DOI:
10.1039/C8SC01942H
(Edge Article)
Chem. Sci., 2018,
9, 5747-5757
Chiral Brønsted acid-catalyzed intramolecular SN2′ reaction for enantioselective construction of a quaternary stereogenic center†
Received
29th April 2018
, Accepted 3rd June 2018
First published on 5th June 2018
Abstract
An enantioselective intramolecular anti-SN2′ cyclization reaction for the construction of a quaternary stereogenic center was accomplished through the activation of the leaving group using a binaphthol-derived phosphoramide as the chiral Brønsted acid catalyst. The present allylic substitution reaction is beneficial not only for the regioselective nucleophilic substitution at the multi-substituted site of the double bond but also for controlling the stereochemical outcome because of using a geometrically defined double bond. Indeed, the reaction afforded synthetically useful amino alcohol derivatives having a tetra-substituted carbon center in a highly enantioselective manner in most cases, in which the modification of the sulfonamide unit of the phosphoramide catalyst was demonstrated to improve the enantioselectivity. Experimental and theoretical elucidation of the reaction mechanism suggested that the reaction proceeds through a synchronous anti-SN2′ pathway, although NMR monitoring of the reaction indicated the formation of the phosphorimidate ester via the SN2 reaction of the catalyst with the substrate, which results in catalyst deactivation. Further theoretical studies of the origin of the stereochemical outcome at the generated quaternary stereogenic center were performed. Structural analysis of the transition states at the enantio-determining step revealed that the distinct discrimination of the substituents attached to the geometrically defined double bond is achieved by the anthryl and sulfonamide substituents of the catalyst through the three-point hydrogen bonding interactions and the T-shaped C–H⋯π interactions.
Introduction
The catalytic enantioselective construction of a quaternary stereogenic center continues to be a challenging issue in organic synthesis (Fig. 1).1 One of the conventional methods for constructing the quaternary stereogenic center is nucleophilic addition to a polarized double bond, such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CN, or CC, having two substituents at the addition site. In fact, the enantioselective nucleophilic addition to a ketone (CO) or a ketimine (CN) as an electrophile has been extensively investigated and a wide variety of chiral catalysts have been employed to control the stereochemical outcome of the addition products in an enantioselective manner (Fig. 1a).2 Among established approaches, organocatalytic protocols have drawn much attention in terms of metal-free conditions.3 However, in general, ketones and ketimines are intrinsically less reactive than their corresponding aldehydes and aldimines due to steric congestion at the reaction site. To enhance the reactivity of these electrophiles, functional and structural modifications, for instance, the introduction of an electron-withdrawing group or a ring system, are necessary. In this regard, electrophiles that are applicable to these nucleophilic additions are rather limited particularly in organocatalytic processes.2b,4 In contrast to these conventional methods, we have directed our attention to an allylic substitution reaction, namely, the SN2′ reaction, which is initiated by the activation of a leaving group using a Brønsted acid catalyst, as an alternative and efficient method for the construction of a quaternary stereogenic center (Fig. 1b). We have anticipated that the allylic substitution reaction under acidic conditions would proceed through a positively charged transition state and hence the introduction of multiple substituents to the reaction site would lead to the stabilization of the transition state, which in turn would facilitate the formation of a tetra-substituted carbon center, despite the increase in steric congestion around the reaction site. In addition, the relative position of the leaving group to the substituents at the reaction site is apparently defined by the geometry of the double bond by which these two substituents are discriminated, i.e., near the leaving group side, R1, and far from it, R2 (Fig. 1b). It can be considered that the discrimination of these two substituents at the reaction site is the key to achieving high enantioselectivity. Consequently, the proposed method is quite feasible for constructing a quaternary stereogenic center in an enantioselective manner. Indeed, it was accomplished by means of an intramolecular SN2′ reaction using a chiral Brønsted acid catalyst. We disclose herein the developed method in detail and discuss the mechanistic insight into the present SN2′ reaction on the basis of experimental and theoretical studies.
