
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic synthesis of functionalized (polar and non-polar) polyolefin block copolymers†
Dylan J.
Walsh
 ,
Eric
Su
and
Damien
Guironnet
*
,
Eric
Su
and
Damien
Guironnet
*
Department of Chemical and Biomolecular Engineering, University of Illinois Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: guironne@illinois.edu
First published on 9th May 2018
Abstract
Herein, we report a methodology for the synthesis of polyolefin containing block-copolymers using a catalytic postpolymerization modification strategy. The most common polyolefin grades are converted into macroinitiators using a cross-metathesis reaction. These functionalized polyolefins are then used to initiate living: coordinative ring opening polymerization of lactide, anionic ring opening polymerization of epoxide, and radical polymerization of styrene to yield the corresponding block copolymers. The high activity of the catalysts employed in the different steps offers improved practicality for scalable synthesis.
Introduction
Over the past few decades, block copolymers have emerged as a class of soft materials with a wide range of technological applications.1–3 Due to the high tunability of their chemical structure (morphology, architecture, and domain size), block copolymers have been utilized as surfactants, thermoplastic elastomers, nano-templates, membranes, etc.4 Controlled polymerizations such as ionic, controlled radical (ATRP, NMP, RAFT), and ring opening polymerizations have been the standard means for producing block copolymers.1 While these methods have proven successful, they lack the ability to homopolymerize the world's two most produced and inexpensive monomers: ethylene and propylene.5,6Polyolefins today are produced industrially via catalytic insertion (co)polymerization of ethylene, propylene, and linear α-olefins on the scale of 70 × 106 metric tons per year.5,7 The tunability of the polymer's crystallinity offers a mixture of properties such as toughness, elasticity, solvent resistance, etc. which are difficult to reproduce economically by other monomers.6,8 Therefore, the incorporation of polyolefins into block copolymers is of great value, as it would further expand the usefulness of the largest family of polymers in our society. However, due to the high oxophilicity of the insertion metal catalysts used in industry, commercial polyolefin block copolymers have been limited to non-polar monomers, which also limits applications.6,7,9 Hence, the synthesis of functionalized block copolymers containing polyolefins remains a modern challenge for synthetic chemists.
Over the years, alternative approaches have been developed to synthesize functionalized polyolefin block copolymers. Nearly all approaches rely on the formation of (semi-) telechelic polymers which can be used to introduce a polar functionality.10,11 The most popular of these approaches employs the anionic polymerization of butadiene quenched by an epoxide, followed by the hydrogenation, of the polymer to yield a hydroxyl terminated linear low density polyethylene (LLDPE) block.12 This approach is efficient, but is not compatible with the synthesis of polypropylene blocks and uses stoichiometric amounts of a pyrophoric initiator. Ring opening metathesis (ROMP) of cyclic alkenes has also been implemented to yield telechelic high density polyethylene (HDPE).13,14 Despite being successful, employing monomers such as butadiene and cyclic alkenes remains less favorable than the direct polymerization of ethylene and their inability to yield block copolymers containing stereoregular polypropylene (PP) is limiting. Living insertion polymerizations have been reported to produce telechelic polyolefins from ethylene and propylene, however, these systems only produce one polymer chain per metal which drastically limits their commercial potential.15 Chain transfer insertion polymerizations have been reported to yield multiple chains of polymer per catalytic site. However, this approach still requires high loadings of metal(loid) chain transfer agents and also provides low stereocontrol.16–19
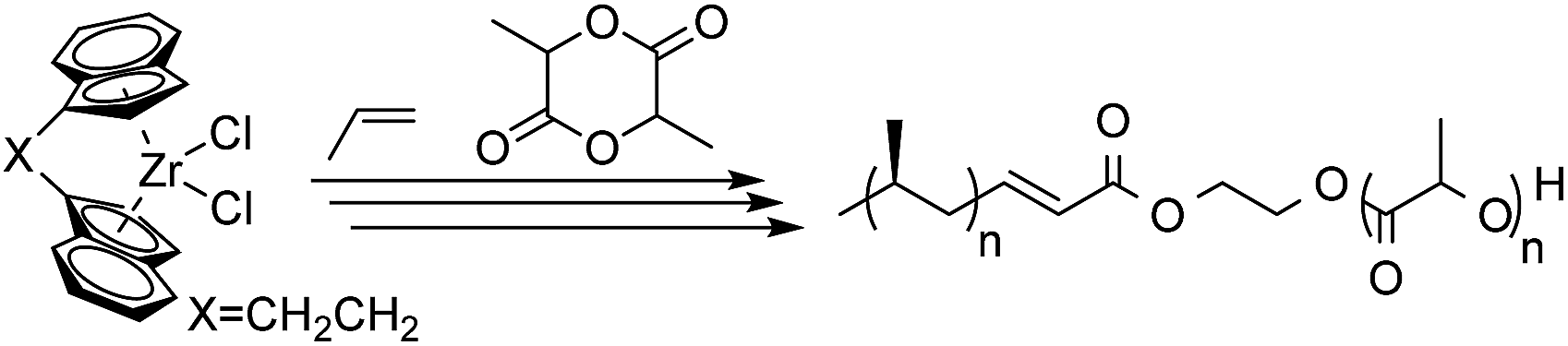
In this work, we develop a general methodology to produce HDPE, PP, LLDPE and HBPE (hyperbranched polyethylene) containing block copolymers (Scheme 1). Our approach consists of performing three consecutive reactions: first a traditional insertion polymerization to yield alkene terminated polyolefins, second a cross metathesis reaction to functionalize the polyolefin end-group, third a controlled living polymerization initiated by the end functionalized polyolefins. Our approach takes advantage of the selectivity, productivity and chemical versatility of the different catalytic reactions employed. In step 1, the catalytic olefin polymerizations yield exclusively olefin terminated polymers through β-X elimination or chain transfer to monomer.20 In step 2, the selectivity of the metathesis reaction between olefins and acrylates quantitatively converts the polyolefins into macroinitiators. In step 3, the living coordinative Ring Opening Polymerization (cROP) of lactide,14 anionic Ring Opening Polymerization (aROP) of epoxide,21 and Atom Transfer Radical Polymerization (ATRP) of styrene22 are employed to quantitatively synthesize the block copolymers from the polyolefin macroinitiators (Scheme 1). Finally, we also demonstrate the advantage of using exclusively catalytic reactions to synthesize olefin containing block copolymers by comparing the overall productivity of our process to previously reported strategies.
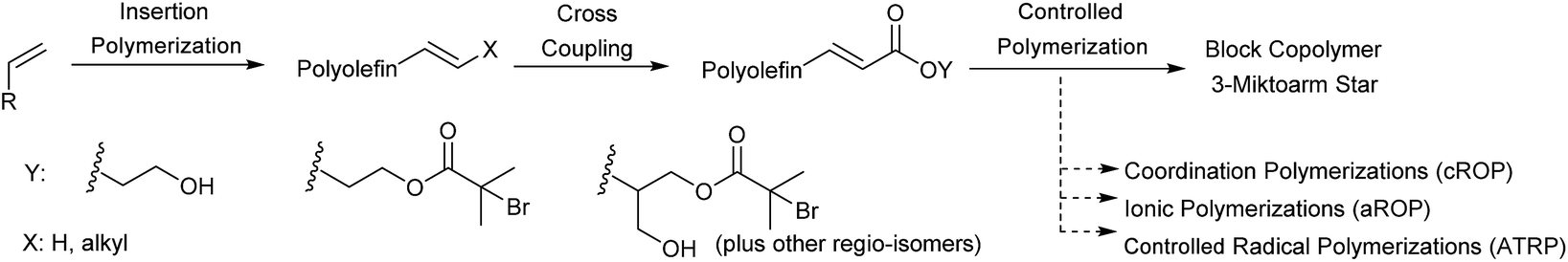 | ||
| Scheme 1 Generalized route for block copolymer synthesis. | ||
Prior to our work, other groups have employed postpolymerization modification techniques to convert vinylic terminated polyolefins into macroinitiators. Hydrosilation,23 thiol-ene,24 hydroalumination25,26 and esterification27 reactions have been applied with moderate to high conversions. It is worth noting that these reactions were only performed on low molecular weight vinylic terminated polyolefins (Mn < 5 kg mol−1). The non-quantitative conversion of most of these reactions and the exclusive reactivity toward the vinyl terminated polymers (which is catalyst dependent and often not the most common end-group) drastically restricts the impact of these previous methods. Additionally, in situ cross coupling compatiblization has been attempted by Duchateau et al. resulting in a process with less than 50% efficiency for producing the desired block copolymer.28 The approach reported here, aims to address these limitations by quantitatively converting mono and di-substituted alkene terminated polyolefins, of any molecular weight into block copolymers.
