
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into the SN2-type reactivity of aryl-Co(III) masked-carbenes for C–C bond forming transformations†
O.
Planas‡
 a,
S.
Roldán-Gómez‡
a,
V.
Martin-Diaconescu
b,
J. M.
Luis
*a,
A.
Company
a and
X.
Ribas
*a
a,
S.
Roldán-Gómez‡
a,
V.
Martin-Diaconescu
b,
J. M.
Luis
*a,
A.
Company
a and
X.
Ribas
*a
aInstitut de Química Computacional i Catàlisi (IQCC), Departament de Química, Universitat de Girona, Campus Montilivi, Girona, E-17003, Catalonia, Spain. E-mail: josepm.luis@udg.edu; xavi.ribas@udg.edu
bInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Avinguda Països Catalans 16, 43007 Tarragona, Catalonia, Spain
First published on 29th May 2018
Abstract
Herein we describe the synthesis and characterization of a family of C-metalated aryl-Co(III) enolates, which can be considered as masked-carbenes, using diazoacetates as coupling partners. These species have been proved to be necessary intermediates in the C(sp2)–C(sp3) bond forming event to obtain cyclic amides, taming the elusive Co(III)-carbene species. The scope of diazoacetates has been exhaustively examined, varying the nature of the ester and the α-substitution, and a clear preference for electron-poor carbene precursors is observed. Exhaustive experimental and theoretical studies indicate that an unprecedented intramolecular SN2-type process governs the formation of the newly formed C–C bond. Furthermore, the key role of several Lewis acids as carboxylate-activating reagents is further explored by DFT calculations.
Introduction
Recently, transition-metal-catalyzed C–H bond activation has emerged as a powerful and versatile tool in organic synthesis.1–3 Most of the achievements have been accomplished with precious metals such as Rh, Ru and Pd.4–10 However, methodologies using more abundant and cost-efficient 3d transition metal catalysts have attracted increasing attention in the field of directed C–H activation and functionalization.11–14 Indeed, this recent interest in first-row transition metal catalysts has allowed the development of new C–H functionalization protocols for the synthesis of several organic molecules using Fe,15–17 Mn18,19 and Ni20,21 as catalysts. Particularly, Co has attracted recent interest probably due to the practicality and efficiency of group 9 metals. Two different approaches coexist in the field of Co-catalyzed C–H functionalization: the low-valent approach,22,23 which has been extensively studied, and the high-valent approach.24The high-valent approach in Co-catalysis has emerged as a tremendously valuable tool for the construction of organic scaffolds through the use of Cp*Co(III) catalysts25–27 as well as N′N-bidentate-chelating directing groups.28 Knowledge of the mechanistic pathways governing these transformations is still in its infancy,29 although an organometallic Co(III) species is generally implicated.30,31 Proof-of-concept protocols reported by Broderick and Legg32,33 and Avilés34 provided early evidence of organometallic Co(III) species obtained through C–H activation. After these two examples the field remained dormant until recently, when several cobaltacycles synthesized through C–H activation were reported.30,31,35 Indeed, cobalt has recently been proved to be a practical metal to perform a vast array of transformations. Particularly, the use of carbene precursors has attracted recent interest due to their versatility as coupling partners in transformations such as carbonylation36,37 and annulation,38–44 including Co(III)-radical-based outer-sphere functionalization (Scheme 1a). Regarding C–H functionalization, the field was pioneered by Miura using Co(II) salts.45 Recently, the use of Cp*Co(III) catalysts allowed several organometallic C–H functionalization reactions utilizing diazo esters as coupling partners (Scheme 1b).46–50 The reactivity observed with Co(III) is currently rationalized through the formation of an elusive aryl-Co(III)-carbene (B, Scheme 1c), which undergoes migratory insertion to furnish the corresponding alkyl-Co(III) intermediate (C, Scheme 1c).51 This rationalization is indeed not surprising, thus migratory insertion into metal–carbon bonds is a common pathway in metal–carbene functionalization reactions.52
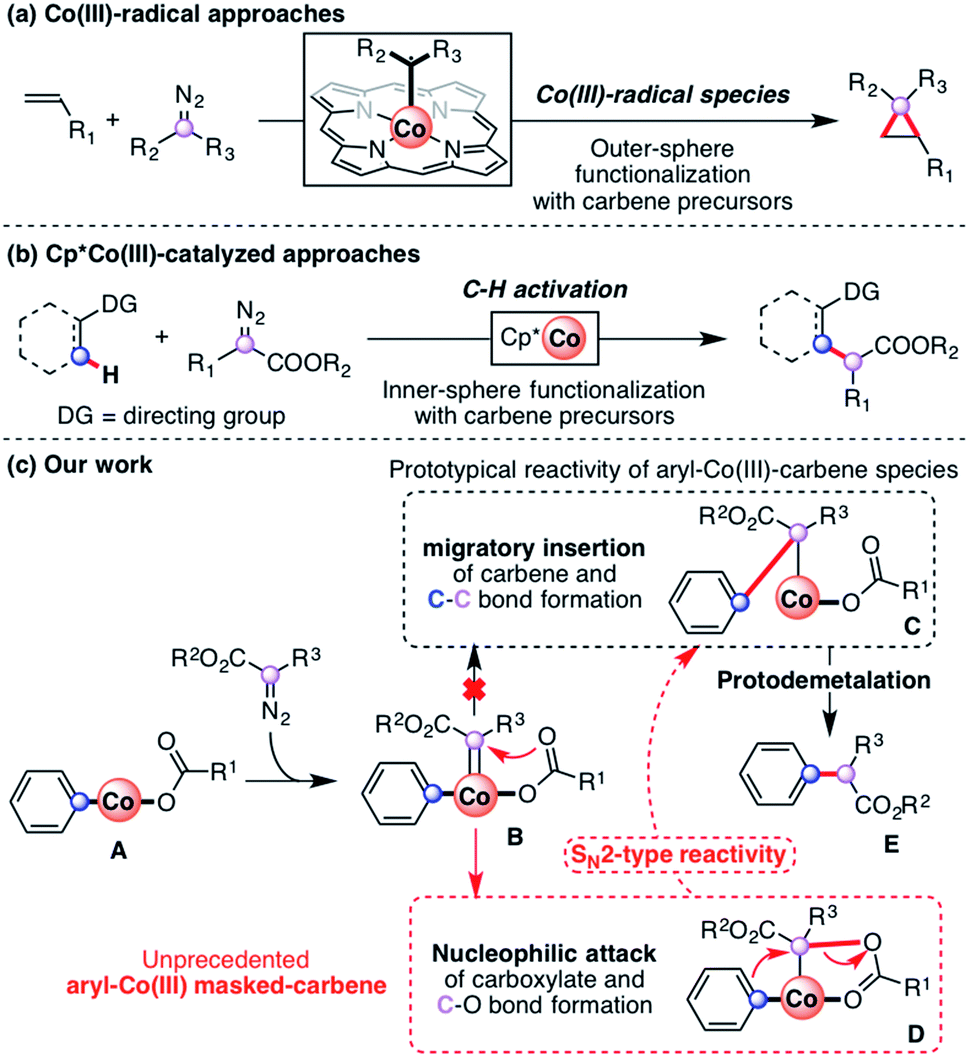 | ||
| Scheme 1 Reactivity of Co(III) species with diazoacetates: (a) functionalization through Co(III)-radical species; (b) C–H activation utilizing Cp*Co(III) catalysts; (c) prototypical migratory insertion mechanism (black) and our work, which proved the existence of new intermediates using macrocyclic model substrates (red). | ||
The mechanistic understanding of metal-catalyzed transformations constitutes a landmark of our research.53–56 To achieve this goal, a variety of macrocyclic model substrates can be utilized, as they proved to be key for the synthesis and characterization of several organometallic intermediates.57–59 Following this line, we have recently published the synthesis and characterization of a family of aryl-Co(III) complexes through C–H activation.60 These complexes were competent intermediates during alkyne annulation reactions, as well as diazoacetate couplings.61 When our aryl-Co(III) complexes reacted with ethyl diazoacetate (EDA) under anhydrous conditions, an unprecedented C-metalated aryl-Co(III) enolate was obtained (D, Scheme 1c). Thus, the nucleophilic attack of the adjacent carboxylate moiety gives a rare cis aryl-Co(III)-alkyl organometallic species, which was never considered until our report and constitutes a unique example in the field. Interestingly, this intermediate D, which can be considered either as a masked-carbene moiety or a carbenoid, evolves to intermediate C (Scheme 1c) through a rare intramolecular SN2-type pathway in which the carboxylate moiety acts as both a relay and a leaving group. Then, after protodemetalation of C the corresponding organic product containing a new C–C bond (E, Scheme 1c) can be released and the Co(III) catalyst is regenerated.
Herein we report on the full understanding of the electronic and steric effects that control this carbene transfer via the masked-carbene strategy. To that end, the synthesis of a family of C-metalated aryl-Co(III) enolates bearing differently substituted carboxylate moieties (R1, Scheme 2), using a macrocyclic ligand as a model substrate, is reported. Moreover, several diazoacetates bearing substituents with different electronic and steric properties will be tested (R2 and R3, Scheme 2). Finally, new pieces of experimental and computational evidence are reported to provide a broader mechanistic insight into the formation of aryl-Co(III) masked-carbenes as well as into the unique intramolecular SN2-type reactivity to obtain new C–C bonds.
