
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-reporting and refoldable profluorescent single-chain nanoparticles†
Tobias S.
Fischer
ab,
Sebastian
Spann
cd,
Qi
An
e,
Burkhard
Luy
cd,
Manuel
Tsotsalas
ce,
James P.
Blinco
 *af,
Hatice
Mutlu
*ab and
Christopher
Barner-Kowollik
*abf
*af,
Hatice
Mutlu
*ab and
Christopher
Barner-Kowollik
*abf
aMacromolecular Architectures, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstraße 18, 76128 Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu
bSoft Matter Synthesis Laboratory, Institut für Biologische Grenzflächen (IBG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Karlsruhe, Germany. E-mail: hatice.mutlu@kit.edu
cInstitut für Organische Chemie, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany
dInstitut für Biologische Grenzflächen 4 – Magnetische Resonanz, Karlsruher Institut für Technologie (KIT), Postfach 3640, 76021 Karlsruhe, Germany
eInstitut für Funktionelle Grenzflächen, Karlsruhe Institue of Technology (KIT), Herrmann-von Helmholtz-Platz 1, 76344, Eggenstein-Leopoldshafen, Germany
fSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology (QUT), 2 George Street, QLD 4000, Brisbane, Australia. E-mail: christopher.barnerkowollik@qut.edu.au; j.blinco@qut.edu.au
First published on 30th April 2018
Abstract
We pioneer the formation of self-reporting and refoldable profluorescent single-chain nanoparticles (SCNPs) via the light-induced reaction (λmax = 320 nm) of nitroxide radicals with a photo-active crosslinker. Whereas the tethered nitroxide moiety in these polymers fully quenches the luminescence (i.e. fluorescence) of the aromatic backbone, nitroxide trapping of a transient C-radical leads to the corresponding closed shell alkoxyamine thereby restoring luminescence of the folded SCNP. Hence, the polymer in the folded state is capable of emitting light, while in the non-folded state the luminescence is silenced. Under oxidative conditions the initially folded SCNPs unfold, resulting in luminescence switch-off and the reestablishment of the initial precursor polymer. Critically, we show that the luminescence can be repeatedly silenced and reactivated. Importantly, the self-reporting character of the SCNPs was followed by size-exclusion chromatography (SEC), dynamic light scattering (DLS), fluorescence, electron paramagnetic resonance (EPR), nuclear magnetic resonance (NMR) and diffusion ordered NMR spectroscopy (DOSY).
Introduction
Single-chain folded nanoparticles have attracted significant interest over the past years, particularly in the precision polymer synthesis community, due to their unique properties and applications in sensing, catalysis, drug delivery and nano-medicine.1–5 In order to generate complex macromolecular designs, synthetic polymer chemists commonly utilize reversible deactivation radical polymerization (RDRP) protocols to synthesize well-defined polymers. These methods are often combined with orthogonal polymer ligation systems to tether the recognition motifs that induce chain folding to the lateral polymer chain.6,7 Generally, single-chain nanoparticles (SCNPs) are accessible by two pathways: (i) via so-called selective point folding, where the folding motifs are attached at pre-selected points along the polymer chain, which leads to well-defined SCNPS or (ii) via the repeating unit approach, leading to less well-defined SCNP structures, due to the random attachment of the folding units along the polymer chain.8 While the collapse of the parent single polymer chain can be induced either by non-covalent9–15 or dynamic covalent16–20 interactions, a vast majority of the cross-linking strategies developed so far falls in the category of covalent chemistry.6,21,22 For instance, attempting to design SCNPs via radical-coupling chemistry, the groups of Suh, Thayumanavan and Mecerreyes reported the collapse of various polymer functionalities via radical-mediated intramolecular covalent cross-linking.23–25 Specifically, azo-bis(isobutyronitrile) (AIBN) was selected as radical initiator for the intramolecular crosslinking of these polymers. On the other hand, Berda and colleagues recently introduced atom transfer radical coupling (ATRC)26 to single-chain folding technology. Intriguingly, SCNP folding by light-induced processes are relatively new to single-chain technology, with only a few reported examples.27–33 Critically, to the best of our knowledge, there exists no example of SCNPs that exploit the combination of light-induced chemistry and persistent radicals (such as nitroxides) to induce single-chain folded structures. In fact, the nitroxide radicals are known as one of the rare classes of radicals capable of being handled under ambient conditions on the laboratory bench and find applications in diverse fields as cathodes for light-weight organic batteries,34–36 antioxidants,37–39 and catalysts40 among others.41–44 Furthermore, the nitroxide radicals are also the most exploited paramagnetic sensors. Indeed, they form the basis of spin probes and spin labels,45 which are well known in the fields of chemical and biological materials science. Hence, we have also recently reported the synthesis of functional self-reporting switch-on systems, where the fluorescence of a fluorophore is silenced by the unpaired spins of nitroxides and restored by the annihilation of the paramagnetic moiety by reduction to yield a paramagnetic hydroxylamine or by trapping of the radical.46–48Generally, polymers bearing an aromatic backbone are known for their fluorescence properties. Polystyrene (PS) is one less known polymer that in solution and in PS particles displays fluorescence due to monomer fluorescence and π-stacking,49,50 although the aromatic rings in PS are smaller compared to pyrene, for example. Hence, in the current contribution, we exploit the concept of nitroxide containing polymers and their quenching of fluorescence (i.e. luminescence) in PS systems for the preparation of profluorescent single-chain nanoparticles and induce the radical coupling reaction employing a UV responsive bifunctional crosslinker. Unlike noncovalent and dynamic cross-linking chemistry, typical covalent cross-linking delivers static nanoparticles that remain stable and unchanged in response to external stimuli. Importantly, the herein established folding process is fully reversible and self-reporting via luminescence, i.e. the folded state does luminesce, while the luminescence of the exited states in the folded polymers is quenched in the open chain. The thus introduced self-reporting SCNPs that emit information about their folding state constitute a critical step forward as reversibly addressable imaging agents.