Results and discussion
A series of single site homogenous insertion catalysts were used to produce a polyolefin library containing HDPE, PP, LLDPE and HBPE (Chart 1).29–32 Two grades of linear polyethylene were synthesized. Low molecular weight linear polyethylene (l-HDPE) with an Mn = 800 g mol−1 was produced from a phosphinosulfonate nickel catalyst,30 and serves as a model system since there is a high concentration of end-groups which eases the characterization of the product by NMR spectroscopy. While a higher molecular weight semi-crystalline HDPE (h-HDPE), Mn = 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, with an industrially relevant melting temperature (Tm = 132 °C) was produced from a phosphinosulfonate palladium complex.29h-HDPE was implemented to show that the methodology is quantitative for various molecular weights and for a variety of polymerization catalysts. A bis(phenoxyimine)titanium dichloride catalyst31 was implemented to synthesize the stereoselective synPP and LLDPE. Finally, a Pd-diimine catalyst32 was used to produce an amorphous polyethylene that we refer to as HBPE, with a comparatively higher molecular weight of Mn = 89000 g mol−1. The molecular structure of this polymer makes it soluble at room temperature in most organic solvents which is advantageous for the characterization of the material as the low concentration of end-group makes it otherwise challenging to quantify conversion.
000 g mol−1, with an industrially relevant melting temperature (Tm = 132 °C) was produced from a phosphinosulfonate palladium complex.29h-HDPE was implemented to show that the methodology is quantitative for various molecular weights and for a variety of polymerization catalysts. A bis(phenoxyimine)titanium dichloride catalyst31 was implemented to synthesize the stereoselective synPP and LLDPE. Finally, a Pd-diimine catalyst32 was used to produce an amorphous polyethylene that we refer to as HBPE, with a comparatively higher molecular weight of Mn = 89000 g mol−1. The molecular structure of this polymer makes it soluble at room temperature in most organic solvents which is advantageous for the characterization of the material as the low concentration of end-group makes it otherwise challenging to quantify conversion.