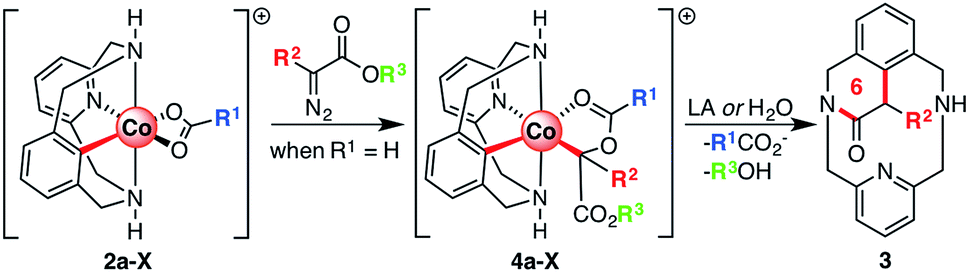 | ||
| Scheme 2 Reactivity of aryl-Co(III) bearing different carboxylate ligands (R1) with diazoacetates with diverse substituents on the α-(R2) and ester (R3) positions. | ||
Results and discussion
Initially, several organometallic aryl-Co(III) intermediates bearing different carboxylate anions (2a-X, X = TFA, TPA (triphenylacetate), OPiv, or OBz-Y; Y = H, Me, OMe, Cl, COMe, CN, or NO2) were synthesized starting from 1a-H, CoBr2 and the corresponding silver carboxylate salts (see the ESI† for more details). Then, 2a-X were tested in the annulation reaction utilizing ethyl diazoacetate (EDA) as a coupling partner to furnish the cyclic amide 3a (Scheme 3). First, following our previous work,61 aryl-Co(III) species bearing alkyl carboxylates were tested using 2.0 equiv. of EDA and 4.0 equiv. of H2O as additives in 2,2,2-trifluoroethanol (TFE) at 100 °C (alkyl, Scheme 3). We recently reported that the reaction of 2a-OAc and 2a-TFA with EDA furnished 3a in excellent yields. To further understand this transformation, we attempted the reaction of EDA with sterically crowded aryl-Co(III) carboxylates such as 2a-OPiv and 2a-TPA. Interestingly, when R1 = tBu (OPiv), 3a was furnished in poor yield (20–23%), even in the presence of Lewis acids such as LiOTf. This result suggests that carboxylates bearing bulky groups inhibit the reaction. Indeed, when 2a-TPA was reacted with EDA, 3a was obtained in trace amounts. It should be noted that triphenylacetate is a weak base and subsequently, a good leaving group. However, the bulky “CPh3” moiety inhibits the formation of 3a, indicating that steric effects play an important role too. The formation of cyclic amide 3a was also studied starting from aryl-Co(III) complexes bearing differently p-substituted benzoate anions (2a-OBz-Y; Scheme 3). In this case, when 4.0 equiv. of H2O were added, 3a was obtained in good yields (71–85%) regardless of the substitution of the carboxylate moiety. Thus, the electronic properties of the carboxylate anions have little impact on the yield of 3a under these particular reaction conditions.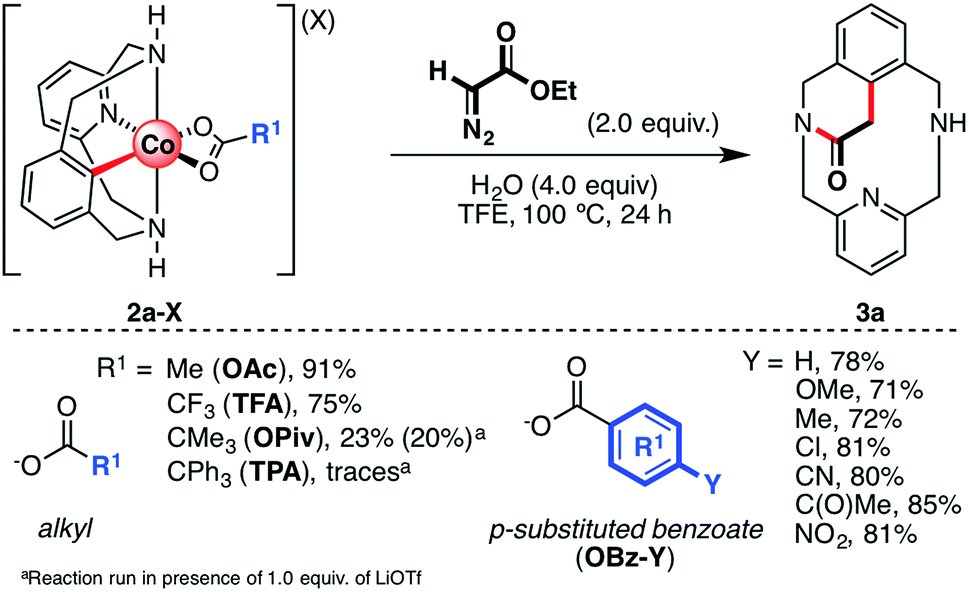 | ||
| Scheme 3 Reaction scope with a variety of alkyl and p-substituted aryl carboxylate ligands. Yields were determined after isolation by silica column chromatography. | ||
Reactivity of 2a-OAc with a variety of substituted diazoesters was also evaluated (Scheme 4) in the presence of 4.0 equiv. of H2O and TFE as the solvent. First, we evaluated the substitution on the ester moiety using a variety of alkyl and aryl diazoacetates (R3, Scheme 4). When the ester moiety bears an alkyl fragment such as a methyl, ethyl, tert-butyl, benzyl or aryl moiety, the reaction proceeds with good to excellent yields, affording macrocyclic amide 3a. In spite of the promising reactivity with EDA, no product was observed when 2a-OAc was reacted with a variety of α-substituted ethyl diazocarboxylate moieties (R2, Scheme 4), in contrast to the described reactivity of Cp*Co(III) catalysts.46–51 Indeed, when a methyl (b) or a benzyl (c) moiety was present in the α-position, only traces of 3b or 3c were detected. Surprisingly, when ethyl diazomalonate (d) was used as a coupling partner, the unexpected formation of 3a was observed in 73% yield after decarboxylation. Thus, only acceptor/acceptor carbene precursors engage in coordination and annulation to furnish product 3. However, trace amounts of products were detected when a highly electron-withdrawing group (trifluoromethyl (e), nitro (f) and cyano (g) groups) was directly attached in the α-position with respect to the diazo carbon, probably due to its high reactivity. Furthermore, when electron-rich aryl moieties such as phenyl (h) and p-OMe-phenyl (i) are attached to the α-position of ethyl diazoacetate, trace amounts of the corresponding cyclic amides 3h and 3i are obtained. However, if the phenyl group contains an electron-withdrawing group, such as a bromide (j), trifluoromethyl (k) or nitro (l) group, the corresponding amides 3j, 3k and 3l are obtained in good yields (65–74%) in the presence of 1.0 equiv. of LiOTf as additive. These results suggest that the formation of aryl-Co(III)-carbenes and their subsequent reactivity depends on both steric and electronic properties of the diazoacetate. Thus, in contrast to the previously reported methodologies,46–51 the reaction with acceptor/donor diazoacetates does not proceed while acceptor/acceptor carbene precursors afford amide 3.
 | ||
| Scheme 4 Reaction scope with several ester-substituted (R3) and α-substituted (R2) diazo compounds. Yields were determined after isolation by silica column chromatography. | ||
In order to further compare the reactivity of different substrates under the optimized reaction conditions, we performed several intermolecular competition reactions (Scheme 5). First, the electronic properties of the carbene precursor were tested through a reaction of 2a-OAc and 4.0 equiv. of ethyl p-substituted-phenyl diazoacetates j and l (Scheme 5a) in the presence of 4.0 equiv. of H2O and 1.0 equiv. of LiOTf as additives. Under these conditions, 3j was obtained in 47% yield while 3l was furnished in 32% yield (79% combined isolated yield). Thus, both electron-poor carbene precursors j and l react similarly with 2a-OAc (ratio 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Thereafter, the steric effects of the diazoacetate coupling partners were evaluated through an intermolecular competition experiment between diazo esters a and j (Scheme 5b). When 4.0 equiv. of each carbene precursor were mixed with 2a-OAc in TFE, exclusive formation of 3a was observed (ratio 3a/3j > 20:1). Thus, this result indicates that the reaction of 2a-OAc with sterically demanding carbene precursors is not favored in the presence of EDA, subsequently forming cyclic amide 3a as a single product. Finally, as the reaction of 2a-OAc with some precursors was not successful, diazo esters c, e and i were tested as poisoning reagents (Scheme 5c) in the reaction with diazo ester l. Under these conditions, the reaction yielded 47–76% of cyclic amide product 3l, which clearly indicates that neither the alkyl-substituted diazoacetates (c and e) nor the electron-rich carbene precursor (i) inhibited the reactivity through either coordination or decomposition of the organometallic 2a-OAc complex.