Results and discussion
As polymer scaffold, we prepared well-defined random copolymers based on styrene and 4-(chloromethyl)styrene (CMS) with various molar concentrations of the CMS monomer in the copolymer (5–30 mol%; see Scheme 1 and Table 1).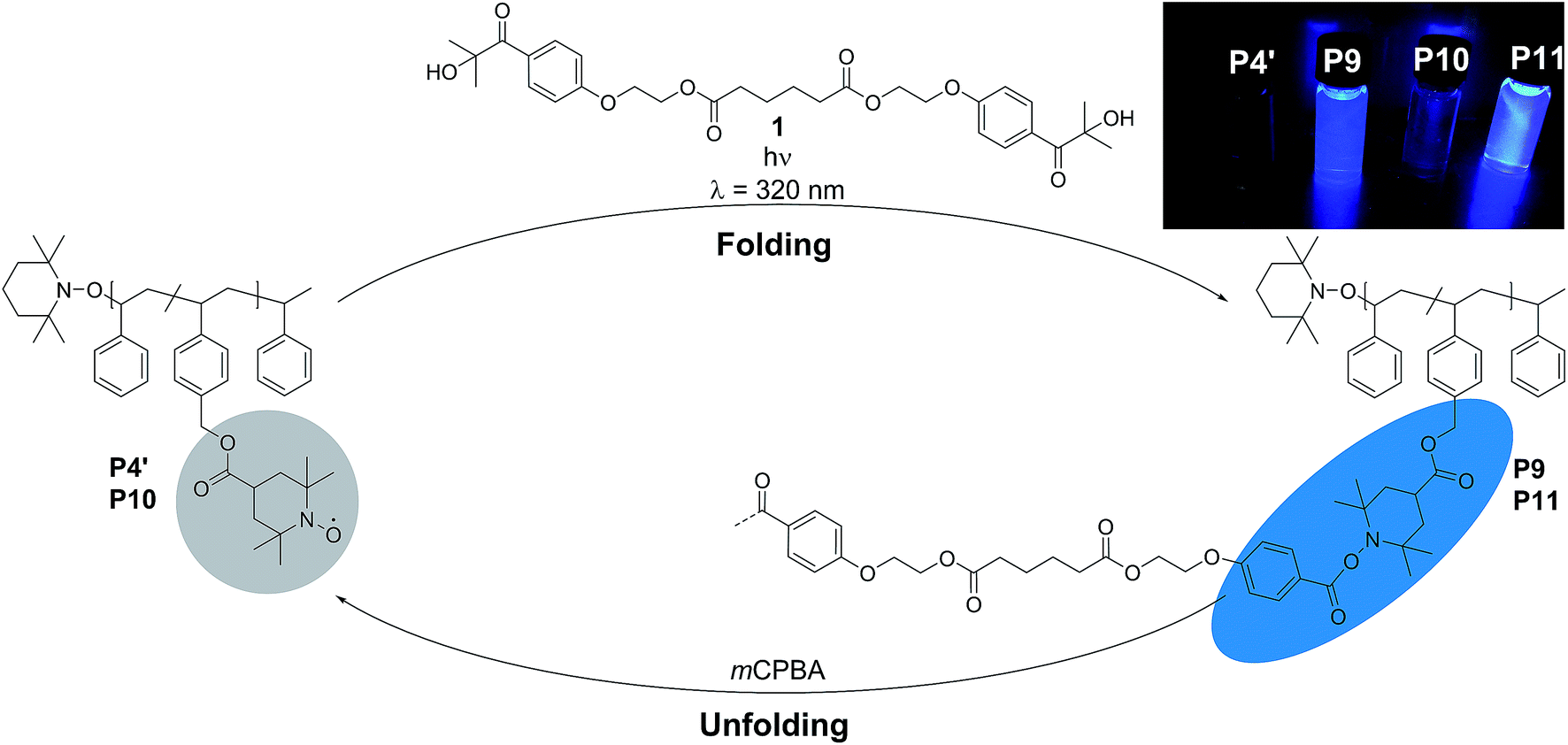 | ||
| Scheme 1 Folding cycle of the parent polymer P4′ with the bifunctional crosslinker 1. The folding is induced by UV-light (λ = 320 nm) resulting in the intramolecularly folded SCNPs P9 and P11. The unfolding of SCNP P9 was induced by mCPBA restoring the initial state (P10). | ||
| ID | M n /g mol−1 | Đ | D h,DLS | D h,DOSY | Density of TEMPOd | 1 (eq.)e |
|---|---|---|---|---|---|---|
| a Determined via SEC in THF (35 °C, 1 mL min−1) as eluent, calibrated with PS standards. b Determined via DLS in DMAc + LiBr (0.3%) as solvent. c Determined via DOSY. d Determined by EPR. e Equivalents were calculated to the corresponding amount of TEMPO. | ||||||
| P4′ | 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
1.3 | 3.0 | 3.4 | 3.89 | — |
| P9 | 18500 |
1.5 | 2.8 | 2.6 | 0.43 | 2 |
| P10 | 33400 |
1.4 | 3.2 | 3.5 | 2.16 | — |
| P11 | 16200 |
1.6 | 2.3 | 2.9 | 0.03 | 2 |
To assess the efficiency of the reaction and to determine the optimum conditions, not only the different molar ratios of CMS, but the chain-length of the copolymer was varied (refer to ESI Table S.1†). The copolymers were prepared by a well-established route in our group, using nitroxide mediated polymerization (NMP)27,39,51 to afford the precursor polymers poly(styrene-stat-4-(chloromethyl)styrene) P1–P4. Subsequently, the copolymers were decorated with 4-carboxy-2,2,6,6-tetramethylpiperidine-1-oxyl (4-carboxy-TEMPO) in a nucleophilic substitution reaction to yield the parent copolymers P1′–P4′ for single-chain folding. Although the information obtained is less comprehensive, we adopted 1H NMR analysis for the nitroxide containing polymers to follow the post-polymerization modification reaction, which results in a characteristic downfield shift of the methylene proton resonance of the CMS group between 4.6 to 5.2 ppm (refer to Fig. S3–S6†). Confirmation of the modification was also achieved via SEC measurements (Fig. S13–S15†), indicating a shift to higher molecular weights after modification. Intriguingly, the PS-stat-CMS based polymer P4 (see Section A.7,† for structural details) displays luminescence (excitation wavelength λ = 240 nm) in the range of about 270–375 nm (refer to the ESI Fig. S24a†), while the luminescence is spin-silenced after the attachment of the nitroxide, yielding the profluorescent parent polymer P4′.