O, CN, or CC, having two substituents at the addition site. In fact, the enantioselective nucleophilic addition to a ketone (CO) or a ketimine (CN) as an electrophile has been extensively investigated and a wide variety of chiral catalysts have been employed to control the stereochemical outcome of the addition products in an enantioselective manner (Fig. 1a).2 Among established approaches, organocatalytic protocols have drawn much attention in terms of metal-free conditions.3 However, in general, ketones and ketimines are intrinsically less reactive than their corresponding aldehydes and aldimines due to steric congestion at the reaction site. To enhance the reactivity of these electrophiles, functional and structural modifications, for instance, the introduction of an electron-withdrawing group or a ring system, are necessary. In this regard, electrophiles that are applicable to these nucleophilic additions are rather limited particularly in organocatalytic processes.2b,4 In contrast to these conventional methods, we have directed our attention to an allylic substitution reaction, namely, the SN2′ reaction, which is initiated by the activation of a leaving group using a Brønsted acid catalyst, as an alternative and efficient method for the construction of a quaternary stereogenic center (Fig. 1b). We have anticipated that the allylic substitution reaction under acidic conditions would proceed through a positively charged transition state and hence the introduction of multiple substituents to the reaction site would lead to the stabilization of the transition state, which in turn would facilitate the formation of a tetra-substituted carbon center, despite the increase in steric congestion around the reaction site. In addition, the relative position of the leaving group to the substituents at the reaction site is apparently defined by the geometry of the double bond by which these two substituents are discriminated, i.e., near the leaving group side, R1, and far from it, R2 (Fig. 1b). It can be considered that the discrimination of these two substituents at the reaction site is the key to achieving high enantioselectivity. Consequently, the proposed method is quite feasible for constructing a quaternary stereogenic center in an enantioselective manner. Indeed, it was accomplished by means of an intramolecular SN2′ reaction using a chiral Brønsted acid catalyst. We disclose herein the developed method in detail and discuss the mechanistic insight into the present SN2′ reaction on the basis of experimental and theoretical studies.
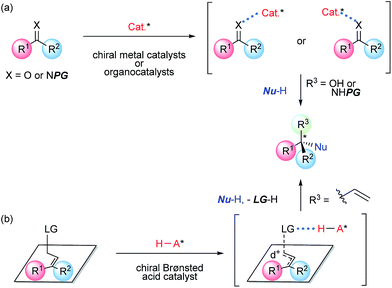 |
| | Fig. 1 Enantioselective construction of quaternary stereogenic center via nucleophilic addition vs. nucleophilic substitution. | |
Results and discussion
Reaction system to be established
The asymmetric allylic substitution reaction, represented by the Tsuji–Trost reaction, has been intensively investigated and a number of examples using chiral catalysts, including transition metals and Lewis acids, have been reported to date.5,6 In contrast, the use of a chiral Brønsted acid catalyst,7 namely, a metal-free process, has largely been unexplored8 and no example has been reported for the catalytic enantio-control of the quaternary stereogenic center despite the marked benefits of the proposed allylic substitution reaction. In this context, taking advantage of the allylic substitution reaction via the activation of the leaving group by a chiral Brønsted acid catalyst, we envisioned a novel approach to the enantioselective construction of a quaternary stereogenic center under metal-free conditions.
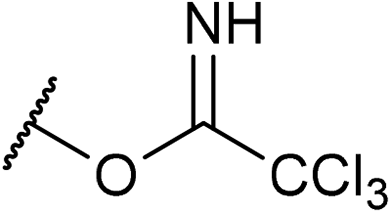
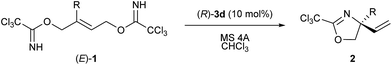
To validate the proposed method, we employed allylic substrate 1 having trichloroacetimidate moieties at both ends of the allylic position (Scheme 1), which can be readily prepared from butyne diol in two steps in the geometrically pure (E)-form.9 Under acidic conditions, bis-trichloroacetimidate 1 undergoes the SN2′ reaction, in which one of the trichloroacetimidate moieties functions as a leaving group whereas the other is utilized as a nucleophile. The reaction is capable of an intramolecular process in which the leaving group and the nucleophilic moiety are assembled into a geometrically well-arranged system and the substitution mode is restricted to an SN2′ fashion. More importantly, despite expecting the formation of regioisomers 2 and 2′ principally in the proposed system, it can be anticipated that the nucleophilic substitution reaction would predominantly proceed through the multi-substituted site, affording regioisomer 2 having a quaternary stereogenic center, because of resonance stabilization of the positively charged transition state by additional substituent R. In this context, bis-trichloroacetimidate 1 (R ≠ H) is an ideal substrate for the enantioselective construction of the quaternary stereogenic center under acidic conditions.