:1). Thereafter, the steric effects of the diazoacetate coupling partners were evaluated through an intermolecular competition experiment between diazo esters a and j (Scheme 5b). When 4.0 equiv. of each carbene precursor were mixed with 2a-OAc in TFE, exclusive formation of 3a was observed (ratio 3a/3j > 20:1). Thus, this result indicates that the reaction of 2a-OAc with sterically demanding carbene precursors is not favored in the presence of EDA, subsequently forming cyclic amide 3a as a single product. Finally, as the reaction of 2a-OAc with some precursors was not successful, diazo esters c, e and i were tested as poisoning reagents (Scheme 5c) in the reaction with diazo ester l. Under these conditions, the reaction yielded 47–76% of cyclic amide product 3l, which clearly indicates that neither the alkyl-substituted diazoacetates (c and e) nor the electron-rich carbene precursor (i) inhibited the reactivity through either coordination or decomposition of the organometallic 2a-OAc complex.
 | ||
| Scheme 5 (a) Competition experiments using diazo esters with different electronic properties. (b) Competition experiments using diazo esters with different steric properties. (c) Poisoning studies of the reaction between 2a-OAc and diazo acetate l, using diazoacetates c, e and i as potential poisons. (d) Deuterium incorporation into 3l using labeled water as the additive. Isolated yields after silica column chromatography; ratios determined by proton NMR. | ||
Deuterium labeling experiments were also performed using diazo ester l as a coupling partner in order to complement the examples that we previously reported (Scheme 5d).61 It was found that when the reaction of 2a-OAc with 4.0 equiv. of l was carried out using 4.0 equiv. of D2O as the additive instead of ordinary water, 3l was obtained in 71% yield and a 21% D-incorporation was observed by 1H NMR analysis (see Fig. S1†). This result is in agreement with the previously reported D-labeling experiments, indicating a possible protodemetalation step prior to the formation of the corresponding cyclic amide 3.
To gain further insight into the reaction mechanism, several C-metalated aryl-Co(III) enolate complexes bearing a variety of carboxylate ligands (4a-X) were synthesized (Scheme 6). When 2a-X was reacted with 2.0 equiv. of EDA at 100 °C for 15 minutes, the corresponding 4a-X intermediates were obtained in moderate to excellent yields (21–89%). Characterization by NMR and HRMS indicated the formation of diamagnetic aryl-Co(III) species bearing an alkyl moiety corresponding to the EDA fragment. Indeed, the structure of intermediates 4a-X was further confirmed by XRD analysis (Fig. 1; X = OPiv (a), OBz (b), OBz-NO2 (c) and OBz-COMe (d)). The solid-state structures of 4a-X showed the formation of an Oh aryl-Co(III) center bearing a carbonyl group trans to the aryl moiety. In the cis-position with respect to the aryl fragment, a C-enolate binds to the metal center as an alkyl ligand (4a-OPiv; Co–C1 = 1.962 Å), thus providing an unusually stable cis-aryl-Co(III)-alkyl organometallic intermediate. Overall, the solid-state structures present similar XRD features and show the formation of rare C-metalated aryl-Co(III) enolates, which are presumably formed through a nucleophilic attack of the carboxylate anion to a putative highly electrophilic Co(III)-carbene. Remarkably, it should be noted that the C2–O2 distance is slightly larger with electron-poor benzoate moieties (4a-OBz, C2–O2 = 1.480 Å; 4a-OBz-NO2, C2–O2 = 1.507 Å), which indicates that species bearing electron-poor fragments might be more prone to evolve to amide 3 through an intramolecular SN2-like mechanism.
 | ||
| Scheme 6 Synthesis of C-metalated aryl-Co(III) enolates bearing a variety of carboxylate anions (4a-X, where X = carboxylate anion). Yields after recrystallization. | ||
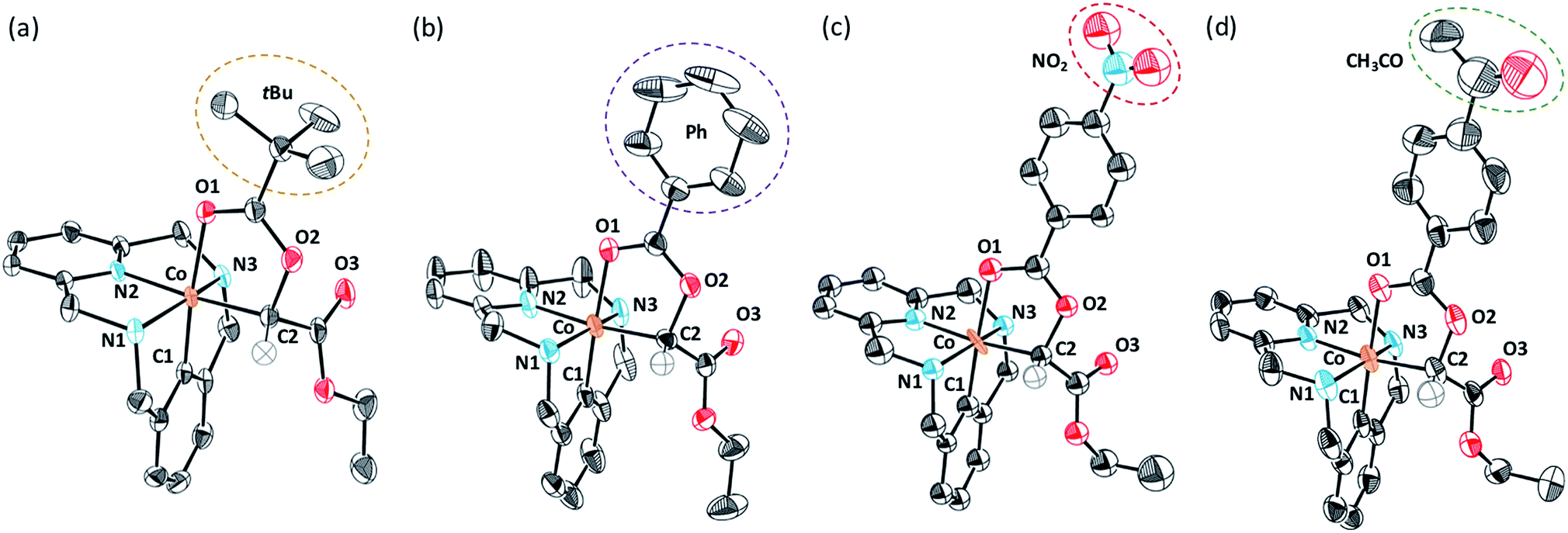 | ||
| Fig. 1 Solid state structures of organometallic aryl-Co(III)-alkyl compounds 4a-X. Hydrogen atoms, carboxylate anions and solvent molecules have been omitted for clarity; only one of the enantiomers is depicted; ellipsoids are set at 50% probability. (a) Crystal data for 4a-OPiv. Selected bond distances [Å] and angles [°]: Co–C(1) 1.840(6), Co–C2 1.962(4), Co–N(1) 2.002(4), Co–N(2) 1.892(3), Co–N(3) 1.994(4), Co–O(1) 2.023(4), C(2)–O(2) 1.480(7); C(1)–Co–C(2) 92.7(2), C(1)–Co–O(1) 174.8(2). (b) Crystal data for 4a-OBz. Selected bond distances [Å] and angles [°]: Co–C(1) 1.853(2), Co–C2 1.971(4), Co–N(1) 2.003(4), Co–N(2) 1.893(3), Co–N(3) 2.008(4), Co–O(1) 1.989(4), C(2)–O(2) 1.480(7); C(1)–Co–C(2) 95.5(2), C(1)–Co–O(1) 177.4(2). (c) Crystal data for 4a-OBz-NO2. Selected bond distances [Å] and angles [°]: Co–C(1) 1.809(2), Co–C2 1.976(4), Co–N(1) 2.002(4), Co–N(2) 1.899(3), Co–N(3) 1.981(4), Co–O(1) 2.000(4), C(2)–O(2) 1.507(7); C(1)–Co–C(2) 94.1(1), C(1)–Co–O(1) 175.3(2). (d) Crystal data for 4a-OBz-COMe. Selected bond distances [Å] and angles [°]: Co–C(1) 1.849(2), Co–C2 1.978(4), Co–N(1) 2.002(4), Co–N(2) 1.886(3), Co–N(3) 2.000(4), Co–O(1) 2.016(4), C(2)–O(2) 1.499 (7); C(1)–Co–C(2) 95.8(1), C(1)–Co–O(1) 177.9(2). | ||
Once the organometallic 4a-X complexes were analyzed and characterized, we evaluated their capability to evolve to product 3a through an intramolecular SN2-type mechanism. Firstly, 4a-OBz-Y complexes were heated at 100 °C for 24 h utilizing TFE as the solvent. Interestingly, 3a was observed regardless of the electronic properties of the para-substituted benzoate ester, albeit higher yields were obtained with electron-withdrawing substituents (see Table S1†). The evolution of 4a-OBz-Y was further studied by means of kinetic analysis. Electronic effects on the C–C bond formation were tested through a Hammett plot prepared following Chatani's method,62,63 in which the evolution of an electronically different set of para-substituted benzoate species (4a-OBz-Y) to cyclic amide 3a was compared at short reaction times (Fig. 2). Thus, 4a-OBz-Y were heated in anhydrous TFE over 3 h and the crude mixture was analyzed by proton NMR using 1,3,5-trimethoxybenzene as the internal standard (Fig. 2a). Then, the amount of 3a obtained starting from 4a-OBz-Y (YY) was compared to the yield obtained from 4a-OBz-H (YH). A linear correlation (R2 = 0.963) from the conversion of the corresponding organometallic complexes vs. the Hammett parameter (σp) led to a positive slope value of ρ = 0.568. This value is consistent with a rate-determining step, the transition state of which builds partial negative charge (Scheme 5). Further evidence of an SN2-type mechanism was obtained when product yields were correlated with the acidity of the leaving group (LG) conjugate acid in a Brønsted plot. As shown in Fig. 2b, a linear correlation (R2 = 0.963) was obtained, leading to a negative value of βLG = −0.573. Thus, in spite of the moderate value obtained (0 > βLG > −1), this result clearly indicates that the leaving group ability plays a role in the rate-determining step of this transformation.64 However, 2a-TPA, whose corresponding enolate corresponds to a good leaving group (pKa(TPAH) = 3.33), is found to be unreactive towards the formation of 3a. This result suggests that steric factors have to be taken into account too. In addition to these experiments, a surprisingly good linear correlation (R2 = 0.973) between the 13C chemical shift of the C-enolate moiety (C2, Fig. 2c) and the corresponding Hammett parameter of 4a-OBz-Y complexes was found. Indeed, the plot in Fig. 2c suggests that the electronic properties of the substituent have a considerable effect on the electron density on C2, subsequently modifying its electrophilic character. Thus, when EWGs are attached to the benzoate moiety, C2 becomes more electrophilic and subsequently more reactive towards a substitution. This trend was also observed when the theoretical activation barriers of the different para-substituted benzoate 4a-OBz-Y complexes were compared with Hammett σp (Gibbs energies were used for the linear correlation vs. Hammett parameters; see the ESI† for full details). The plot in Fig. 2d shows that 4a-OBz-NO2 has a lower activation barrier (ΔG‡ = 30.0 kcal mol−1) compared to the electron-rich 4a-OBz-OMe (ΔG‡ = 27.3 kcal mol−1) species and the trend observed (R2 = 0.949) is in agreement with the experimental linear correlations mentioned above. Further evidence for an SN2-type event was obtained by evaluating the impact of the nucleophile strength on the activation barrier, i.e. by computing different p-aryl substitutions on the ligand backbone (Fig. 3 and S30†). Starting from the electron-rich 4b-OAc (R = p-OMe) complex, the SN2-type barrier decreases by around 2 kcal mol−1 (TS3b; 27.0 kcal mol−1) compared to the previously reported activation energy starting from 4a-OAc (TS3a; 28.9 kcal mol−1).61 Contrariwise, when the SN2-type barrier is calculated for a poorer nucleophilic aryl-Co(III) complex such as 4c-OAc (R = p-NO2), a substantial increase can be observed (TS3c; 34.7 kcal mol−1). Thus, these theoretical studies suggest that the nucleophilic character of the aryl-Co(III) bond plays a key role in the C–C bond forming/C–O cleavage event.