The use of light is a powerful tool to drive reactions, since light is readily available and readily controlled in intensity and wavelength. α-Hydroxyalkyl phenyl ketones undergo a clean Norrish-type I cleavage upon irradiation to produce a benzoyl and the corresponding ketyl radical with high efficiency and quantum yields.52,53 Depending on the substituents in the para position of the aryl group, the cleavage of the C–C bond can occur either from the singlet or the triplet excited state, and the photocleavage can involve either the n–π* or π–π* excited state. Thus, inspired by the photo-induced ligation of nitroxide radicals with the photoinitiator (2-hydroxy-1-(4-(2-hydroxyethoxy)phenyl)-2-methylpropan-1-one) (Irgacure 2959, an α-hydroxyalkyl phenyl ketone derivative or a photoactive PS-based polymer),52,54 we targeted the intramolecular collapse induced by UV-light (λmax = 320 nm) of a nitroxide containing polymer through a novel bifunctional crosslinker 1 equipped with 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959) functionalities on both termini. 1 was prepared in a one-step synthesis by esterification of adipic acid with commercially available Irgacure 2959 (refer to the ESI for the synthesis (Section A.2†) and detailed characterization (Section C.1†)). Folding of P1′–P4′ was initially conducted by employing a stoichiometric amount of 1 (refer to ESI† for the SEC traces of P1′–P4′ and P5–P8, Fig. S17–S20†). After irradiation by UV light, SEC of the polymers P5–P8 shows the characteristic shift towards higher retention times, due to the reduced hydrodynamic volume of the single-chain architectures (Fig. S17–S20†). In addition, the influence of the crosslinker equivalents on the SCNPs formation was investigated with regard to the pendant TEMPO groups, specifically for polymer P4′ (Fig. 1a). Here, we observed a similar behaviour as the Berda team noted in their recent study.55 With increasing amount of crosslinker, a major peak shift towards higher retention times is detected. By employing 1 in large excess (25 eq.), interchain coupling of 1 is witnessed as a side reaction (refer to Fig. 1a, green curve). Thus, once the optimum folding conditions were established, 2 equivalents of 1 were used to affect a pronounced collapse visible in the SEC. As expected, the shift in the SEC chromatogram was detected (refer to Fig. 1b, red curve) resulting from the decrease of the hydrodynamic volume caused by the single-chain collapse. Importantly, the resulting SCNPs display inherent luminescence (excitation wavelength λ = 240 nm) in the range of about 380–550 nm, establishing a self-reporting reversible SCNPs folding system (compare the black and red lines in Fig. 1c). Critically, compared to other reported fluorescent SCNPs,56–59 it is not necessary to install a fluorophore before or after the crosslinking process, since the crosslinking reaction between the styryl TEMPO derivative and the α-hydroxyalkyl phenyl ketones of the linker 1 generates the luminescence. The 1H NMR spectrum of the folded SCNPs P9 displays the characteristic CH2 resonances of 1 at 4.21 and 4.44 ppm (Fig. S10†). In order to underpin the observation of luminescence switch-on (excitation wavelength λ = 240 nm) after the crosslinking reaction, a monofunctional Irgacure 2959 derivative 2 was prepared and reacted with P4′ to yield P9′ (refer to Fig. 2). Interestingly, Jokusch et al. observed phosphorescence for the Irgacure 2959 moiety,60 giving a similar spectrum compared to the recorded luminescence spectra for the SCNPs P9 and P11. Furthermore, small molecule studies reveal no luminescence for the folded polymers P9 and P11. The luminescence is only detectable in case 1 or 2 are attached to the polymer (refer to Fig. 1c, 2b and S22;† for further information for the small molecule study refer to the ESI Section A and C†). The peak at 480 nm is an artefact at double the wavelength that was used for excitation (240 nm), which was proven by the measurement of pure DCM (refer to Fig. S22 in the ESI†). Additional confirmation of the folded nature of the product P9 was obtained by DOSY measurements. Applying the Stokes–Einstein equation (eqn (1) in the ESI†), the hydrodynamic diameter (Dh,DOSY) of the SCNPs of P9 is calculated to be 2.6 nm compared to 3.4 nm of the unfolded state of P4′, which is in agreement with the DLS analysis (refer to Table 1, Fig. 1d, and Section C7–C8 in the ESI† for spectra and further details). The parent polymer and the respective SCNPs were further characterized by EPR in toluene to determine the radical structure as well as spin density of the samples (Fig. 2, black and red lines, respectively). The g value and hyperfine coupling constants (aN) for P4′ (refer to Table S2†) agree with the classic TEMPO-based nitroxide structure.61 Thus, the EPR measurements of the folded sample P9 underpin the almost quantitative reaction of the nitroxide moieties with 1.
