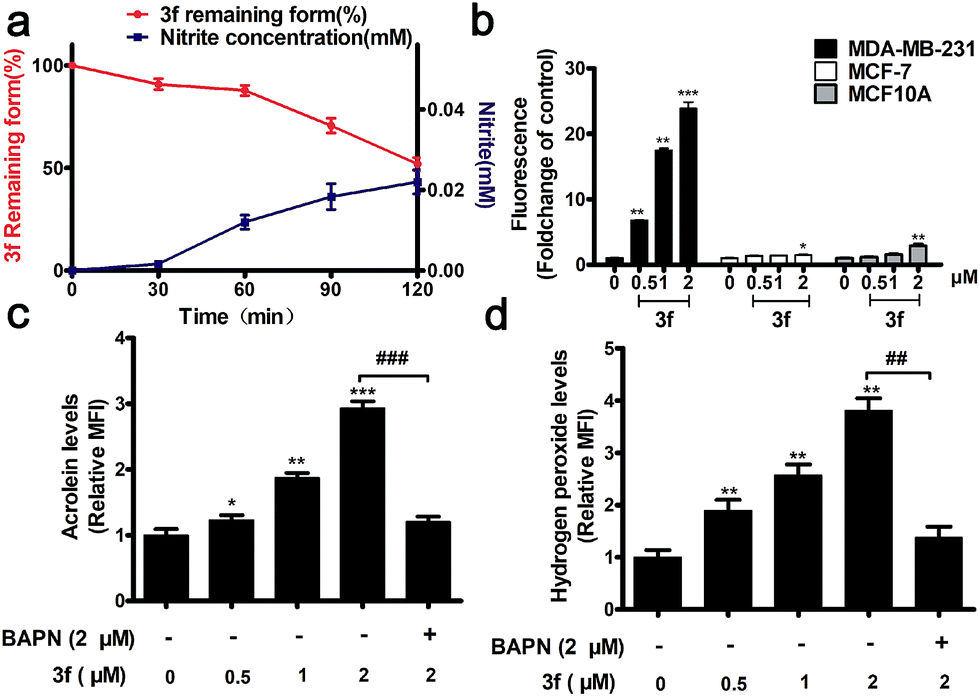 | ||
| Fig. 2 (a) The correlations between NO releasing behaviors (blue) and decomposition (red) of 3f at the initial concentration of 150 μM. (b) NO released from 3f in MDA-MB-231, MCF-7 and MCF10A cells determined by an NO probe (DAF-FM DA). *P < 0.05, **P < 0.01, ***P < 0.001 vs. the control group. (c) 3f dose-dependently promoted acrolein release in MDA-MB-231 cells. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the control group, ###P < 0.001 vs. the 3f + BAPN group. (d) H2O2 released from 3f in MDA-MB-231 determined by an H2O2 probe (BES–H2O2–Ac). **P < 0.01 vs. the control group, ##P < 0.01 vs. the 3f + BAPN group. Data are presented as means ± SD from three independent experiments. | ||
Interestingly, 3f more potently inhibited the proliferation of MDA-MB-231 cells than the NO releasing moiety 1f as well as H2O2 and acrolein alone or any two or three of them in combination at equimolar dose (Table S3‡). In contrast, 3f displayed 20-and 14-fold less inhibitory activity than H2O2, acrolein and 1f in combination at equimolar dose on MCF-7 cells and MCF10A cells with relatively low levels of LOX expression, respectively (Table S4‡). Moreover, pre-treatment with BAPN, carboxy-PTIO (an NO scavenger),18 or N-acetylcysteine (NAC, a ROS scavenger)19 substantially mitigated the anti-proliferative activity of 3f (Fig. S9,‡3a and 3b). These results suggest that the inhibitory activity of 3f might be attributable to the synergistic effects of the three toxic molecules.