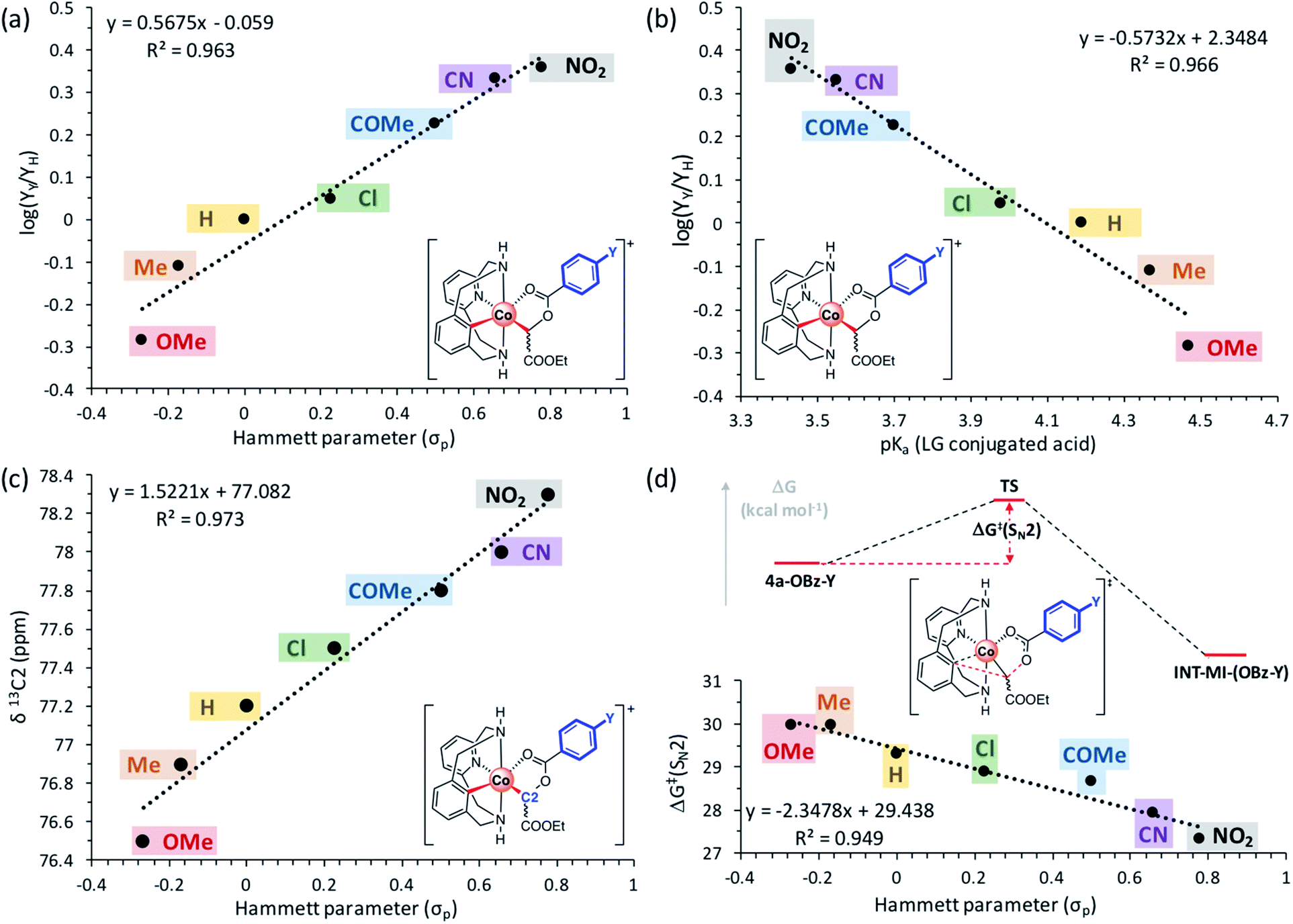 | ||
| Fig. 2 (a) Hammett plot (R2 = 0.963) of para-substituted benzoate C-metalated aryl-Co(III) enolate complexes (4a-OBz-Y; Y = OMe, Me, H, Cl, COMe, CN, or NO2) with a slope value of ρ = 0.568 (plot constructed comparing yields of 3a obtained at 3 h from 4a-OBz-Yversus4a-OBz-H). (b) Nucleofugality Brønsted plot (R2 = 0.966) of 4a-OBz-Y (Y = OMe, Me, H, Cl, COMe, CN, NO2) complexes with a slope value of βLG = −0.573. (c) Linear correlation (R2 = 0.973) between the 13C chemical shift of C2 and Hammett parameters of 4a-OBz-Y (Y = OMe, Me, H, Cl, COMe, CN, or NO2) complexes. (d) Linear correlation (R2 = 0.949) between Gibbs energy barriers of the SN2-type transition state (ΔG‡(SN2), full details can be found in Table S9 and Fig. S20†) and Hammett parameters of 4a-OBz-Y (Y = OMe, Me, H, Cl, COMe, CN, or NO2) complexes; yields calculated by 1H NMR analysis using 1,3,5-trimethoxybenzene as the internal standard. | ||
 | ||
| Fig. 3 Gibbs energy profile of the SN2-type event of p-substituted C-metalated aryl-Co(III) complex enolates 4x-OAc (R = H (x = a), OMe (x = b) and NO2 (x = c)). Relative Gibbs energy values are given in kcal mol−1 (see Fig. S30† for full profiles). | ||
All these results taken together indicate a transition state in the rate-determining step which builds partial negative charge (ρ > 0), depends on the para-substituents of the benzoate moiety and presents a considerable sensitivity to both the leaving group ability (βLG < 0) and the nucleophilicity of the aryl-Co(III) moiety (Fig. 3). Indeed, these results strongly suggest a plausible transition state in which a simultaneous C–C bond formation/C–O cleavage event is occurring in a concerted intramolecular SN2-type reaction.