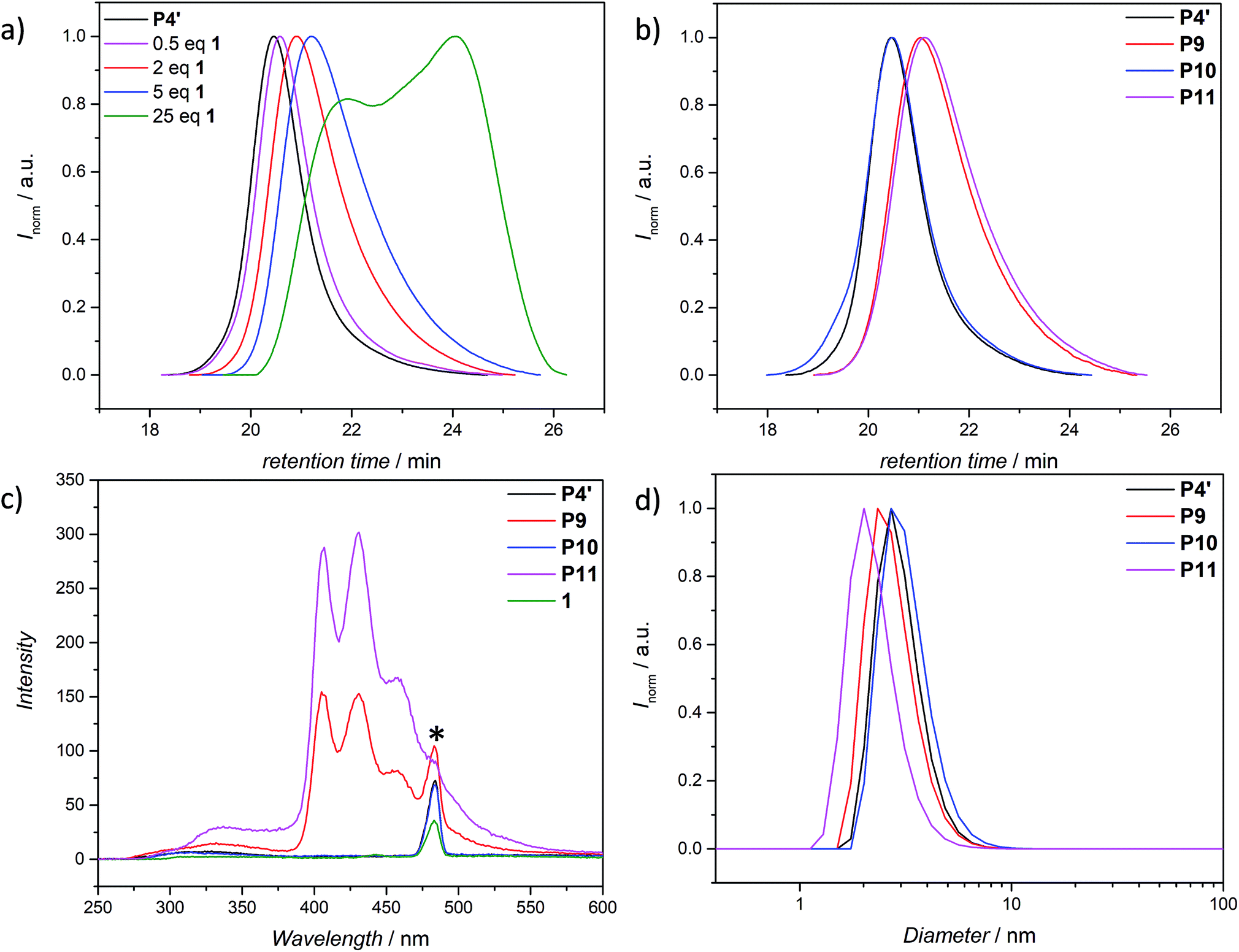 | ||
| Fig. 1 (a) SEC chromatogram of P4′ (black curve) and the SEC curves of the folded polymer P9 as a function of the amount of added crosslinker 1. (b) Folding cycle of the TEMPO polymer P4′. The black curve shows the initial precursor polymer, red curve shows the folded polymer, the blue curve shows the unfolded SCNP followed by the refolding (pink curve). (c) Emission spectra (λexc. = 240 nm) of the polymers P4′, P9–P11 as well as the crosslinker 1, measured in DCM (c = 0.2 mg mL−1). The peak marked with a star is a systematic artefact at double the excited wavelength. (d) Analysis of P4′, P9–P11via dynamic light scattering (DLS) in DMAc + 0.3% LiBr, the average (minimum of 3 measurements per sample) number distributions are shown. | ||
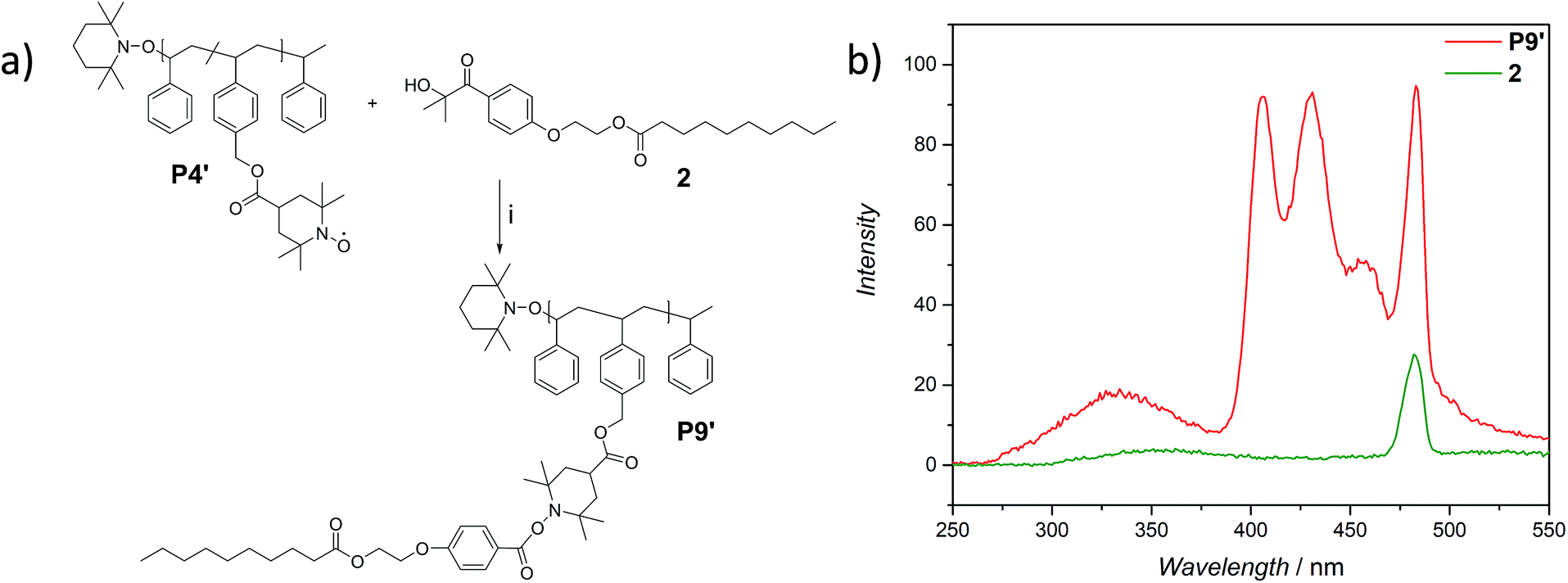 | ||
| Fig. 2 Synthesis of a non-collapsed analogue to P9 (P9′) via the UV-light induced reaction of the precursor polymer P4′ and the photo-active monofunctional crosslinker 2. (a) (i) hν (λmax = 320 nm), ambient temperature, 20 h. (b) Fluorescence spectra of the mono-functional crosslinker 2 and P9′, demonstrating the fluorescent nature of the molecular construct. | ||
Based on the work of Chalmers et al., describing the deprotection of methoxy-protected TEMPO derivatives by meta-chloroperbenzoic acid (mCPBA) affording the hydroxyl amine which is then oxidized to the nitroxide radical, we reactivate the capped TEMPO species P4′ by reoxidation yielding the hydroxylamine moiety, which subsequently reforms the nitroxide (refer to Scheme 1 and Fig. 1b, blue curve, and refer to Section A.11 in the ESI† for the deprotection conditions).62 Gratifyingly, the SEC chromatogram displays an almost identical molecular weight distribution for P10 compared to the original parent polymer P4′. Furthermore, the EPR data for P10 also suggest quantitative unfolding by restoring the initial amount of nitroxides in the polymeric backbone, confirming the results obtained by SEC (compare the values of the radical densities for TEMPO moiety shown in Table 1, and Fig. 3). In addition, the fluorescence spectrum of P10 depicts an identical shape as the unfolded precursor polymer P4′ and no luminescence was monitored (Fig. 1c). Thus, the switch on/off behaviour of the luminescence is perfectly suited to assess the folded state of the SCNPs.