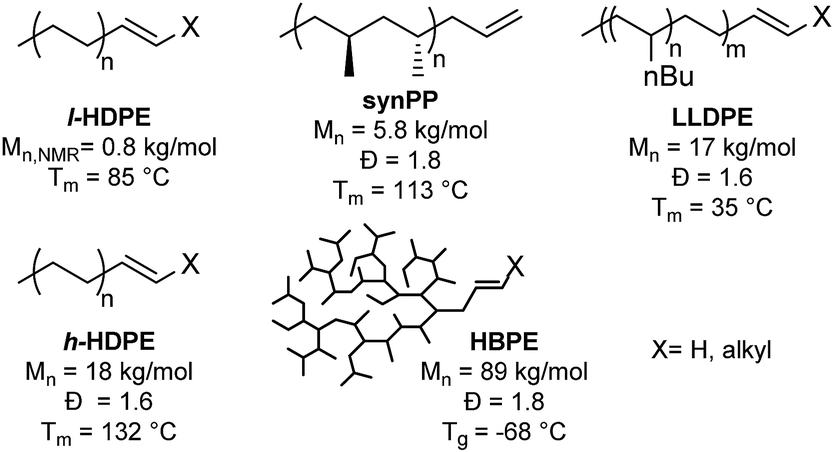 | ||
| Chart 1 Polyolefin library. | ||
As recently illustrated by Mecking et al., implementing a ruthenium catalyzed cross coupling metathesis represents an efficient approach to functionalizing amorphous polyolefins.33 However, in order to implement cross coupling metathesis for the functionalization of the full library of polyolefins, it was necessary to identify reaction conditions compatible with the elevated temperature (120 °C) necessary to solubilize semi-crystalline polyolefins. We found that by adding 1 mol% of Hoveyda–Grubbs II catalyst34 (HG-catalyst) simultaneously with a cross coupling partner, 2-hydroxyethyl acrylate (HEA) or 2-(2-bromoisobutyryloxy) ethyl acrylate (BIEA), over 30 minutes was highly effective for converting l-HDPE end groups. Slow feeding of the olefin metathesis catalyst was necessary to thwart its thermal deactivation. Fig. 1 presents the 1H NMR for l-HDPE, illustrating quantitative conversion (see ESI† Section 9 for sensitivity experiments) of both terminal (5.9 ppm and 5.0 ppm) and internal olefins (5.5 ppm).33
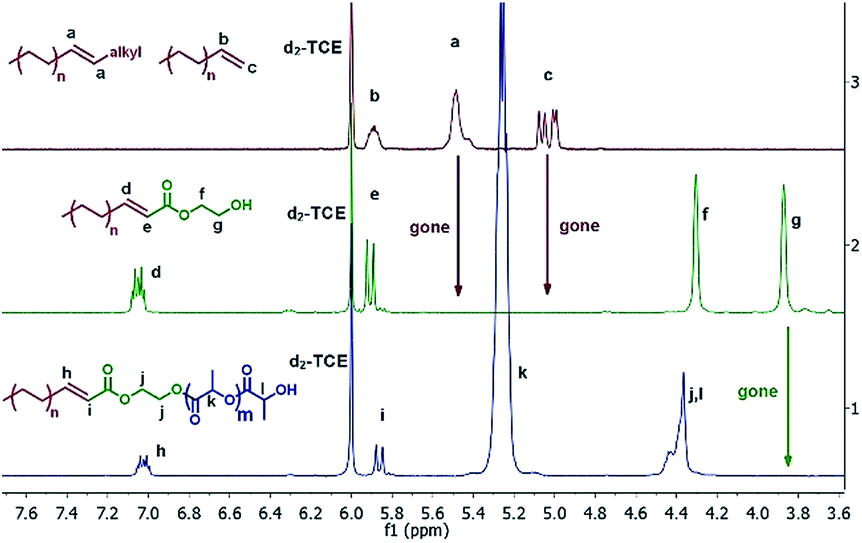 | ||
| Fig. 1 Spectrum of l-HDPE progressing through the methodology. Top: alkene functionalized l-HDPE. Middle: l-HDPE after cross metathesis, Table 1 entry 1-1. Bottom: l-HDPE-b-PLA, Table 2 entry 2-1. | ||
Next, we applied similar reaction conditions to the full library of polyolefins with only minor adjustments to HEA feed procedure depending on the ratio of internal to terminal double bonds present. Quantitative conversions of the polyolefin double bonds were achieved for all cross coupling reactions for all polyolefins as determined by 1H NMR (Table 1). Stacked spectrums for the different polymers can be found in the ESI† (S1–S8). The absence of internal olefinic double bonds in the 1H NMR spectrum at 5.5 ppm confirms that no polyolefin homo-polymer remained (see ESI† Section 8 for homopolymer 1H NMR). The original polyolefins may participate in homo-coupling, but the homo-coupling product will be easily re-activated and will eventually be paired with HEA. Moreover, acrylates are ideal coupling partners because electron deficient olefins are known to be slow at homodimerization.35 This is confirmed by our own experiments when we subjected only HEA to our reaction conditions and did not observe any homo-coupling product formation.