Further experimental evidence of a unique SN2-type intramolecular mechanism was obtained when para-substituted 4a-OBz-Y were studied by high-resolution mass spectrometry (HRMS) (Fig. 4). Indeed, the corresponding organometallic product of an intramolecular substitution (INT-MI′) was detected when MS/MS analysis was performed for the mass peak corresponding to 4a-OBz-Y, irrespective of the nature of substituent Y (Y = OMe, Me, H, Cl, COMe, CN, or NO2). A single peak which corresponds to a fragment involving the loss of the corresponding carboxylic acid ([INT-MI-RCOOH]+ = C19H21CoN3O2+; simulated m/z = 382.0960; Fig. 4) was observed regardless of the carboxylate moiety present in the isolated compounds, which suggests the same product for each 4a-OBz-Y species. Furthermore, the relative intensity of INT-MI′ at the same collision energy (15 eV, see also Fig. S6–S13†) is higher when an electron-poor p-substituted benzoate is present in 4a-OBz-Y (rel. intensity of 55.6% and 65.3% when Y = NO2 and CN, respectively) compared to electron-rich substituents (rel. intensity of 10.9% and 14.8% when Y = OMe and Me, respectively). This trend strongly supports the nucleofugality trend of the para-substituted leaving groups (Fig. 2b) as well as the electrophilic character of C2 (Fig. 2c), indicating that with electron-poor benzoates the C–C bond formation/C–O bond cleavage event is easier and subsequently faster.
 | ||
| Fig. 4 Detection of INT-MI′ (simulated m/z = 382.0960) by MS/MS studies of organometallic 4a-OBz-Y complexes: 4a-OBz-OMe (m/z = 534.1428), 4a-OBz-Me (m/z = 518.1478), 4a-OBz (m/z = 504.1351), 4a-OBz-Cl (m/z = 538.0949), 4a-OBz-COMe (m/z = 546.1429), 4a-OBz-CN (m/z = 529.1268) and 4a-OBz-NO2 (m/z = 538.0949). | ||
At this point our interest was focused on the reaction of α-substituted diazoacetates, particularly on the reaction of 2a-OAc with electron-rich (i) and electron-poor (l) carbene precursors. As shown in Scheme 5b, the reaction of α-phenyl diazoacetates is slower when compared to that of ethyl diazoacetate, which suggests steric influence or even other reaction pathways not considered before. Furthermore, it has been shown that electron-rich diazoacetates do not inhibit the reaction with other carbene precursors (Scheme 5c). Thus, 2a-OAc was reacted with 2.0 equiv. of diazoacetates i and l over 60 min (Scheme 7), and the reaction crudes were analyzed by HRMS. With electron-rich diazoacetate i, the corresponding organometallic 4a-OAc-PhOMe intermediate was not detected in agreement with the reactivity observed in Scheme 4 (see Fig. S14†). In contrast, when electron-poor diazoacetate l was reacted with 2a-OAc, a peak at m/z = 563.1335 corresponding to 4a-OAc-PhNO2 was observed (C27H28N4O6Co+; simulated m/z = 563.1335, Fig. S15†) indicating the formation of a similar C-metalated aryl-Co(III) enolate which can be considered either as a carbenoid or a masked-carbene. Further evidence of the formation of this particular cobaltacycle was found when MS/MS analysis of the 563.1335 peak for 4a-OAc-PhNO2 was performed. A single peak with m/z = 503.1128 was observed (20 eV), which corresponds to a fragment involving the loss of acetic acid (C25H24N4O4Co+, simulated m/z = 503.1124; Fig. S16†), similarly to when MS/MS analysis was applied to para-substituted 4a-OBz-Y species in Fig. 4. Indeed, this species corresponds to the previously observed analogous INT-MI′ with EDA derivatives, thus indicating the formation of a C-metalated aryl-Co(III) enolate when α-phenyl diazoacetates are utilized as coupling partners.
 | ||
| Scheme 7 Reaction of 2a-OAc with α-substituted ethyldiazoacetates and HRMS analysis of reaction crudes. | ||
The effect of several additives on the SN2-type pathway was also evaluated (see Scheme 8). It has been proven both experimentally and theoretically that Lewis acids assist the C–C bond formation/C–O cleavage event and accelerate the reaction, particularly in the case of LiOTf.61 Furthermore, recent studies demonstrate that Lewis acids have beneficial effects on C–H activation methodologies using Co(III) as a catalyst, both assisting C–H activation and carbene formation.46 Based on these pioneering studies, we explored the possibility of adding other similar Lewis acids such as MOTf (M = H, Li, Na, or K), M(OTf)2 (M = Mg or Ba) and other larger but highly acidic compounds such as tris(pentafluorophenyl)borane (Scheme 8). Thus, 4a-OAc was mixed with 1.0 equiv. of Lewis acid at 100 °C. After 24 h, good to excellent yields of 3a were obtained with organic acids such as HOTf (82%) and metal cations such as LiOTf (89%), Mg(OTf)2 (67%), NaOTf (71%) and KOTf (72%). Interestingly, a poor yield (34%) was obtained when Ba(OTf)2 was used as an additive. Finally, with boron-based Lewis acids such as B(C6F5)3, 3a was observed only in 26% yield. This latter result suggests that the evolution of 4a-OAc to 3a depends both on Lewis acid strength and bulkiness.
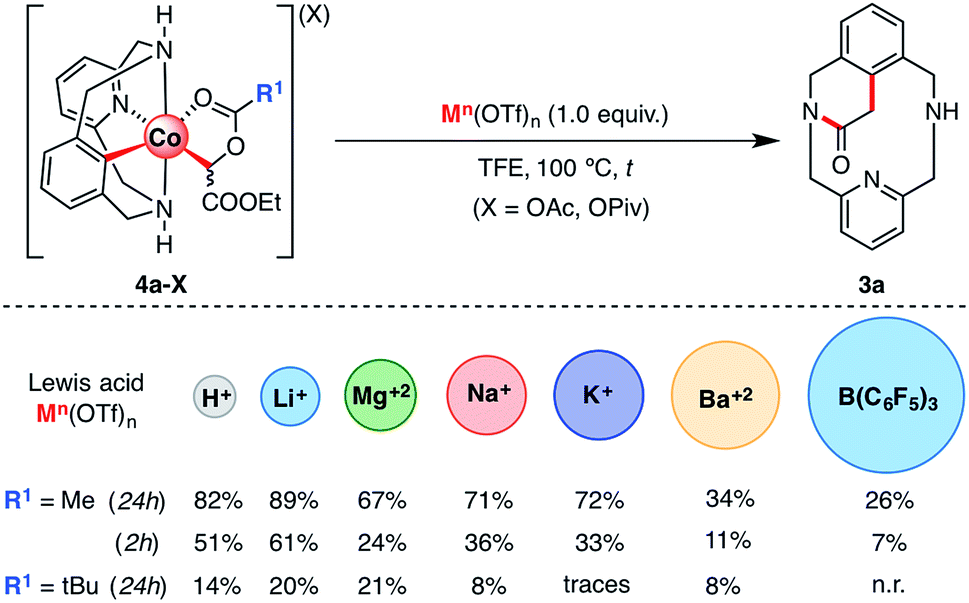 | ||
| Scheme 8 Evolution of organometallic 4a-X (X = OAc and OPiv) intermediates to 3a in the presence of several Lewis acids as additives (see Scheme S14 and Table S6† for details). | ||
In order to gain more information about the effects of the different additives used to promote the evolution of 4a-OAc to 3a, the same reaction was run over a shorter time period. After 2 h, 3a was obtained in good yields using acids such as LiOTf (61%) and HOTf (51%). However, a moderate-to-poor yield of 3a was obtained when larger and less acidic metal cations such as NaOTf (36%), KOTf (33%), Mg(OTf)2 (24%) and Ba(OTf)2 (11%) were added. In addition, following the reactivity trend observed after 24 h, a low yield (7%) of 3a was obtained when tris(pentafluorophenyl)borane was added, which suggests that large Lewis acids hinder the interaction between the Lewis acid and the organometallic 4a-OAc complex, hence preventing its activation towards an intramolecular SN2-type reaction. Indeed, similar results were obtained when 4a-OPiv was subjected to the same reaction conditions. In this case, however, poor yields were obtained even with hard Lewis acids such as LiOTf (20%) or HOTf (14%) and the reaction was completely inhibited with larger Lewis acids such as KOTf, Ba(OTf)2 or B(C6F5)3. Indeed, steric hindrance is a plausible reason for the lack of reactivity of 2a-TPA with EDA even in the presence of additives such as LiOTf or H2O (see Scheme 3 for further details).
To gain further insight into the interaction of different Lewis acids (LA) with 4a-OAc we performed computational studies focused on the intramolecular LA-assisted SN2-type event (see the ESI† for computational details). To do so, we considered the explicit inclusion of cationic metal ions M+ (M = Li, Na, and K) in the computational analysis of the C–C bond formation/C–O cleavage event. In order to properly determine the Gibbs energy of 4a-OAc-M(TFE)3 adduct formation taking into account the solvent environment of the alkali cations, at least three explicit TFE molecules had to be included in the DFT calculations (Fig. 5 and S23†). This event is exergonic for the three alkali metals.
 | ||
| Fig. 5 (a) DFT Gibbs energy of the adduct formation when 4a-OAc is mixed with M+ cationic Lewis acids (M = Li, Na and K) taking into account the presence of three explicit TFE molecules. Relative Gibbs energy values are given in kcal mol−1; concentration of the solvated M+ = 0.039 mol L−1 (see Fig. S22–S24† for details). | ||
In agreement with experimental results, the activation barrier for the SN2-type C–C bond formation/C–O cleavage event decreases compared to the same step in the absence of additives (ΔG‡ = 28.9 kcal mol−1, see Fig. 3).61 Interestingly, a clear trend in agreement with the reactivity observed in Scheme 6 was found (Fig. 6). In fact, the increase of the activation energy of the LA-assisted SN2 step correlates with the size and strength of the corresponding Lewis acid: ΔG‡(Li+) = 22.6 < ΔG‡(Na+) = 24.7 < ΔG‡(K+) = 25.2 kcal mol−1. Thus, when Lewis acids are added to the reaction media, they coordinate to 4a-OAc (O2 and O3, Fig. 1), turning the carboxylate moiety into a better leaving group through a LA-mediated carboxylate activation. However, as calculations show in Fig. 6 and reactivity studies suggest (Scheme 8), the carboxylate activation depends on both the strength of the Lewis acid and its size.