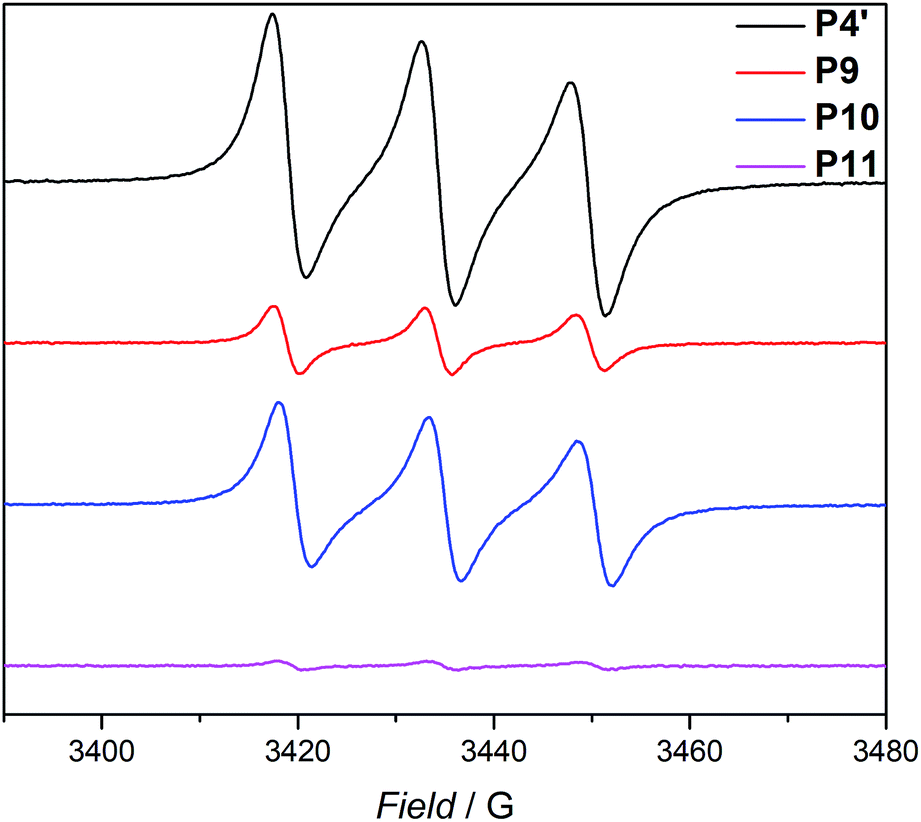 | ||
| Fig. 3 EPR signals for the parent polymer P4′, the folded SCNP P9, the unfolded P10 and refolded SCNP P11. All samples were measured in toluene at ambient temperature. | ||
The dynamic-covalent folding/unfolding is similarly reflected in the DLS derived hydrodynamic diameter (Dh), which reverts back to larger hydrodynamic diameters (i.e., 3.2. nm), indicating the successful unfolding (refer to Fig. 1d, blue curve and Table 1). In addition, the Dh,DOSY derived value of the SCNP for P10 is close to 3.5 nm, which is in agreement with the DLS analysis (Table 1 and Fig. S36–S37†). The 1H NMR spectrum of the unfolded SCNPs P10 (refer to Fig. S9†) shows a decrease of the characteristic CH2 resonances of 1 at 4.21 and 4.44 ppm by close to 82%. These results are congruent with the reported yields for the deprotection of nitroxide moieties by Chalmers et al.62 Hence, not all nitroxide radicals are completely recovered, although the unfolding appears to be complete. We assume that only one functionality of the bifunctional crosslinker is cleaved, while the second functionality is still attached to the polymer. The latter assumption is confirmed by above discussed SEC and DLS results, where slightly higher apparent molecular weight and diameter values were recorded for P10 compared to P4′. In addition, the slight decrease in the signal intensity of P10 in comparison to P4′ observed during the EPR measurements (refer Fig. 3, blue and black lines, respectively) indicate a slightly lower spin density, thus reaffirming the aforementioned assumption of the cleavage of one crosslink site, while the second crosslink is still attached to the nitroxide.
After the SCNPs P9 has been successfully unfolded, we implemented the UV-light induced refolding reaction of 1 with P10 to unambiguously prove the reformation of the nitroxide radicals (refer to Scheme 1). In a similar manner to the initial SCNPs formation studies, 2 equivalents of 1 were used in order to effect a pronounced collapse visible in the SEC. Indeed, the expected shift in the SEC chromatogram was detected (refer to Fig. 1b, pink curve), resulting from the decrease of the hydrodynamic volume caused by the single-chain collapse. Surprisingly, the shift of the refolded SCNP P11 is more pronounced as the initial SCNPs P9. In fact, mCPBA is often used as oxidizing agent for the generation of nitroxides from piperidine derivatives46 as well as deprotection agent as reported by Chalmers et al. Consequently, the unfolding reaction may regenerate some deactivated nitroxide radicals that were not accessible for the first folding reaction as well as restore the crosslinked moieties. The SEC data underpinning the second folding cycle are corroborated by fluorescence and DLS data. The fluorescence spectrum (refer to Fig. 1c) shows higher intensity (pink curve) compared to the initial folded state (red curve), which indicates more crosslinking points are formed, allowing for a more distinct luminescence. The fluorescence quantum yield after the second folding is increased by close to a factor of 150 (refer to Fig. S27 in the ESI†). Importantly, the DLS derived Dh values are also congruent with the above results: the initially folded SCNPs (Fig. 1d, red curve) are less compact than the refolded system (Fig. 1d, pink curve), i.e. Dh going from 2.8 nm to 2.3 nm after the first and second folding, respectively. In addition, the size reduction after the second folding was monitored by DOSY measurements (refer Table 1, Fig. S38–S39†). Similar to DLS and SEC, DOSY provides information about the hydrodynamic radii by measuring the diffusion coefficient, which should typically increase upon folding, thus resulting in a smaller Dh value. Based on the Stokes–Einstein equation, the calculated Dh,DOSY shows the same trend, thus a decreasing Dh from 3.5 to 2.9 nm for the refolded P11 as beforehand accessed by DLS. Finally, by the virtue of the EPR experiments, a very significant decrease in the signal intensity (Fig. 2, pink line) upon the SCNPs folding process is evident, which implies the superior folding of polymer P10 to SCNPs P11.