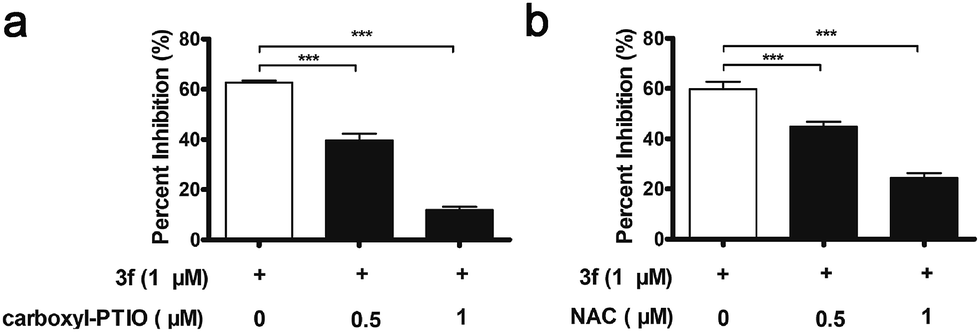 | ||
| Fig. 3 MDA-MB-231 cells were pre-treated with or without an NO scavenger (carboxy-PTIO), or a ROS scavenger (NAC) for 1 h, and then treated with 3f (1 μM) for 72 h. (a) Carboxy-PTIO diminished inhibitory activity of 3f against MDA-MB-231 cells, ***P < 0.001 vs. the control group. (b) NAC diminished inhibitory activity of 3f against MDA-MB-231 cells, ***P < 0.001 vs. the control group. | ||
It is known that NO can react rapidly with superoxide to form peroxynitrite, which acts as an inducer of cytotoxicity and apoptosis.20 Thus, the potency of 3f may also be attributed, at least in part, to the contribution of peroxynitrite. In addition, the IC50 value of 1f on MDA-MB-231 (5.18 μM) was significantly lower than that of H2O2 (26.01 μM) or acrolein (13.25 μM) (Table S3‡). And the amounts of NO released intracellularly were quite well associated with the antiproliferative activity of 3a–f, and the most active compound 3f released the highest levels of NO (Fig. S6a‡). These results suggest that the contribution of NO may be the most important among the three toxic molecules.
Given that the adhesion of tumor cells to vascular endothelial cells is crucial for effective invasion and metastasis,21 we tested the effects of 3f on the adhesion of MDA-MB-231 cells to human umbilical vein endothelial cells (HUVECs) by the fluorescence assay. MDA-MB-231 cells were stained with Rhodamine 123 and co-cultured for 1 h on confluent HUVECs that had been pre-treated with IL-1β22 for 4 h and after being washed, the adhered cells were counted under a fluorescence microscope. Treatment with 3f significantly inhibited the IL-1β-induced adhesion of MDA-MB-231 cells to HUVECs (Fig. 4a). We also observed that treatment with 3f attenuated the TGF-β-induced23 migration (Fig. 4b), invasion (Fig. 4c) and lateral migration (Fig. 4d) of MDA-MB-231 cells in a dose-dependent manner (Fig. S10‡). Thus, 3f inhibited the migration and invasion of TNBC cells in vitro.
 | ||
| Fig. 4 (a) 3f inhibited adhesion of MDA-MB-231 cells to HUVECs: fluorescence microscopy showed MDA-MB-231 cells (green) adhered to the HUVECs. **P < 0.01. (b) 3f inhibited migration of MDA-MB-231 cells. (c) 3f inhibited invasion of MDA-MB-231 cells. (d) 3f inhibited lateral migration of MDA-MB-231 cells. Data are shown as mean ± SD from each group. Scale bars, 100 μm. | ||
Acute toxicity assays revealed that following intravenous treatment with 3f, the LD50 value of 3f was 147.20 mg kg−1 in BALB/c nude mice throughout the 14 day observation (Table S5 and Fig. S11‡). To evaluate the in vivo anti-TNBC efficacy of 3f, individual BALB/c nude mice were inoculated subcutaneously with MDA-MB-231 cells. After the establishment of solid tumors, the mice were randomized and treated intravenously with the vehicle (saline) or 3f at 2, 4, or 8 mg kg−1 every other day for 21 consecutive days. Compared with the vehicle control, treatment with 3f markedly inhibited the tumour growth and reduced the tumor volumes and weights in a dose-dependent manner (Fig. 5a–c). Significantly, treatment with 3f at 8 mg kg−1 inhibited the tumour growth by 76.2% (w/w) and did not affect the survival and body weights in mice (Fig. 5d). Hence, 3f was a relatively safe compound and effectively inhibited the growth of TNBC in vivo.