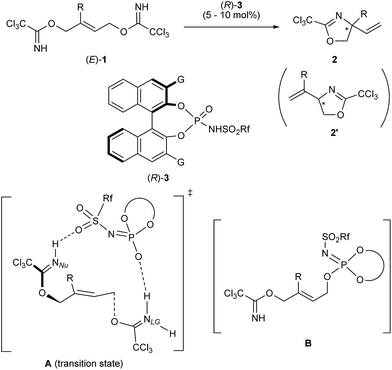 |
| | Scheme 1 Enantioselective allylic substitution reaction of bis-trichloroacetimidate 1 catalyzed by chiral phosphoramide derivative 3. | |
Transition metal complexes as well as achiral Lewis and Brønsted acids have been used as efficient catalysts for this type of transformation.10 In addition, an enantioselective variant using a chiral palladium catalyst has been reported,11 although substrate 1 has no substituent (R = H) at the vinyl position, affording the product having a tertiary stereogenic center. It should be emphasized here that the enantio-control of the quaternary stereogenic center has never been reported despite providing a practical access to synthetically useful amino alcohol derivatives 2 (R ≠ H) in an enantio-enriched form. We successfully demonstrated that chiral phosphoramide derivative 3 functioned as an efficient catalyst for the intramolecular SN2′ reaction of bis-trichloroacetimidate 1, affording enantio-enriched amino alcohol derivatives 2 bearing a quaternary stereogenic center (R ≠ H). Furthermore, the mechanistic elucidation of the present SN2′ reaction was also thoroughly investigated. It was experimentally and theoretically clarified that the reaction proceeded through synchronous anti-SN2′ pathway A, as illustrated in Scheme 1, whereas the formation of phosphorimidate ester B was also detected by 1H and 31P NMR measurements during the course of the allylic substitution reaction. However, ester B, which was generated by the nucleophilic substitution reaction, namely, the SN2 reaction, of catalyst 3 with 1 at the terminal allylic position far from R group, was proven to be the less active species. Thus, catalyst 3 was deactivated by this side reaction, although covalently linked phosphate esters were recently reported as an active intermediate in chiral phosphoric acid catalysis.12 The proposed reaction pathway, which involves the deactivation pathway, was also confirmed by theoretical studies of a model system. These theoretical studies also indicated that the sulfonamide unit introduced to the phosphorus center is the key to accelerating the proposed synchronous anti-SN2′ reaction through transition state A. Thus, the oxygen atom of the sulfonamide group interacts with the proton attached to the nucleophilic imidate moiety through the hydrogen bond and increases the nucleophilicity of the nitrogen atom NNu. Prior to the activation of the nucleophilic site NNu, the anti-SN2′ reaction is initiated through the protonation of imidate nitrogen NLG of the leaving group by the acidic H–O moiety of 3, giving rise to the substitution product 2 in a highly enantioselective manner in most cases.