 | ||
| Fig. 6 Gibbs energy profile of the SN2-type event in the presence of several M+ cationic Lewis acids (M = Li, Na and K). Relative Gibbs energy values are given in kcal mol−1 (see Fig. S21† for details). | ||
The mechanism of the corresponding C–C bond formation and C–O bond cleavage event for the electron-poor α-p-NO2-phenyl diazoacetate (l) was further studied by means of DFT calculations (for full details see the ESI†). The reaction profile in Fig. 7 shows that 2a-OAc reacts with diazoacetate l to form an adduct (Adduct, ΔG = 11.8 kcal mol−1) in which the acetate ligand is coordinated in a monodentate fashion. Then, the adduct with the corresponding diazoacetate evolves to 4a-OAc-PhNO2 (ΔG = −26.9 kcal mol−1) through an activation barrier of ΔG‡ = 28.4 kcal mol−1 (TS1) in a highly exergonic process. Interestingly, the formation of a transient carbene species was not observed computationally and 4a-OAc-PhNO2 was directly obtained from the adduct with diazoacetate l. As mentioned before, experimental evidence of the formation of 4a-OAc-PhNO2 species was also obtained by HRMS studies (Fig. 4), which agrees with the DFT calculations presented in Fig. 7. Then, 4a-OAc-PhNO2 could evolve to INT-MI-PhNO2 (ΔG = −9.3 kcal mol−1) through a highly asynchronous late transition state (TS2, ΔG = 9.9 kcal mol−1; see Fig. 8, right). Interestingly, TS2 is strongly asynchronous, contrariwise to the corresponding TS for 4a-OAc (Fig. 8, left) which shows a clearly synchronous transition state. Thus, although the SN2-event occurs in a concerted manner for both 4a-OAc and 4a-OAc-PhNO2, the IRC calculation connecting 4a-OAc-PhNO2, TS2 and INT-MI-PhNO2 (see Fig. S27†) shows a first C–O bond breaking followed by the formation of the C–C bond, the latter determining the Gibbs energy and geometry of TS2. The high activation barrier of this process (ΔG‡ = 36.8 kcal mol−1) hampers this transformation, which agrees with the experimental results obtained when 2a-OAc reacts with diazoacetate l in the absence of additives (Table S2,† entry 13). Despite the highly energetic transition state (TS2) and the endergonic process from 4a-OAc-NO2 to INT-MI-NO2, the latter species could be detected by HRMS when MS/MS analysis was applied to 4a-OAc-NO2 with a high collision energy (20 eV). Thus, the need for a high collision energy in MS/MS analysis agrees with the theoretical results depicted in Fig. 7.
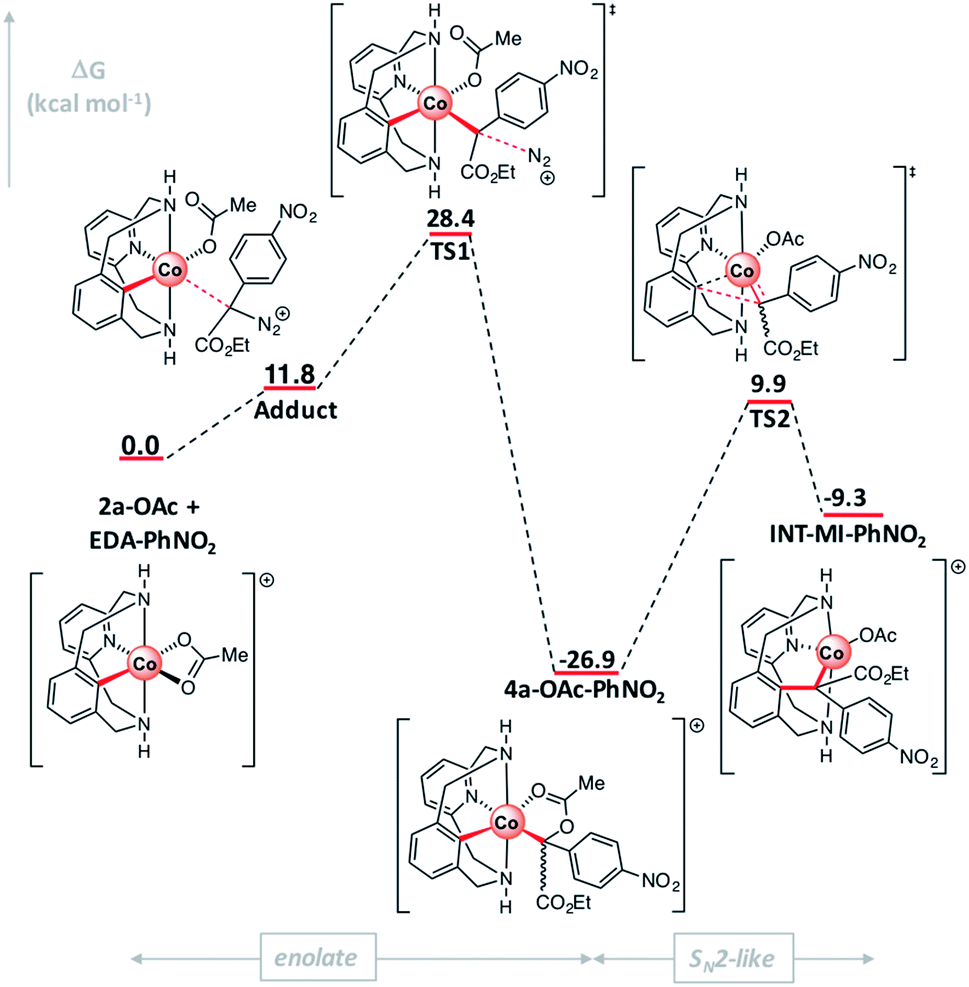 | ||
| Fig. 7 Gibbs energy profile of C-metalated aryl-Co(III) enolate formation and the SN2-type event using diazoacetate l. Relative Gibbs energy values are given in kcal mol−1 (see Fig. S26† for details). | ||
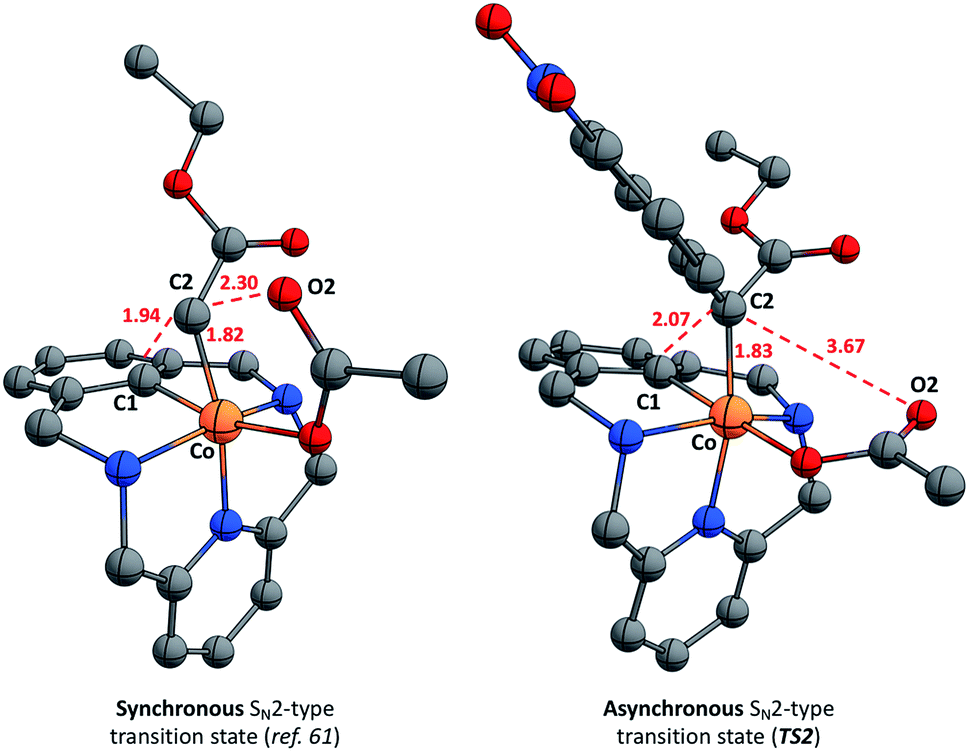 | ||
| Fig. 8 Computed transition state structures of the corresponding concerted SN2-type C–C bond forming step with ethyl diazoacetate (left, synchronous) and diazoacetate l (right, asynchronous). Selected bond distances are depicted in red (Å). | ||
Experimental results clearly indicate that in the presence of 1.0 equiv. of Li(OTf) the reaction proceeded with excellent yields while in its absence poor yields were obtained (see Scheme 4, 3l). Thus, we considered the explicit inclusion of a lithium cation in the computational analysis of the C–O cleavage/C–C bond formation event (Fig. 9). Again, the interaction of the solvated Li+ with the complex is exergonic (ΔG = −2.4 kcal mol−1, see Fig. S29†).