 | ||
| Fig. 5 Inhibitory effects of 3f on the growth of MDA-MB-231 tumors in vivo. (a) Images of tumours for each group. (b) The volumes of tumors. (c) The weights of tumours. (d): The nude mouse body weights were measured every 2 days. Data are shown as means ± SD from each group of mice (n = 8). ****P < 0.0001 vs. the control group. | ||
We next determined the effect of 3f on the lung metastasis (a common location of distant metastasis) of TNBC in a mouse model. Treatment with 3f every other day significantly reduced the numbers of lung tumor nodules in BALB/c nude mice in a dose-dependent manner at 6 weeks post inoculation (Fig. 6a and c). All of the mice receiving 3f at 2 or 4 mg kg−1 survived with an obvious gain in body weights while only 5 mice in the control group (n = 8) receiving vehicle treatment survived and they significantly lost their body weights (Fig. 6b and d). Such data clearly demonstrated that treatment with 3f inhibited the lung metastasis of TNBC in mice.
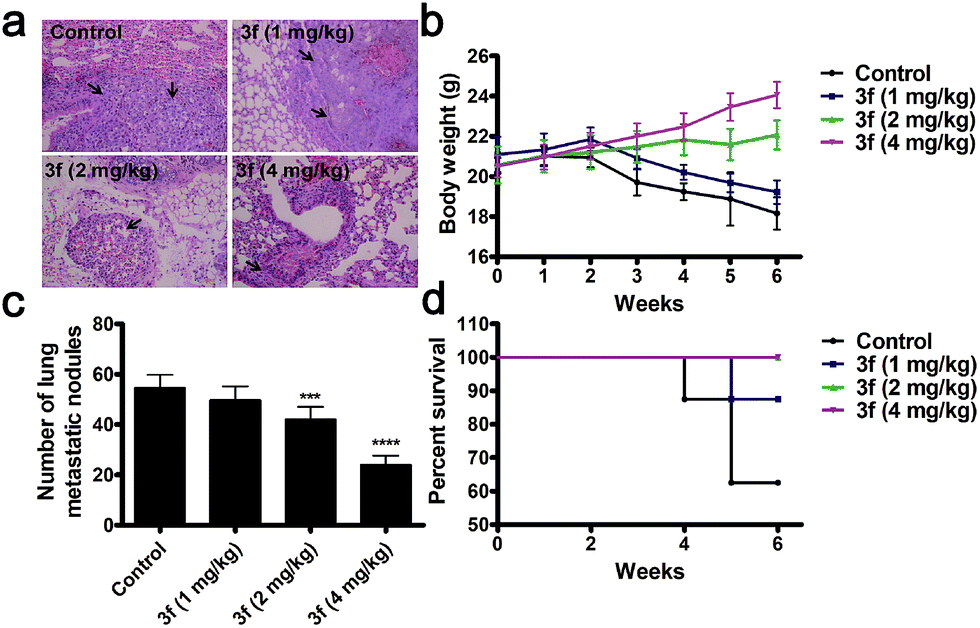 | ||
| Fig. 6 Compound 3f inhibits the lung metastasis of MDA-MB-231 cells in mice. (a) Representative images of lung metastasis from mice in different groups (n = 8) (scale bar: 100 μm). (b) Numbers of lung metastatic nodules from mice in different groups. (c) Body weight. (d) Survival rate. n = 5 in the control group; n = 7 in the 3f (1 mg kg−1) group; n = 8 in the other 2 groups. Values are means ± S.D. ***P < 0.001, ****P < 0.0001 vs. the control group. | ||
To examine whether 3f could inhibit the production of TNBC cell-derived MVs, budding MVs at the MDA-MB-231 cell surface were directly imaged by immunofluorescence. MDA-MB-231 cells were treated with the vehicle or 3f (20 nM) in the presence or absence of 40 nM PTIO for 1 h and stained with FITC-anti-human TGM2 and DAPI. While an obvious fluorescence signal of anti-TGM2 staining was observed in the control cells, a very weak anti-TGM2 signal was detected in the cells treated with 3f. Treatment with PTIO partially rescued the fluorescence signal in MDA-MB-231 cells (Fig. 7a). Given that the focal adhesion (FAs) can stimulate cell motility that is required for the invasion and metastasis of cancer cells,4 we further investigated the effect of 3f on the MV-mediated FAs by immunofluorescence. MDA-MB-231 cells were pre-treated with, or without, carboxy-PTIO for 1 h and then treated with 3f. Their supernatants containing the shed MVs were collected and used to stimulate the formation of FAs in MDA-MB-231 cells. Following staining with Alexa Fluor 568-conjugated phalloidin and FITC-anti-vinculin antibody, we observed lower numbers of FAs in the cells that had been treated with the conditional medium from the 3f-treated cells than those in the control cells, which was completely rescued by pre-treatment with PTIO (Fig. 7b). Quantitative analysis revealed that treatment with 3f significantly reduced the numbers of FAs and their total areas in (Fig. S12‡). These three independent lines of evidence demonstrated that treatment with 3f significantly reduced the formation of MVs, dependent on NO production in MDA-MB-231 cells, contributing to the anti-TNBC activity.