Conclusions
In summary, we pioneer a refoldable, self-reporting single-chain nanoparticle platform technology based on the photoligation of nitroxide radicals with a novel bifunctional crosslinker bearing a photoreactive functionality. The initially folded, luminescent SCNPs are unfolded under oxidative conditions, resulting in luminescence switch-off and the reestablishment of the initial precursor polymer. Subsequently, the unfolded polymer was refolded by the same photo-induced reaction, resulting in the restoration of the luminescence (refer to Scheme 1 and Fig. 3) and the characteristic molecular and hydrodynamic diameter changes. We submit that the introduced photochemical reversible SCNP folding system is the first to indicate its folded or unfolded state by a simple luminescence readout. The technology presented herein may thus serve as a guiding principle to readout the state of SCNPs in more complex scenarios. Furthermore, the presented principle could be adopted in other fields to distinguish between small molecular (non-luminescent) species and polymeric luminescent structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. B.-K. and B. L. acknowledges funding for this project from the Sonderforschungsbereich 1176 (project A1) funded by the German Research Council (DFG). Additional long term support from the Karlsruhe Institute of Technology (KIT), the Helmholtz association via the BioInterfaces in Technology and Medicine (BIFTM) and the Science and Technology of Nanosystems (STN) programs is acknowledged. C. B.-K. acknowledges continued support from the Queensland University of Technology (QUT) and the Australian Research Council (ARC) in the form of a Laureate fellowship enabling his photochemical research program. The authors acknowledge Prof. M. Meier and Dr P. Levkin (both KIT) respectively for the access to ATR-IR and DLS equipment.References
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2012, 33, 958–971 CrossRef CAS PubMed.
- C. K. Lyon, A. Prasher, A. M. Hanlon, B. T. Tuten, C. A. Tooley, P. G. Frank and E. B. Berda, Polym. Chem., 2015, 6, 181–197 RSC.
- M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917–924 CrossRef CAS PubMed.
- A. M. Hanlon, C. K. Lyon and E. B. Berda, Macromolecules, 2016, 49, 2–14 CrossRef CAS.
- M. Gonzalez-Burgos, A. Latorre-Sanchez and J. A. Pomposo, Chem. Soc. Rev., 2015, 44, 6122–6142 RSC.
- R. K. Roy and J.-F. Lutz, J. Am. Chem. Soc., 2014, 136, 12888–12891 CrossRef CAS PubMed.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS PubMed.
- O. Altintas and C. Barner-Kowollik, Macromol. Rapid Commun., 2016, 37, 29–46 CrossRef CAS PubMed.
- T. S. Fischer, D. Schulze-Sünninghausen, B. Luy, O. Altintas and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2016, 55, 11276–11280 CrossRef CAS PubMed.
- O. Altintas, M. Artar, G. Ter Huurne, I. K. Voets, A. R. A. Palmans, C. Barner-Kowollik and E. W. Meijer, Macromolecules, 2015, 48, 8921–8932 CrossRef CAS.
- E. A. Appel, J. Dyson, J. Delbarrio, Z. Walsh and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 4185–4189 CrossRef CAS PubMed.
- N. Hosono, M. A. J. Gillissen, Y. Li, S. S. Sheiko, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 501–510 CrossRef CAS PubMed.
- F. Wang, H. Pu and X. Che, Chem. Commun., 2016, 52, 3516–3519 RSC.
- S. Mavila, I. Rozenberg and N. G. Lemcoff, Chem. Sci., 2014, 5, 4196–4203 RSC.
- A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439–443 CrossRef CAS.
- B. T. Tuten, D. Chao, K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068–3071 RSC.
- N. Wedler-Jasinski, T. Lueckerath, H. Mutlu, A. S. Goldmann, A. Walther, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2017, 6–9 Search PubMed.
- B. S. Murray and D. A. Fulton, Macromolecules, 2011, 44, 7242–7252 CrossRef CAS.
- J. Zhang, J. Tanaka, P. Gurnani, P. Wilson, M. Hartlieb and S. Perrier, Polym. Chem., 2017, 8, 4079–4089 RSC.
- A. Sanchez-Sanchez, D. A. Fulton and J. A. Pomposo, Chem. Commun., 2014, 50, 1871–1874 RSC.
- R. Lambert, A.-L. Wirotius and D. Taton, ACS Macro Lett., 2017, 6, 489–494 CrossRef CAS.
- J. Zhang, G. Gody, M. Hartlieb, S. Catrouillet, J. Moffat and S. Perrier, Macromolecules, 2016, 49, 8933–8942 CrossRef CAS.
- K. S. Park, D. Y. Kim, S. K. Choi and D. H. Suh, Jpn. J. Appl. Phys., 2003, 42, 3877–3880 CrossRef CAS.
- J. Jiang and S. Thayumanavan, Macromolecules, 2005, 38, 5886–5891 CrossRef CAS.
- D. Mecerreyes, V. Lee, C. J. Hawker, J. L. Hedrick, A. Wursch, W. Volksen, T. Magbitang, E. Huang and R. D. Miller, Adv. Mater., 2001, 13, 204–208 CrossRef CAS.
- A. M. Hanlon, R. Chen, K. J. Rodriguez, C. Willis, J. G. Dickinson, M. Cashman and E. B. Berda, Macromolecules, 2017, 50, 2996–3003 CrossRef CAS.
- J. Willenbacher, K. N. R. Wuest, J. O. Mueller, M. Kaupp, H.-A. Wagenknecht and C. Barner-Kowollik, ACS Macro Lett., 2014, 3, 574–579 CrossRef CAS.
- T. K. Claus, J. Zhang, L. Martin, M. Hartlieb, H. Mutlu, S. Perrier, G. Delaittre and C. Barner-Kowollik, Macromol. Rapid Commun., 2017, 38, 1700264 CrossRef PubMed.
- C. Heiler, J. T. Offenloch, E. Blasco and C. Barner-Kowollik, ACS Macro Lett., 2016, 6, 56–61 CrossRef.
- O. Altintas, J. Willenbacher, K. N. R. Wuest, K. K. Oehlenschlaeger, P. Krolla-Sidenstein, H. Gliemann and C. Barner-Kowollik, Macromolecules, 2013, 46, 8092–8101 CrossRef CAS.
- P. G. Frank, B. T. Tuten, A. Prasher, D. Chao and E. B. Berda, Macromol. Rapid Commun., 2014, 35, 249–253 CrossRef CAS PubMed.