Optimization of reaction conditions and chiral phosphoric acid catalyst
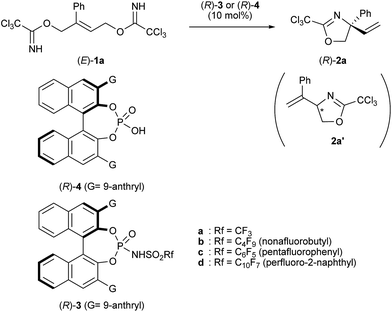
Initially, chiral phosphoric acid 4 (G = anthryl) was employed to confirm the efficiency of the parent catalyst. As shown in Table 1, the reaction of geometrically pure (E)-1a was conducted using catalyst (R)-4 (10 mol%) in dichloromethane at room temperature. However, only a trace amount of substitution product 2a was formed presumably due to the low acidity of catalyst 4 (Table 1, entry 1).13 In order to enhance the acidity of the catalyst, we employed phosphoramide derivative (R)-3.14 As expected, catalyst 3a having a triflate group, the most representative phosphoramide catalyst having a high acidity, accelerated the reaction markedly in dichloromethane at room temperature. Desired cyclized product 2a bearing a quaternary stereogenic center was obtained exclusively in good yield and no regioisomer 2a′ was formed at all, albeit the low enantioselectivity (27% ee) (Table 1, entry 2).15 Further screening for the reaction solvent revealed that chloroform was best in terms of catalytic efficiency and slight enhancement of the enantioselectivity was detected (Table 1, entries 3–5), although it was still not high enough (36% ee). Lowering the reaction temperature to −40 °C dramatically enhanced the enantioselectivity (80% ee) (Table 1, entry 6). However, a significant amount of starting bis-trichloroacetimidate 1a remained unchanged even after 24 h. This serious problem was surmounted by adding molecular sieves (MS) 4A (Table 1, entry 7)16 and product 2a was formed in excellent yield within 6 h without marked loss of enantioselectivity (Table 1, entry 7 vs. 6). In the presence of MS 4A, catalyst loading could be reduced to 5 mol% with no decrease in yield or enantioselectivity of 2a (Table 1, entry 8). To further enhance the enantioselectivity, structural modification of the chiral acid catalyst was examined. Chiral phosphoric acids and their derivatives were commonly modified at structurally adjustable sites, such as the introduction of substituent G at 3,3′-positions, the chiral framework, and the acidic functionality. Among the approaches reported, 3,3′-substituent G has been widely investigated. However, the enantioselectivity could not be improved despite extensive screening for substituents G in the present reaction.17 The screening for the chiral frameworks was also unsuccessful.17 The anthryl substituent and the BINOL framework were a worthwhile combination in terms of enantioselectivity; however, these still gave unsatisfactory results (80% ee). We then turned our attention to the acidic functionality. Although it has been investigated in terms of an increase in catalyst acidity,18 it has been anticipated that the introduction of a suitable substituent to the acidic functionality would be an intriguing approach for enantioselectivity enhancement.18d Indeed, as shown in entries 9–11, Table 1, marked influence on the enantioselectivity was noted by changing the substituent at the sulfonamide moiety.19 Although the introduction of a longer perfluoroalkyl chain, namely, nonafluorobutyl, instead of trifluoromethyl, resulted in the reduction of the enantioselectivity (Table 1 entry 9), the catalyst having a perfluoroaryl group enhanced the enantioselectivity (Table 1, entries 10 and 11) and, in particular, the perfluoro-2-naphthyl substituent markedly improved the enantioselectivity. Further reduction of the temperature to −60 °C resulted in the formation of 2a with the highest enantioselectivity (95% ee) among the conditions tested (Table 1, entry 12). The absolute configuration of 2a was determined to be (R) by derivatization into a structurally known compound.20
Table 1 Screening for reaction conditions and catalyst in intramolecular allylic substitution reactiona
|
|
| Entry |
Catalyst |
Solvent |
Conditions |
Yieldb (%) |
eec (%) |
|
Unless otherwise noted, all reactions were carried out using 0.01 mmol of (R)-3 or (R)-4 (10 mol%), 0.1 mmol of (E)-1a, and MS 4A (50 mg) in the indicated solvent (1.0 mL).
Isolated yield.
ee was determined by chiral stationary phase HPLC analysis.
MS 4A was used.