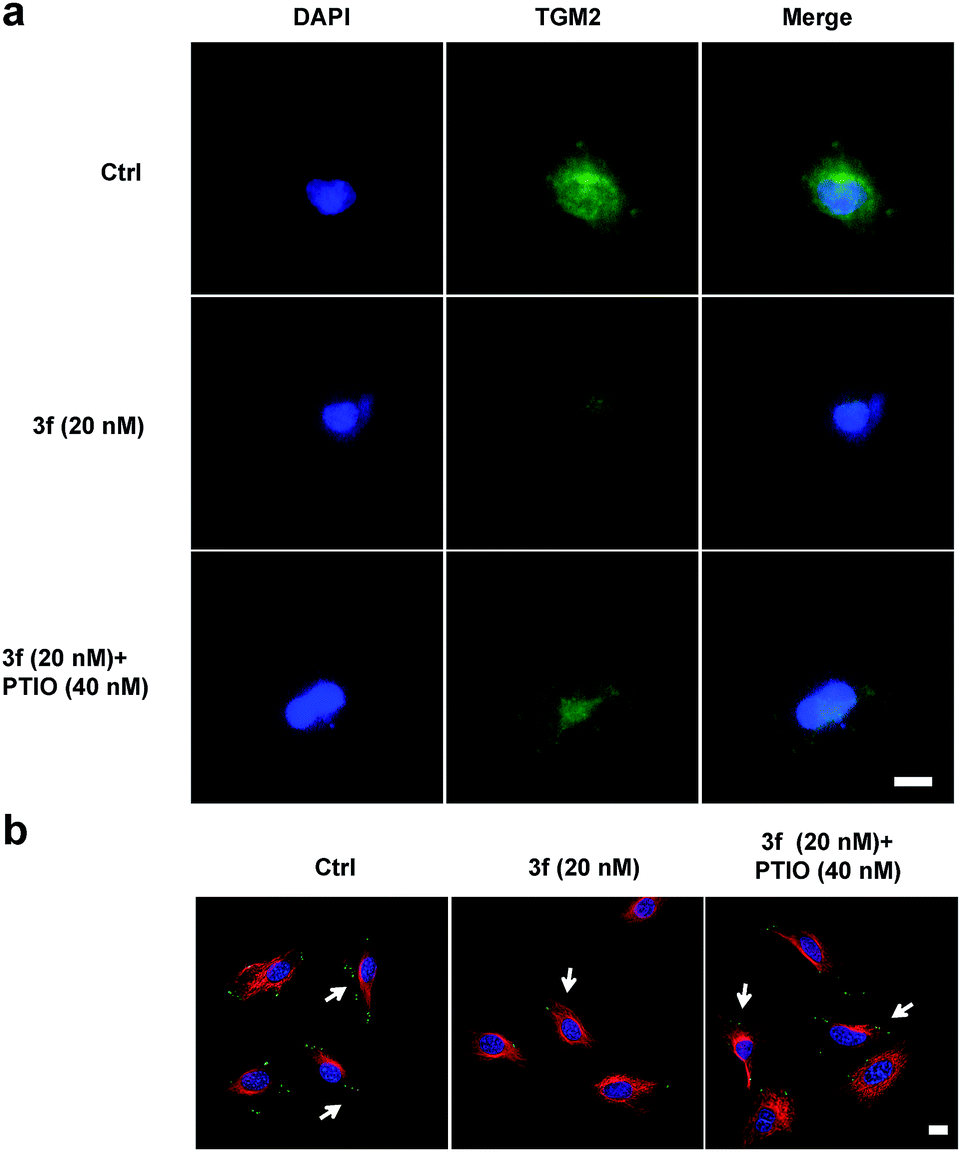 | ||
| Fig. 7 NO released by 3f inhibited TNBC cell-derived MV formation. (a) 3f inhibited formation of MVs: TMG2 and FITC-conjugated goat anti-mouse IgG2a (green) labeled budding MVs on the surface of the cells. DAPI stained the cells to visualize the nucleus (blue). (b) Alexa Fluor 568-conjugated phalloidin detected F-actin stress fibers (red), vinculin antibody detected focal adhesions (FAs; green), and DAPI detected nuclei (blue). FAs were denoted by arrowheads (white). | ||
A previous study has shown that RAB22A is necessary for the shedding of MVs in TNBC cells.4 The gene expression can be regulated by epigenetic modifications, such as DNA methylation and microRNA regulation.24 Indeed, the expression of the RAB22A gene can be down-regulated by microRNA-203 (miR-203), but up-regulated by the miR-203 specific inhibitor or hypoxia (1% O2),4,25,26 Accordingly, the shedding MVs are enhanced by RAB22A, but down-regulated by miR-203 in TNBC cells. It is notable that NO as an epigenetic regulator has been reported to upregulate the expression of miR-203.27 To gain more insights into how NO affects the generation of MVs, we assessed the effect of 3f on the miR-203 and RAB22A expression in MDA-MB-231 cells by quantitative RT-PCR. It was observed that NO produced by 3f treatment increased the relative levels of miR-203 but decreased the levels of RAB22A mRNA transcription in MDA-MB-231 cells (Fig. 8a and b). The modulatory effects of 3f on the levels of miR-203/RAB22A expression were significantly mitigated by pre-treatment with carboxy-PTIO or transfection with the miR-203 inhibitor. It was also observed that