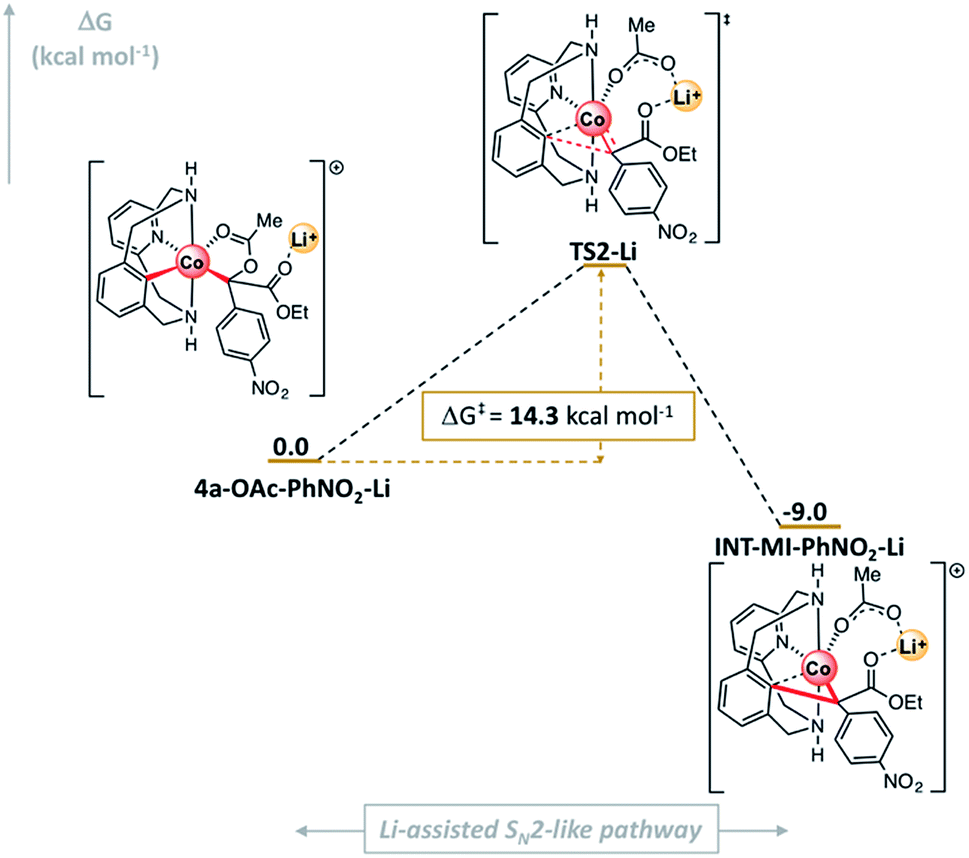 | ||
| Fig. 9 Gibbs energy profile of the SN2-type event using diazoacetate l in the presence of Li+. Relative Gibbs energy values are given in kcal mol−1 (see Fig. S28† for details). | ||
Then, 4a-OAc-PhNO2-Li evolves to INT-MI-NO2-Li (ΔG = −9.0 kcal mol−1; Fig. 9) overcoming a barrier of 14.3 kcal mol−1 in an asynchronous exergonic process. Again, the C–O bond cleavage is followed by the C–C bond formation, which determines the structure of TS2-Li. Strikingly, when a Li+ ion interacts with the organometallic 4a-OAc-NO2 complex, the activation barrier for the SN2-like pathway decreases by 22.5 kcal mol−1 (ΔΔG‡ = 36.8 (TS2, Fig. 7) – 14.3 (TS2-Li, Fig. 9) kcal mol−1). Thus, when Lewis acids are added to the reaction media, they coordinate to the ester and carboxylate moieties in 4a-OAc-PhNO2, turning the latter into better leaving groups through a LA-mediated carboxylate activation and subsequently favoring the formation of the cyclic amide product 3l.
It should be noted that the SN2-type event occurs in an electrophilic tertiary carbon in an asynchronous manner, which constitutes a rare example of bimolecular substitution reaction. Subsequently, after the unprecedented Co(III)-mediated intramolecular SN2-type C–C bond formation, INT-MI-PhNO2 evolves to 3 through a protodemetalation step and a subsequent LA-assisted cyclization (Scheme 2), as reported in previously reported Co(III)-catalyzed diazoacetate coupling protocols.47,48,51,61
After studying the effect of Lewis acids on the formation of cyclic amides 3, we turned our attention to the final cobalt species generated after reaction completion. In previous studies, organometallic 2a-OAc and 4a-OAc were successfully applied as catalysts for the functionalization of 1a-H with EDA under an inert atmosphere, which excludes reaction pathways that imply oxidation state changes.61 To obtain further evidence of a plausible redox-neutral reactivity, XAS spectroscopy was applied to determine the oxidation state of cobalt at the end of the reaction, as well as its possible coordination environment (Scheme 9). The XAS spectrum of the final reaction crude shows a rising edge at 7720 eV with a pre-edge centered at 7710.5, consistent with that of previously reported Co(III) species from our group.60,61 These XAS features suggest that the final cobalt species is a Co(III) compound, indicating a redox neutral mechanism in which cobalt does not change its oxidation state. Indeed, this redox-neutral mechanism was previously proposed in similar transformations with Cp*Co(III) catalysts.46–51
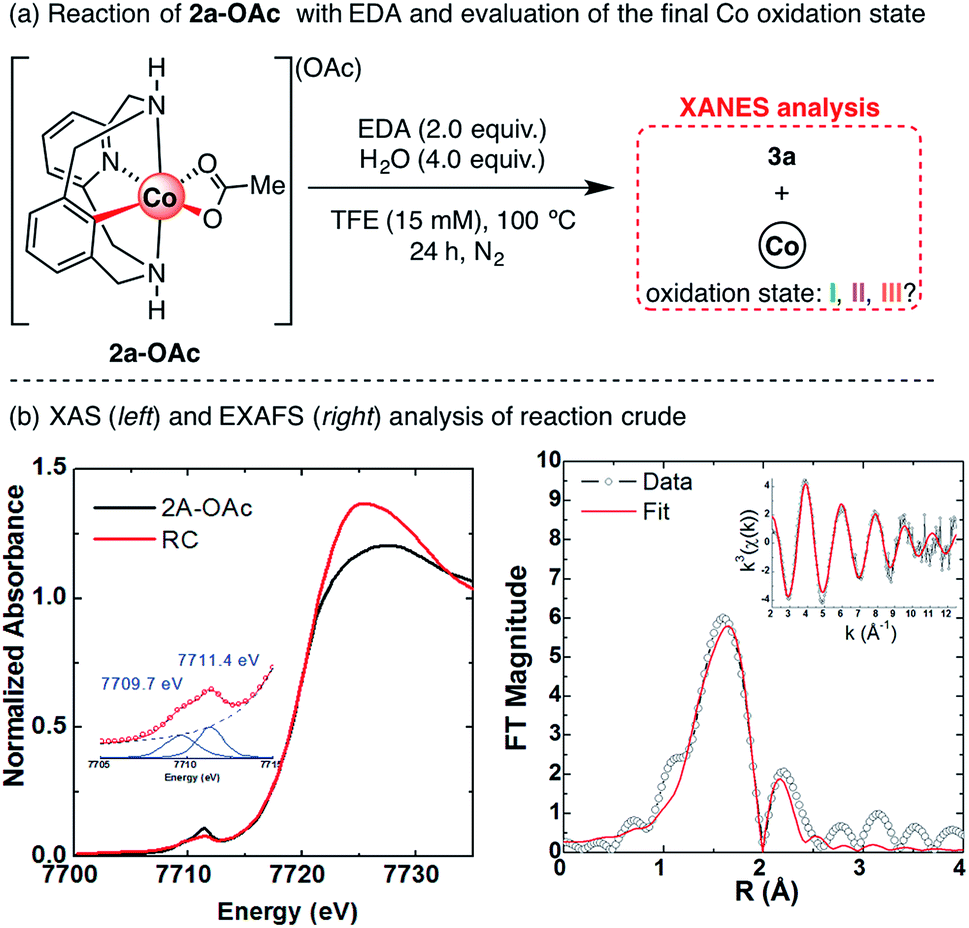 | ||
| Scheme 9 (a) Evolution of 4a-OAc to 3a and (b) XAS of the crude reaction mixture. Left: XANES region showing the rising edge of final “Co+n” species overlaid with the 2a-OAc spectrum previously reported; inset: fitted pre-edge of final “Co+n” species. Right: Fourier-transformed EXAFS spectra of final “Co+n” species; inset: k3-weighted unfiltered EXAFS spectra. | ||
Further EXAFS analysis (Scheme 9b, right) shows a first coordination sphere consisting of two shells of scattering atoms with two/three N/O atoms in the 2.00–2.04 Å range and three/four N/O atoms in the 2.14–2.18 Å range. Furthermore, taking into account that scattering paths from possible acetate ligands bound in a bidentate fashion significantly improve the fit, the presence of two acetate ligands coordinating the Co(III) centre is plausible (see Table S7†). The shorter scattering shell is consistent with what might be expected for a Co(III) coordination sphere; however, the longer scattering shell suggests some decomposition of Co(III) to Co(II) due to the previously reported reductant effect of ethyl diazoacetate.65 Indeed, Co(II) acetate has a pre-edge centered at ∼7709.5 eV,66 similar to the 7709.7 eV fitted pre-edge peak in Scheme 9. Thus, the obtained XANES analysis results, together with previously reported examples,45–50,60 suggest a redox-neutral mechanism in which Co(III) does not change its oxidation state.