5 mol% of (R)-3 was used.
|
| 1 |
(R)-4 |
CH2Cl2 |
RT, 24 h |
Trace |
n.d. |
| 2 |
(R)-3a |
CH2Cl2 |
RT, 0.5 h |
92 |
27 |
| 3 |
(R)-3a |
CHCl3 |
RT, 0.5 h |
95 |
36 |
| 4 |
(R)-3a |
Toluene |
RT, 24 h |
34 |
24 |
| 5 |
(R)-3a |
MeCN |
RT, 0.5 h |
72 |
<1 |
| 6 |
(R)-3a |
CHCl3 |
−40 °C, 24 h |
29 |
80 |
| 7d |
(R)-3a |
CHCl3 |
−40 °C, 6 h |
95 |
79 |
| 8d,e |
(R)-3a |
CHCl3 |
−40 °C, 24 h |
94 |
80 |
| 9d,e |
(R)-3b |
CHCl3 |
−40 °C, 24 h |
79 |
65 |
| 10d,e |
(R)-3c |
CHCl3 |
−40 °C, 24 h |
92 |
83 |
| 11d,e |
(R)-3d |
CHCl3 |
−40 °C, 24 h |
96 |
89 |
| 12d |
(R)-3d |
CHCl3 |
−60 °C, 48 h |
85 |
95 |
Scope of substrates
The substrate scope of the present transformation was demonstrated in the reaction using a series of aryl- or alkynyl-substituted bis-trichloroacetimidate 1 and optimized catalyst (R)-3d. The reactivity of 1 was markedly dependent on the properties of the R substituent, such as the electronic nature and the steric hindrance. As expected, the reaction of the substrate having an electron-donating group at the aryl moiety proceeded rapidly. In contrast, the substrate having an electron-withdrawing group at the aryl moiety decelerated the reaction markedly. Therefore, the reaction temperature was optimized for each substrate. As shown in Table 2, substrate 1 having a meta- or para-substituted aryl moiety underwent the intramolecular substitution reaction efficiently under the optimized reaction conditions, affording SN2′-substituted product 2 in good yield with high enantioselectivity (around 90% ee), irrespective of the electronic nature of the aryl moieties (entries 1–7). In contrast, ortho-substitution at the aryl moiety had a negative effect on the enantioselectivity or the chemical yield (entries 8–10). In particular, an ortho-methyl substituent retarded the reaction significantly and only a trace amount of the desired product was formed (entry 9).21 Although an alkynyl substituent exhibited moderate enantioselectivity (entry 12), substrates having a fused aryl, namely, 2-naphthyl (entry 11), and thiophen-3-yl (entry 13) as the heteroaryl compounds were also applicable to the present reaction, affording the substitution products in good enantioselectivities. In contrast, the use of a substrate having an alkyl substituent, such as a benzyl group (R = Bn), resulted in no reaction. The substrate-dependent reactivity observed in the present reaction suggests that the conjugation between the R substituent and the allylic moiety is essential for the progress of the reaction.
Table 2 Substrate scope of enantioselective intramolecular SN2′ reactiona
|
|
| Entry |
1 and 2(R) |
Conditions |
Yieldb (%) |
eec (%) |
|
Unless otherwise noted, all reactions were carried out using 0.01 mmol of (R)-3d (10 mol%), 0.1 mmol of (E)-1, and MS 4A (50 mg) in chloroform (1.0 mL).
Isolated yield.
ee was determined by chiral stationary phase HPLC analysis.
5 mol% of (R)-3d was used.