transfection with the miR-203 specific inhibitor or culture of cells under an extrinsic hypoxia condition (1% O2) significantly increased the RAB22A transcription. The results were consistent with previous reports.4,26 However, the promoting effects by an extrinsic hypoxic condition (1% O2) on the levels of RAB22A transcription were mitigated by treatment with 3f (Fig. 8a and b). Such results indicated that high levels of NO produced by 3f treatment lowered the RAB22A transcription by increasing miR-203 expression in MDA-MB-231 cells. Subsequently, we determined the numbers of shedding MVs in the supernatants of cultured cells by purifying the MVs using serial centrifugation and nanoparticle tracking analysis. Firstly, transfection with the miR-203 specific inhibitor or culture of cells under an extrinsic hypoxia condition (1% O2) significantly increased the numbers of MVs in the supernatants of cultured MDA-MB-231 cells (Fig. 8c). These results were consistent with a previous report4 and indicated that the inhibition of endogenous miR-203 enhanced the production of MVs in TNBC cells. However, the promoting formation of MVs in the cells cultured under an extrinsic hypoxic condition (1% O2) was significantly mitigated by treatment with 3f. Secondly, treatment with 3f significantly reduced the numbers of MVs in the supernatants of cultured MDA-MB-231 cells, which was almost completely abrogated by pre-treatment with carboxy-PTIO or transfection with the miR-203 specific inhibitor (Fig. 8c). These results suggest that 3f may inhibit the production of MVs in MDA-MB-231 cells by epigenetic modification of the miR-203/RAB22A expression in an NO-dependent manner.