| Entrya | Polyolefin | Acrylate | Conv.b (%) | M n (kg mol−1) | Đ |
|---|---|---|---|---|---|
| a See ESI for conditions. b Determined from 1H NMR of precipitated product. c Determined from GPC or HT-GPC. d Determined by NMR. e Molecular weight too low for GPC analysis. | |||||
| 1-1 | l-HDPE | HEA | Quant | 0.9d | —e |
| 1-2 | h-HDPE | Quant | 19 | 1.5 | |
| 1-3 | synPP | Quant | 5.5 | 1.8 | |
| 1-4 | LLDPE | Quant | 17 | 1.5 | |
| 1-5 | HBPE | Quant | 95 | 1.7 | |
| 1-6 | l-HDPE | BIEA | Quant | 1.1d | —e |
HEA-functionalized polyolefins were used as macroinitiators for cROP of lactide and aROP of tert-butyl glycidyl ether (tBuGE), while BIEA functionalized HDPE was used to initiate the ATRP of styrene (Table 2). The implementation of controlled polymerizations ensures that all polyolefin chains are converted into block copolymers and that no homopolymer is formed. We performed the immortal cROP of lactide on l-HDPE, h-HDPE, synPP, LLPDE, and HBPE (Table 2 entry 2-1 to 2-5). Sub-stoichiometric loadings of Sn(Oct)2 were used to catalyze the cROP. Evidence of complete conversion of the macroinitiator was demonstrated by the total disappearance of the methylene protons adjacent to the hydroxyl group (3.8 ppm) in 1H NMR, Fig. 1.
| Entrya | Block copolymer | Conv.b (%) | f a | M n (kg mol−1) | Đ |
|---|---|---|---|---|---|
| a See ESI for conditions. b Determined from 1H NMR of precipitated product. c Mole fraction of polyolefin. d Determined from GPC or HT-GPC. e Unable to determine due to overlapping signals in the 1H NMR. | |||||
| 2-1 | l-HDPE-PLA | Quant. | 0.54 | 4.2 | 1.4 |
| 2-2 | h-HDPE-PLA | Quant. | 0.47 | 69 | 1.3 |
| 2-3 | synPP-PLA | Quant. | 0.58 | 15 | 1.6 |
| 2-4 | LLDPE-PLA | Quant. | 0.72 | 29 | 1.2 |
| 2-5 | HBPE-PLA | Quant. | 0.84 | 170 | 1.8 |
| 2-6 | l-HDPE-PS | Quant. | 0.46 | 5.0 | 1.1 |
| 2-7 | l-HDPE-P(tBuGE) | —e | 0.29 | 7.5 | 1.2 |
To further showcase the versatility of our approach, we performed ATRP of styrene resulting in quantitative conversion of BIEA functionalized l-HDPE to yield l-HDPE-PS block copolymer (entry 2-6). Additionally, we carried out the immortal aROP of an epoxide monomer by using sub-stoichiometric amounts of KN(TMS)2 (entry 2-7).21 However, to perform this anionic polymerization, it was necessary to reduce the acrylate linker of l-HDPE-HEA into a primary alcohol to avoid side reactions.33
More advanced architectures can be achieved beyond simple diblock copolymers with this methodology. An example of this is a 3-miktoarm star. Star polymers have gained much attention in literature recently for their unique ability to yield highly asymmetric lamellae domains, which are not accessible by linear diblock copolymers.36 By cross coupling a polyolefin to a dual initiating coupling partner, one that contains an alcohol and 2-bromoisobutyrate functionality, both cROP of lactide and ATRP of styrene can be done resulting in a l-HPDE-PLA-PS 3-miktoarm star (Fig. 2). Efficient synthesis of the 3 arm star is demonstrated by the increase in molecular weight (GPC chromatograms in ESI Fig. 57†) after each step.