|
| 1d |
b: 4-MeOC6H4– |
−60 °C, 12 h |
98 |
79 |
| 2d |
c: 4-MeC6H4– |
−60 °C, 12 h |
96 |
90 |
| 3 |
d: 4-ClC6H4– |
−50 °C, 48 h |
90 |
91 |
| 4 |
e: 3-MeOC6H4– |
−50 °C, 12 h |
90 |
90 |
| 5 |
f: 3-MeC6H4– |
−60 °C, 48 h |
97 |
91 |
| 6 |
g: 3-ClC6H4– |
−40 °C, 36 h |
88 |
88 |
| 7 |
h: 3-CF3C6H4– |
−40 °C, 48 h |
86 |
89 |
| 8 |
i: 2-MeOC6H4– |
−40 °C, 12 h |
90 |
60 |
| 9 |
j: 2-MeC6H4– |
rt, 24 h |
Trace |
— |
| 10 |
k: 2-FC6H4– |
−20 °C, 12 h |
87 |
87 |
| 11d |
l: 2-Naphthyl |
−60 °C, 12 h |
85 |
86 |
| 12 |
m: Ph–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– C– |
−40 °C, 48 h |
55 |
60 |
| 13d |
n: Thiophen-3-yl |
−60 °C, 8 h |
95 |
91 |
Derivatization of product
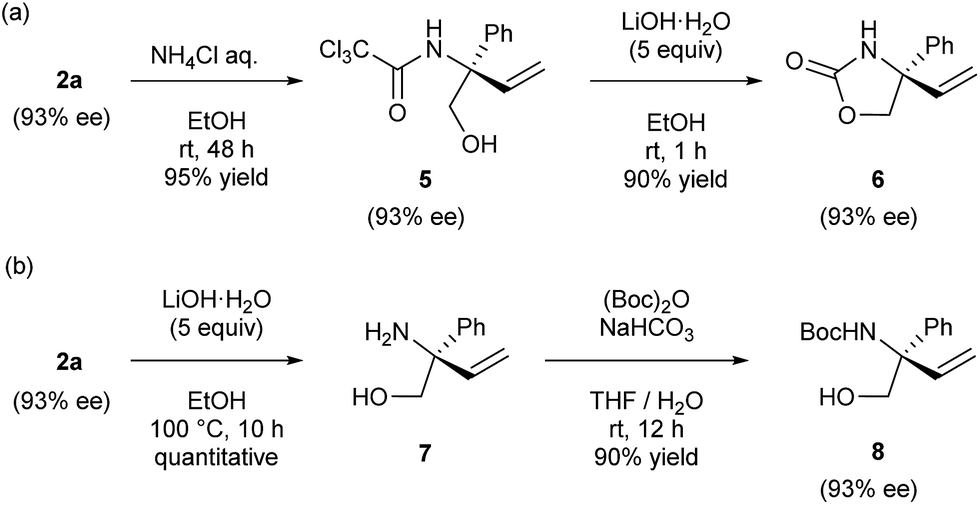
The derivatization of product 2a was next demonstrated (Scheme 2). Under acidic conditions, the oxazoline moiety of product 2a readily underwent partial hydrolysis, affording corresponding amide 5. Subsequent treatment with lithium hydroxide furnished cyclic carbamate 6 in good yield (Scheme 2a). Under basic conditions, the complete hydrolysis of the oxazoline moiety of product 2a took place and this was followed by protection at the nitrogen atom of resultant amino alcohol 7 with Boc group to furnish alcohol 8 in good yield (Scheme 2b). During the course of these derivatizations, no loss of enantiomeric purity was observed in both cases.
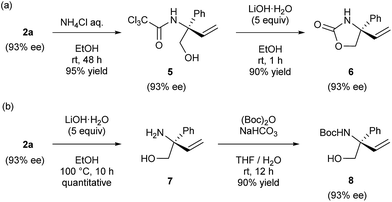 |
| | Scheme 2 Derivatization of product 2a. | |
Experimental elucidation of reaction mechanisms
Mechanistic study 1 (using deuterated substrate).