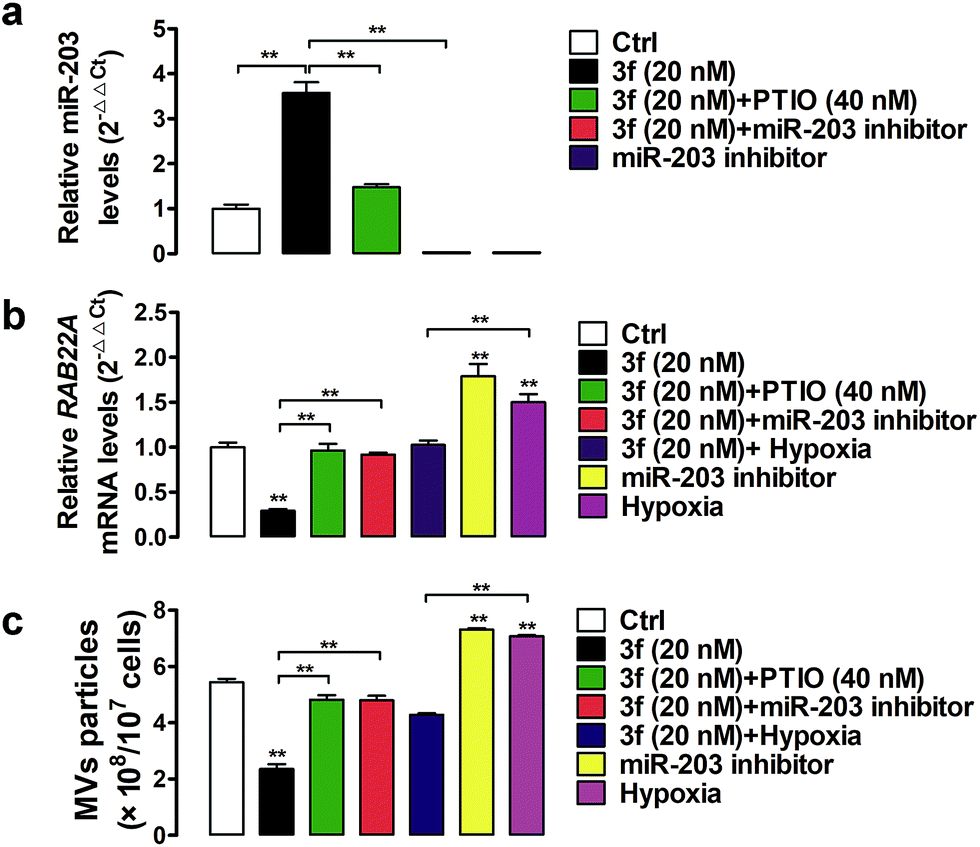 | ||
| Fig. 8 Real-time PCR analysis of miR-203 (a) and RAB22A gene (b) expression in MDA-MB-231 cancer cells treated with the indicated compounds. (c): Shed MVs were isolated, and the number of MV particles per 107 cells was determined by nanoparticle tracking analysis. The data are expressed as means ± SD of three independent experiments. **P < 0.01. | ||
Conclusions
In conclusion, we provide the first evidence of an NO donor acting as an epigenetic modulator of miR-203 and RAB22A expression to inhibit the MV formation, and the proliferation, invasion and metastasis of TNBC. The compounds like 3f may be valuable for the intervention of TNBC or other highly aggressive cancers.Live subject statement
Metastatic triple-negative breast cancer cells (MDA-MB-231, MDA-MB-468 and MDA-MB-436), metastatic melanoma B16F10 cells, metastatic prostate cancer PC-3 cells, metastatic glioma U251 cells, poorly invasive breast cancer MCF-7 cells and human mammary epithelial MCF10A cells were purchased from the American Tissue Culture Collection (ATCC). All animal experimental protocols were approved by the Administration Committee of Experimental Animals in Jiangsu Province and the Ethics Committee of China Pharmaceutical University.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 21372261, 81673305, 21472244 and 81273378) and Jiangsu Province Funds for Distinguished Young Scientists (BK20160033).Notes and references
- D. R. Robinson, Y. M. Wu, R. J. Lonigro, P. Vats, E. Cobain, J. Everett, X. Cao, E. Rabban, C. Kumar-Sinha, V. Raymond, S. Schuetze, A. Alva, J. iddiqui, R. Chugh, F. Worden, M. M. Zalupski, J. Innis, R. J. Mody, S. A. Tomlins, D. Lucas, L. H. Baker, N. Ramnath, A. F. Schott, D. F. Hayes, J. Vijai, K. Offit, E. M. Stoffel, J. S. Roberts, D. C. Smith, L. P. Kunju, M. Talpaz, M. Cieslik and A. M. Chinnaiyan, Nature, 2017, 548, 297 CrossRef PubMed.
- M. Feng, Y. Bao, Z. Li, J. Li, M. Gong, S. Lam, J. Wang, D. M. Marzese, N. Donovan, E. Y. Tan, D. S. Hoon and Q. Yu, J. Clin. Invest., 2014, 124, 5291 CrossRef PubMed.
- A. Becker, B. K. Thakur, J. M. Weiss, H. S. Kim, H. Peinado and D. Lyden, Cancer Cell, 2016, 30, 836 CrossRef PubMed.
- T. Wang, D. M. Gilkes, N. Takano, L. Xiang, W. Luo, C. J. Bishop, P. Chaturvedi, J. J. Green and G. L. Semenza, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3234 CrossRef PubMed.