Conclusions
In summary, a family of C-metalated aryl-Co(III) enolates or masked-carbenes has been synthesized using diazoacetates and they have been characterized through several techniques including XRD. The scope of diazoacetates has been evaluated, showing a clear preference for electron-poor carbene precursors to furnish the corresponding macrocyclic amide products through a proposed SN2-type pathway. Indeed, exhaustive experimental and theoretical studies indicate that an intramolecular SN2-type process occurs from 4a-X complexes, which are necessary intermediates to deliver the alkyl fragment to construct the new C–C bond. Further studies with α-substituted diazoesters also suggest an unprecedented intramolecular asynchronous SN2-type pathway occurring in tertiary carbons. Furthermore, the experimental key role of Lewis acids is reflected in theoretical studies, unveiling a LA-carboxylate-activation event that considerably favours the C–O bond cleavage/C–C bond formation. The profound mechanistic understanding of this carboxylate-assisted taming of otherwise metastable carbenes is expected to become a new tool in the design of novel carbene-based metal-catalyzed C–C transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the European Research Council for the Starting Grant Project ERC-2011-StG-277801 to X. R. and from MINECO of Spain for project CTQ2016-77989-P to A. C. and X. R. and project CTQ2014-52525-P to J. M. L. We also thank the Generalitat de Catalunya for projects 2017SGR264 and 2014SGR931. We thank the MECD and MINECO of Spain for a FPU grant to O. P. and a FPI grant to S. R. X. R. also thanks ICREA for an ICREA Acadèmia award, and COST CHAOS (CA15106). We acknowledge SOLEIL for provision of synchrotron radiation facilities and we would like to thank Dr Landrot Gautier for assistance in using beamline SAMBA. We also thank Dr Laura Gómez (HRMS) and Xavier Fontrodona (XRD) from Serveis Tècnics de Recerca of the Universitat de Girona.Notes and references
- T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef PubMed.
- L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC.
- P. Thansandote and M. Lautens, Chem.–Eur. J., 2009, 15, 5874–5883 CrossRef PubMed.
- G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC.
- T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212–11222 CrossRef PubMed.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef PubMed.
- S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912–4924 RSC.
- L. Ackermann, Modern Arylation Methods, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009 Search PubMed.
- L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef PubMed.
- L. Wang and L. Ackermann, Chem. Commun., 2014, 50, 1083–1085 RSC.
- J. Miao and H. Ge, Eur. J. Org Chem., 2015, 2015, 7859–7868 CrossRef.
- G. Pototschnig, N. Maulide and M. Schnürch, Chem.–Eur. J., 2017, 23, 9206–9232 CrossRef PubMed.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef PubMed.
- J. E. Zweig, D. E. Kim and T. R. Newhouse, Chem. Rev., 2017, 117, 11680–11752 CrossRef PubMed.
- L. Ilies and E. Nakamura, in C-H Bond Activation and Catalytic Functionalization II, ed. P. H. Dixneuf and H. Doucet, Springer International Publishing, Cham, 2016, pp. 1–18 Search PubMed.
- M. Su, C. Li and J. Ma, J. Chin. Chem. Soc., 2016, 63, 828–840 CrossRef.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef PubMed.
- W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef.
- W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef PubMed.
- J. Yamaguchi, K. Muto and K. Itami, Top. Curr. Chem., 2016, 374 Search PubMed.
- R. Becker and W. D. Jones, in Catalysis without Precious Metals, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010, pp. 143–164 Search PubMed.
- K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef PubMed.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef.
- D. Wei, X. Zhu, J.-L. Niu and M.-P. Song, ChemCatChem, 2016, 8, 1242–1263 CrossRef.
- P. G. Chirila and C. J. Whiteoak, Dalton Trans., 2017, 46, 9721–9739 RSC.
- S. Wang, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2017, 53, 3165–3180 RSC.
- T. Yoshino and S. Matsunaga, Adv. Synth. Catal., 2017, 359, 1245–1262 CrossRef.
- Y. Kommagalla and N. Chatani, Coord. Chem. Rev., 2017, 350, 117–135 CrossRef.
- O. Planas, P. G. Chirila, C. J. Whiteoak and X. Ribas, Adv. Organomet. Chem., 2018, 69, 209–282 CrossRef.
- S. Maity, R. Kancherla, U. Dhawa, E. Hoque, S. Pimparkar and D. Maiti, ACS Catal., 2016, 6, 5493–5499 CrossRef.
- X. Yu, K. Chen, S. Guo, P. Shi, C. Song and J. Zhu, Org. Lett., 2017, 19, 5348–5351 CrossRef PubMed.
- K. Kanamori, W. E. Broderick, R. F. Jordan, R. D. Willett and J. I. Legg, J. Am. Chem. Soc., 1986, 108, 7122–7124 CrossRef.
- W. E. Broderick, K. Kanamori, R. D. Willett and J. I. Legg, Inorg. Chem., 1991, 30, 3875–3881 CrossRef.
- T. Avilés, A. Dinis, M. J. Calhorda, P. Pinto, V. Félix and M. G. B. Drew, J. Organomet. Chem., 2001, 625, 186–194 CrossRef.
- L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209–10212 CrossRef PubMed.
- Y. Baek, S. Kim, B. Jeon and P. H. Lee, Org. Lett., 2016, 18, 104–107 CrossRef PubMed.
- K. Ramakrishna and C. Sivasankar, Eur. J. Org. Chem., 2017, 2017, 4035–4043 CrossRef.
- B. G. Das, A. Chirila, M. Tromp, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2016, 138, 8968–8975 CrossRef PubMed.
- Y. Wang, X. Wen, X. Cui, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2017, 139, 1049–1052 CrossRef PubMed.
- T. Ikeno, I. Iwakura and T. Yamada, J. Am. Chem. Soc., 2002, 124, 15152–15153 CrossRef PubMed.
- H. Lu, W. I. Dzik, X. Xu, L. Wojtas, B. de Bruin and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 8518–8521 CrossRef PubMed.
- S. Roy, S. K. Das and B. Chattopadhyay, Angew. Chem., Int. Ed., 2018, 57, 2238–2243 CrossRef PubMed.
- C. te Grotenhuis, N. van den Heuvel, J. I. van der Vlugt and B. de Bruin, Angew. Chem., Int. Ed., 2018, 57, 140–145 CrossRef PubMed.
- C. te Grotenhuis, B. G. Das, P. F. Kuijpers, W. Hageman, M. Trouwborst and B. de Bruin, Chem. Sci., 2017, 8, 8221–8230 RSC.
- T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 775–779 CrossRef PubMed.
- J. H. Kim, S. Greßies and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5577–5581 CrossRef PubMed.
- D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508–4511 CrossRef PubMed.
- J. Li, M. Tang, L. Zang, X. Zhang, Z. Zhang and L. Ackermann, Org. Lett., 2016, 18, 2742–2745 CrossRef PubMed.
- S.-Y. Yan, P.-X. Ling and B.-F. Shi, Adv. Synth. Catal., 2017, 359, 2912–2917 CrossRef.
- X.-G. Liu, S.-S. Zhang, J.-Q. Wu, Q. Li and H. Wang, Tetrahedron Lett., 2015, 56, 4093–4095 CrossRef.
- S. Qu and C. J. Cramer, J. Org. Chem., 2017, 82, 1195–1204 CrossRef PubMed.
- F. Hu, Y. Xia, C. Ma, Y. Zhang and J. Wang, Chem. Commun., 2015, 51, 7986–7995 RSC.
- M. Font, F. Acuña-Parés, T. Parella, J. Serra, J. M. Luis, J. Lloret-Fillol, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 4373 CrossRef PubMed.
- A. Casitas, M. Canta, M. Solà, M. Costas and X. Ribas, J. Am. Chem. Soc., 2011, 133, 19386–19392 CrossRef PubMed.
- A. Casitas, A. E. King, T. Parella, M. Costas, S. S. Stahl and X. Ribas, Chem. Sci., 2010, 1, 326 RSC.
- M. Rovira, S. Roldán-Gómez, V. Martin-Diaconescu, C. J. Whiteoak, A. Company, J. M. Luis and X. Ribas, Chem.–Eur. J., 2017, 23, 11662–11668 CrossRef PubMed.
- X. Ribas and M. Devillard, Chem.–Eur. J., 2018, 24, 1222–1230 CrossRef PubMed.
- W. Zhou, S. Zheng, J. W. Schultz, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2016, 138, 5777–5780 CrossRef PubMed.
- W. Zhou, J. W. Schultz, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2015, 137, 7604–7607 CrossRef PubMed.
- O. Planas, C. J. Whiteoak, V. Martin-Diaconescu, I. Gamba, J. M. Luis, T. Parella, A. Company and X. Ribas, J. Am. Chem. Soc., 2016, 138, 14388–14397 CrossRef PubMed.
- O. Planas, S. Roldán-Gómez, V. Martin-Diaconescu, T. Parella, J. M. Luis, A. Company and X. Ribas, J. Am. Chem. Soc., 2017, 139, 14649–14655 CrossRef PubMed.
- Y. Aihara and N. Chatani, Chem. Sci., 2013, 4, 664–670 RSC.
- A. Yokota, Y. Aihara and N. Chatani, J. Org. Chem., 2014, 79, 11922–11932 CrossRef PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, 2006, vol. 69 Search PubMed.
- J. R. Wolf, C. G. Hamaker, J.-P. Djukic, T. Kodadek and L. K. Woo, J. Am. Chem. Soc., 1995, 117, 9194–9199 CrossRef.
- M. Mrak, N. N. Tušar, A. Ristić, I. Arčon, F. Thibault-Starzyk and V. Kaučič, Microporous Mesoporous Mater., 2002, 56, 257–266 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 1825084 (4a-OPiv), 1825085 (4a-OBz), 1825086 (4a-OBz-NO2), and 1825087 (4a-OBz-COMe) contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc00851e |
| ‡ Equally contributing author. |
| This journal is © The Royal Society of Chemistry 2018 |