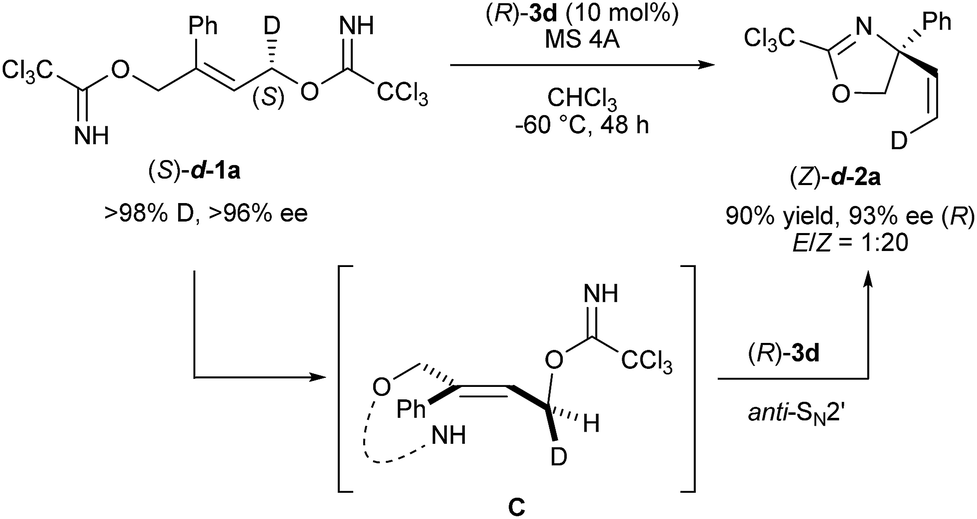
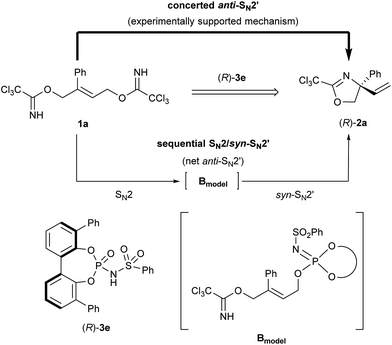
As high enantioselectivities were achieved in the intramolecular SN2′ reaction using chiral phosphoramide catalyst 3d, the mechanistic elucidation of the present reaction became our next focus. At the outset of mechanistic studies, we employed enantiomerically pure bis-trichloroimidate (S)-d-1a, in which deuterium was introduced at the methylene position near the leaving group,22 to identify the concerted process (Scheme 3). In the proposed deuterated substrate study, the stereochemical relationship between the chirality at the deuterated carbon of starting (S)-d-1a and the geometry of the migrated double bond, coupled with the newly generated quaternary stereogenic center, conveys crucial information of whether the reaction proceeds through the SN2′ or SN1 mechanism. As shown in Scheme 3, enantiomerically enriched (Z)-d-2a (93% ee) was obtained in high geometrical selectivity, clearly suggesting that the reaction proceeded through the concerted mechanism, namely, the SN2′ pathway, and not the stepwise mechanism by way of an allyl cation intermediate. More importantly, product (Z)-d-2a had (R)-absolute configuration with 93% ee. The observed stereochemical relationship indicates that the nucleophilic imidate and the leaving one are in an anti-relationship C, as illustrated in Scheme 3, and hence the anti-SN2′ pathway is considered to be the rational mechanism of the present substitution reaction.
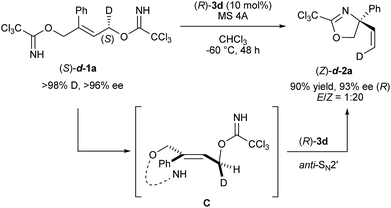 |
| | Scheme 3 Mechanistic study using enantio-enriched deuterated substrate (S)-d-1a. | |
Mechanistic study 2 (NMR monitoring of reaction mixture).