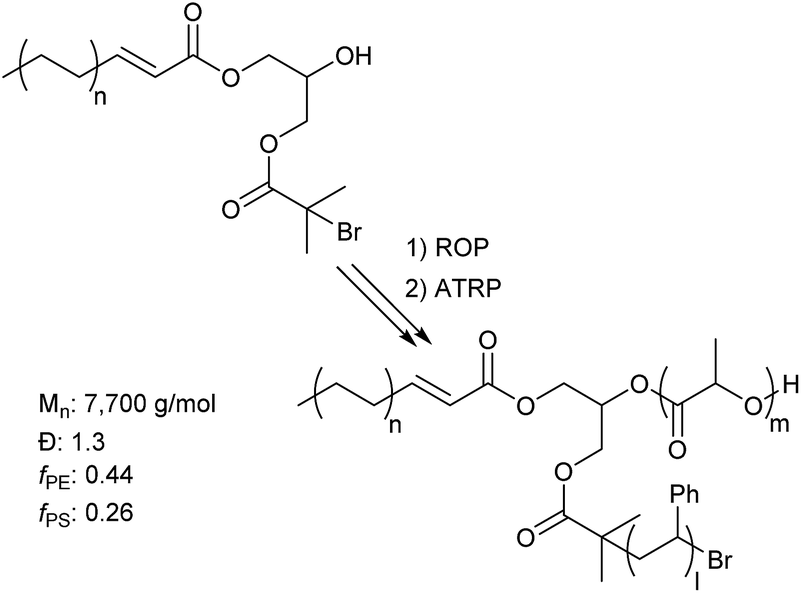 | ||
| Fig. 2 Synthesis of 3-mikoarm star. | ||
In addition to chemical versatility, our approach offers another significant benefit, productivity. The scalability of any material synthesis is a key consideration when thinking about applications.37 In which, one of the main limitations preventing the broader application of block copolymers comes from the overall low productivity of the living polymerization methods. They require using stoichiometric amounts of initiator per polymer chain and/or the use of expensive monomers/initiator. Our system addresses this limitation by implementing catalytic reactions and using industrially relevant monomers. We compared the productivity of our methodology to other published methods for the synthesis of h-HDPE-PLA with a mole fraction of 0.50 and a Mn = 64 kg mol−1 (Table 3). Without significant optimization of the reaction conditions, we were able to achieve an overall productivity of 20000 g of polymer per gram of metal, which is over one order of magnitude more productive than previously reported systems. This productivity could be further improved by applying the methodology to isotactic polypropylene made from a metallocene catalyst.38 Indeed, the high productivity of the catalyst enabled us to achieve an overall productivity of 125000 g of polymer per gram of metal for the synthesis of isoPP-PLA with a mole fraction of 0.50 and a Mn = 71 kg mol−1. To put these numbers into perspective, industrially produced polypropylene is predominantly made with Ziegler–Natta catalysts which produces 600000 grams of polymer per gram of metal.39 However this improvement in productivity comes at the expense of conversion. Indeed the vinylidene terminated polymer chains were unreactive toward the metathesis reaction (30 mol%) resulting in the presence of homopolymer of iPP in the block copolymer.