- W. Fan, X. Tong, Q. Yan, S. Fu and Y. Zhao, Chem. Commun., 2014, 50, 13492–13494 RSC.
- I. Perez-Baena, I. Asenjo-Sanz, A. Arbe, A. J. Moreno, F. Lo Verso, J. Colmenero and J. A. Pomposo, Macromolecules, 2014, 47, 8270–8280 CrossRef CAS.
- J. Winsberg, T. Janoschka, S. Morgenstern, T. Hagemann, S. Muench, G. Hauffman, J. F. Gohy, M. D. Hager and U. S. Schubert, Adv. Mater., 2016, 28, 2238–2243 CrossRef CAS PubMed.
- J. Winsberg, S. Muench, T. Hagemann, S. Morgenstern, T. Janoschka, M. Billing, F. H. Schacher, G. Hauffman, J.-F. Gohy, S. Hoeppener, M. D. Hager and U. S. Schubert, Polym. Chem., 2016, 7, 1711–1718 RSC.
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed.
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed.
- C. L. Rayner, S. E. Bottle, G. A. Gole, M. S. Ward and N. L. Barnett, Neurochem. Int., 2016, 92, 1–12 CrossRef CAS PubMed.
- T. S. Fischer, J. Steinkoenig, H. Woehlk, J. P. Blinco, K. Fairfull-Smith and C. Barner-Kowollik, Polym. Chem., 2017, 8, 5269–5274 RSC.
- L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS PubMed.
- T. Janoschka, A. Teichler, A. Krieg, M. D. Hager and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1394–1407 CrossRef CAS.
- R. K. Roy, A. Meszynska, C. Laure, L. Charles, C. Verchin and J.-F. Lutz, Nat. Commun., 2015, 6, 7237–7239 CrossRef PubMed.
- K. Zhang, M. J. Monteiro and Z. Jia, Polym. Chem., 2016, 7, 5589–5614 RSC.
- J. P. Blinco, D. J. Keddie, T. Wade, P. J. Barker, G. A. George and S. E. Bottle, Polym. Degrad. Stab., 2008, 93, 1613–1618 CrossRef CAS.
- V. Vitzthum, F. Borcard, S. Jannin, M. Morin, P. Miéville, M. A. Caporini, A. Sienkiewicz, S. Gerber-Lemaire and G. Bodenhausen, ChemPhysChem, 2011, 12, 2929–2932 CrossRef CAS PubMed.
- H. Mutlu, C. W. Schmitt, N. Jasinski, H. Woehlk, K. Fairfull-Smith, J. P. Blinco and C. Barner-Kowollik, Polym. Chem., 2017, 8, 6199–6203 RSC.
- E. M. Simpson, Z. D. Ristovski, S. E. Bottle, K. E. Fairfull-Smith and J. P. Blinco, Polym. Chem., 2015, 6, 2962–2969 RSC.
- K. Hansen, K. E. Fairfull-Smith, S. E. Bottle and J. P. Blinco, Macromol. Chem. Phys., 2016, 217, 2330–2340 CrossRef CAS.
- A. Kuo, Chem, 2011, 1, 80–86 Search PubMed.
- A. Kuo, Chem, 2011, 1, 40–51 Search PubMed.
- J. Willenbacher, O. Altintas, V. Trouillet, N. Knöfel, M. J. Monteiro, P. W. Roesky and C. Barner-Kowollik, Polym. Chem., 2015, 6, 4358–4365 RSC.
- A. Mardyukov and A. Studer, Macromol. Rapid Commun., 2013, 34, 94–101 CrossRef CAS PubMed.
- F. Mäsing, A. Mardyukov, C. Doerenkamp, H. Eckert, U. Malkus, H. Nüsse, J. Klingauf and A. Studer, Angew. Chem., Int. Ed., 2015, 54, 12612–12617 CrossRef PubMed.
- G. Delaittre, M. Dietrich, J. P. Blinco, A. Hirschbiel, M. Bruns, L. Barner and C. Barner-Kowollik, Biomacromolecules, 2012, 13, 1700–1705 CrossRef CAS PubMed.
- A. M. Hanlon, I. Martin, E. R. Bright, J. Chouinard, K. J. Rodriguez, G. E. Patenotte and E. B. Berda, Polym. Chem., 2017, 8, 5120–5128 RSC.
- Y. Bai, H. Xing, G. A. Vincil, J. Lee, E. J. Henderson, Y. Lu, N. G. Lemcoff and S. C. Zimmerman, Chem. Sci., 2014, 5, 2862–2868 RSC.
- S. K. Hamilton and E. Harth, ACS Nano, 2009, 3, 402–410 CrossRef CAS PubMed.
- P. Wang, H. Pu, J. Ge, M. Jin, H. Pan, Z. Chang and D. Wan, Mater. Lett., 2014, 132, 102–105 CrossRef CAS.
- J. De-La-Cuesta, E. Gonzalez and J. A. Pomposo, Molecules, 2017, 22, 1819 CrossRef PubMed.
- S. Jockusch, M. S. Landis, B. Freiermuth and N. J. Turro, Macromolecules, 2001, 34, 1619–1626 CrossRef CAS.
- W. Snipes, J. Cupp, G. Cohn and A. Keith, Biophys. J., 1974, 14, 20–32 CrossRef CAS PubMed.
- B. A. Chalmers, J. C. Morris, K. E. Fairfull-Smith, R. S. Grainger and S. E. Bottle, Chem. Commun., 2013, 49, 10382–10384 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01009a |
| This journal is © The Royal Society of Chemistry 2018 |