- D. Vasudevan, R. C. Bovee and D. D. Thomas, Nitric Oxide, 2016, 59, 54 CrossRef PubMed.
- H. Cheng, L. Wang, M. Mollica, A. T. Re, S. Wu and L. Zuo, Cancer Lett., 2014, 353, 1 CrossRef PubMed.
- J. R. Hickok, S. Sahni, Y. Mikhed, M. G. Bonini and D. D. Thomas, J. Biol. Chem., 2011, 286, 41413 CrossRef PubMed.
- Y. Lu, T. Yu, H. Liang, J. Wang, J. Xie, J. Shao, Y. Gao, S. Yu, S. Chen, L. Wang and L. Jia, Sci. Rep., 2014, 4, 4344 CrossRef PubMed.
- L. K. Keefer, ACS Chem. Biol., 2011, 6, 1147 CrossRef PubMed.
- Z. Huang, J. Fu and Y. Zhang, J. Med. Chem., 2017, 60, 7617 CrossRef PubMed.
- J. Finney, H. J. Moon, T. Ronnebaum, M. Lantz and M. Mure, Arch. Biochem. Biophys., 2014, 546, 19 CrossRef PubMed.
- H. E. Barker, T. R. Cox and J. T. Erler, Nat. Rev. Cancer, 2012, 12, 540 CrossRef PubMed.
- D. A. Kirschmann, E. A. Seftor, S. F. Fong, D. R. Nieva, C. M. Sullivan, E. M. Edwards, P. Sommer, K. Csiszar and M. J. Hendrix, Cancer Res., 2002, 62, 4478 Search PubMed.
- J. T. Erler, K. L. Bennewith, M. Nicolau, N. Dornhofer, C. Kong, Q. T. Le, J. T. Chi, S. S. Jeffrey and A. J. Giaccia, Nature, 2006, 440, 1222 CrossRef PubMed.
- J. E. Saavedra, A. Srinivasan, C. L. Bonifant, J. Chu, A. P. Shanklin, J. L. Flippen-Anderson, W. G. Rice, J. A. Turpin, K. M. Davies and L. K. Keefer, J. Org. Chem., 2001, 66, 3090 CrossRef PubMed.
- C. S. Cheung, K. K. Chung, J. C. Lui, C. P. Lau, P. M. Hon, J. Y. Chan, K. P. Fung and S. W. Au, Cancer Lett., 2007, 253, 224 CrossRef PubMed.
- T. R. Cox, A. Gartland and J. T. Erler, Cancer Res., 2016, 76, 188 CrossRef PubMed.
- K. Hirano, A. Hosoi, H. Matsushita, T. Iino, S. Ueha, K. Matsushima, Y. Seto and K. Kakimi, OncoImmunology, 2015, 4, e1019195 CrossRef PubMed.
- I. Downs, J. Liu, T. Y. Aw, P. A. Adegboyega and M. N. Ajuebor, PLoS One, 2012, 7, e38051 CrossRef PubMed.
- A. J. Burke, F. J. Sullivan, F. J. Giles and S. A. Glynn, Carcinogenesis, 2013, 34, 503 CrossRef PubMed.
- C. L. Chaffer and R. A. Weinberg, Science, 2011, 331, 1559 CrossRef PubMed.
- C. Busch, J. Krochmann and U. Drews, PLoS One, 2013, 8, e53970 CrossRef PubMed.
- C. Bai, R. Xue, J. Wu, T. Lv, X. Luo, Y. Huang, Y. Gong, H. Zhang, Y. Zhang and Z. Huang, Chem. Commun., 2017, 53, 5059 RSC.
- B. R. Silverman and J. Shi, Int. J. Mol. Sci., 2016, 17, 2138 CrossRef PubMed.
- D. Yang, G. Liu and K. Wang, PLoS One, 2015, 10, e0132225 CrossRef PubMed.
- F. Su, Y. Chen, S. Zhu, F. Li, S. Zhao, L. Wu, X. Chen and J. Su, Oncotarget, 2016, 7, 71744 Search PubMed.
- W. Li, W. Han, Y. Ma, L. Cui, Y. Tian, Z. Zhou and H. Wang, Free Radical Biol. Med., 2015, 85, 105 CrossRef PubMed.
Footnotes |
| † Dedicated to Professor Sixun Peng on the occasion of his 100th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc00167g |
| This journal is © The Royal Society of Chemistry 2018 |