|
|
|||
|---|---|---|---|
| Polymer | Method | g poly/g cat | g poly/g metal |
| a See ESI for calculations, Section 10. b Mn = 64 kg mol−1, fHDPE = 0.5 (mole frac.). c Contains 20% homopolymer. d Mn = 71 kg mol−1, fPP = 0.5, contains 30% homopolymer. e MgCl2/ester/TiCl4/AlEt3/PhSi(OEt)3. | |||
| h-HDPE-PLAb | Anionic/Pd/AlEt3 (ref. 12) | 4 | 70 |
| ROMP/Pt/Sn(Oct)2 (ref. 14) | 34 | 1900 | |
| CTA/O2/Sn(Oct)2,c (ref. 16) | 50 | 420 | |
| This work | 3200 | 20000 |
|
| isoPP-PLAd | This work | 29000 |
125000 |
| isoPP | 4th Gen. Ziegler Natta,e (ref. 39) | — | 600000 |
Conclusions
In summary, we have developed a facile method to synthesize polyolefin containing block copolymers by implementing three catalytic reactions subsequently. A series of olefin terminated polyolefins were synthesized by catalytic insertion polymerizations. These polymers were then functionalized by cross metathesis reactions to yield polyolefin macroinitiators, which could further be converted into various polyolefin containing block copolymers by three different controlled polymerizations. The absence of homopolymer impurities showcases the high conversion of each step of the transformation. The range of non-polar and polar block-copolymers that are accessible by coordinative polymerizations,40,41 anionic polymerizations,42 and controlled radical polymerization43 make this system a powerful platform to generate a wide range of polyolefin containing materials. The use of highly active catalysts in each step of the process results in an overall process with unprecedented productivity lending itself to potential industrial applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Umicore for the generous gift of Grubbs catalyst and the Department of Chemical and Biomolecular Engineering at the University of Illinois at Urbana−Champaign for funding this research. Major funding for the 500 MHz Bruker CryoProbe was provided by the Roy J. Carver Charitable Trust (Muscatine, Iowa; Grant #15-4521) to the School of Chemical Sciences NMR Lab.Notes and references
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- A.-V. Ruzette and L. Leibler, Nat. Mater., 2005, 4, 19–31 CrossRef CAS PubMed.
- N. Hadjichristidis, S. Pispas and G. Floudas, Block Copolymers: Synthetic Strategies, Physical Properties, and Applications, John Wiley & Sons, Inc., Hoboken, USA, 2002 Search PubMed.
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef PubMed.
- M. C. Baier, M. A. Zuideveld and S. Mecking, Angew. Chem., Int. Ed. Engl., 2014, 53, 9722–9744 CrossRef CAS PubMed.
- J. M. Eagan, J. Xu, R. Di Girolamo, C. M. Thurber, C. W. Macosko, A. M. LaPointe, F. S. Bates and G. W. Coates, Science, 2017, 355, 814–816 CrossRef CAS PubMed.
- C. De Rosa and F. Auriemma, Polym. Chem., 2011, 2, 2155 RSC.
- A. Hotta, E. Cochran, J. Ruokolainen, V. Khanna, G. H. Fredrickson, E. J. Kramer, Y.-W. Shin, F. Shimizu, A. E. Cherian, P. D. Hustad, J. M. Rose and G. W. Coates, Proc. Natl. Acad. Sci., 2006, 103, 15327–15332 CrossRef CAS PubMed.
- Y. Zhao, L. Wang, A. Xiao and H. Yu, Prog. Polym. Sci., 2010, 35, 1195–1216 CrossRef CAS.
- N. M. G. Franssen, J. N. H. Reek and B. de Bruin, Chem. Soc. Rev., 2013, 42, 5809–5832 RSC.
- Y. Wang and M. A. Hillmyer, J. Polym. Sci., Part A-1: Polym. Chem., 2001, 39, 2755–2766 CrossRef CAS.
- L. M. Pitet and M. A. Hillmyer, Macromolecules, 2009, 42, 3674–3680 CrossRef CAS.
- L. M. Pitet, M. A. Amendt and M. A. Hillmyer, J. Am. Chem. Soc., 2010, 132, 8230–8231 CrossRef CAS PubMed.
- K. Zhang, Z. Ye and R. Subramanian, Macromolecules, 2008, 41, 640–649 CrossRef CAS.
- S. K. T. Pillai, W. P. Kretschmer, M. Trebbin, S. Förster and R. Kempe, Chem.–Eur. J., 2012, 18, 13974–13978 CrossRef CAS PubMed.
- R. Briquel, J. Mazzolini, T. Le Bris, O. Boyron, F. Boisson, F. Delolme, F. D'Agosto, C. Boisson and R. Spitz, Angew. Chem., Int. Ed., 2008, 47, 9311–9313 CrossRef CAS PubMed.
- I. German, W. Kelhifi, S. Norsic, C. Boisson and F. D'Agosto, Angew. Chem., Int. Ed., 2013, 52, 3438–3441 CrossRef CAS PubMed.
- A. L. Mogstad and R. M. Waymouth, Macromolecules, 1992, 25, 2282–2284 CrossRef CAS.
- G. Cecchin, G. Morini and F. Piemontesi, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2003, pp. 502–554 Search PubMed.
- M. Erberich, H. Keul and M. Möller, Macromolecules, 2007, 40, 3070–3079 CrossRef CAS.
- Q. Ma and K. L. Wooley, J. Polym. Sci., Part A-1: Polym. Chem., 2000, 38, 4805–4820 CrossRef CAS.
- K. Matyjaszewski, J. Saget, J. Pyun, M. Schlögl and B. Rieger, J. Macromol. Sci. Part A, 2002, 39, 901–913 CrossRef.
- S. Norsic, C. Thomas, F. D'Agosto and C. Boisson, Angew. Chem., Int. Ed., 2015, 54, 4631–4635 CrossRef CAS PubMed.
- H. Kaneko, S. Kojoh, N. Kawahara, S. Matsuo, T. Matsugi and N. Kashiwa, J. Polym. Sci., Part A-1: Polym. Chem., 2005, 43, 5103–5118 CrossRef CAS.
- H. Kaneyoshi and K. Matyjaszewski, J. Appl. Polym. Sci., 2007, 105, 3–13 CrossRef CAS.
- Y. Inoue and K. Matyjaszewski, J. Polym. Sci., Part A-1: Polym. Chem., 2004, 42, 496–504 CrossRef CAS.
- C. Descour, T. Macko, I. Schreur-Piet, M. P. F. Pepels and R. Duchateau, RSC Adv., 2015, 5, 9658–9666 RSC.
- D. Zhang, D. Guironnet, I. Göttker-Schnetmann and S. Mecking, Organometallics, 2009, 28, 4072–4078 CrossRef CAS.
- D. Guironnet, T. Rünzi, I. Göttker-Schnetmann and S. Mecking, Chem. Commun., 2008, 4965–4967 RSC.
- A. E. Cherian, E. B. Lobkovsky and G. W. Coates, Macromolecules, 2005, 38, 6259–6268 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- T. Wiedemann, G. Voit, A. Tchernook, P. Roesle, I. Göttker-Schnetmann and S. Mecking, J. Am. Chem. Soc., 2014, 136, 2078–2085 CrossRef CAS PubMed.
- S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168–8179 CrossRef CAS.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- W. Shi, A. L. Hamilton, K. T. Delaney, G. H. Fredrickson, E. J. Kramer, C. Ntaras, A. Avgeropoulos and N. a. Lynd, J. Am. Chem. Soc., 2015, 137, 6160–6163 CrossRef CAS PubMed.
- T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265–273 CrossRef CAS.
- L. Resconi, F. Piemontesi, I. Camurati, O. Sudmeijer, I. E. Nifant'ev, P. V. Ivchenko and L. G. Kuz'mina, J. Am. Chem. Soc., 1998, 120, 2308–2321 CrossRef CAS.
- R. Mülhaupt, in Ziegler Catalysts, Springer Berlin Heidelberg, Berlin, Heidelberg, 1995, pp. 35–55 Search PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- A. Duda, ROP of Cyclic Esters. Mechanisms of Ionic and Coordination Processes, Polymer Science: A Comprehensive Reference, Elsevier, 2012, vol. 4 Search PubMed.
- S. Penczek and A. Duda, Makromol. Chem., Macromol. Symp., 1993, 67, 15–42 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, GPC chromatogram, DSC traces. See DOI: 10.1039/c8sc00450a |
| This journal is © The Royal Society of Chemistry 2018 |