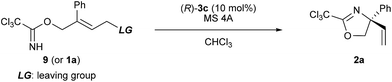
Recently, some examples have emerged indicating that the chiral phosphoric acid undergoes a substitution reaction with the substrate, where the acid catalyst functions as a nucleophile, to generate the corresponding phosphate ester as the reactive intermediate.12 As depicted in Scheme 4, in principle, phosphorimidate B, which was generated through the SN2 reaction of catalyst 3 with 1 at the terminal allylic position far from R group, might be involved as the reactive intermediate in the present reaction. Because the SN2 reaction, giving B with inversion at the substitution site, followed by the syn-SN2′ reaction through D, namely, the net anti-SN2′ pathway, also fulfils the requirement of the experimental evidence obtained in the deuterated substrate experiment as shown in Scheme 3. Considering recent reports12 and the sequential SN2/syn-SN2′ pathway, 31P NMR monitoring was conducted to detect the formation of phosphorimidate B. NMR monitoring was performed using 10 mol% of catalyst 3c and 1a in the presence of MS 4A in CDCl3 at −40 °C (Fig. 2).23 At the beginning of the monitoring, phosphorimidate B (R = Ph, Rf = C6F5) was detected (Fig. 2a),24 although phosphoramide catalyst 3c was not completely transformed into B and a considerable amount of 3c remained even after 4 h (Fig. 2b).25
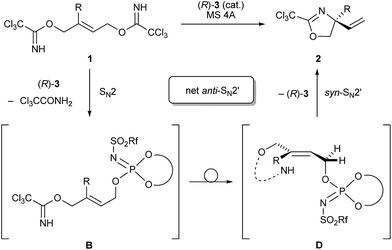 |
| | Scheme 4 Sequential SN2/syn-SN2′ mechanism as a plausible pathway for the formal anti-SN2′ process. | |
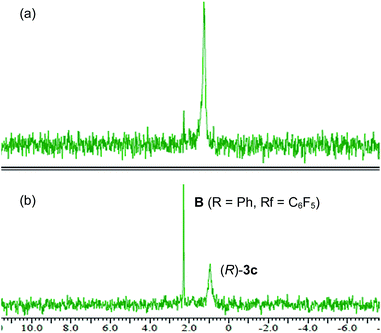 |
| | Fig. 2
31P NMR spectra for the reaction of 1a catalyzed by (R)-3c at −40 °C after (a) 15 min and (b) 4 h. | |
Mechanistic study 3 (substrate having a different leaving group).
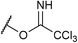
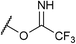
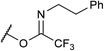
If the reaction would proceed through phosphorimidate B, the leaving group, namely, the imidate moiety far from R group, is not involved in the enantio-determining step (EDS). This is because the quaternary stereogenic center is formed from B, in which the imidate leaving group has been cleaved off from the substrate. We then turned our attention to the use of substrate 9 having a different leaving group (LG). As shown in Table 3, LG significantly influenced the enantioselectivity of product 2a.26 These results reveal that the leaving group is involved in the EDS and hence phosphorimidate B would not be an active intermediate in the actual reaction pathway.
Table 3 Substrate having different leaving groupa
| Entry |
LG
|
Substrate |
Conditions |
Yieldb (%) |
eec (%) |
|
Unless otherwise noted, all reactions were carried out using 0.005 mmol of (R)-3c (10 mol%), 0.05 mmol of (E)-9 or (E)-1a, and MS 4A (25 mg) in chloroform (0.5 mL).
Isolated yield.
ee was determined by chiral stationary phase HPLC analysis.
5 mol% of (R)-3c was used.
Not detected.
|
| 1d |
|
1a
|
−40 °C, 24 h |
92 |
83 |
| 2 |
|
9a
|
−40 °C, 18 h |
90 |
60 |
| 3 |
|
9b
|
−40 °C, 20 h |
90 |
31 |
| 4 |
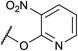
|
9c
|
rt, 24 h |
NDe |
— |
|
Theoretical studies of reaction mechanisms
As the concerted anti-SN2′ pathway is supported by the above experimental studies of the mechanistic elucidation, we further conducted DFT calculation to confirm the actual reaction pathway and elucidate the mechanism of the concerted anti-SN2′ process and the origin of the stereochemical outcome.
Model system.
To identify the energy profile of the present SN2′ reaction, the model system of phosphoramide 3e and standard substrate 1a was employed to streamline the calculation (Scheme 5).27 In the model system, phosphoramide catalyst 3 was simplified to 3e having phenylsulfonamide instead of perfluoroarylsulfonamide and a biphenyl backbone generating an axially chiral structure in which (R)-chirality was introduced in accordance with the actual reaction system. In addition, the generation of major enantiomer (R)-2a was calculated as observed in the actual stereochemical outcome. For the calculation, the energy profile of the sequential SN2/syn-SN2′ pathway, namely, the net anti-SN2′, involving phosphorimidate Bmodel as the intermediate was also considered and compared with that of the concerted anti-SN2′ pathway that is supported by the above experimental results.28 All calculations were performed with the Gaussian 16 package (Revision B.01).29 Geometries were optimized and characterized using frequency calculations at the B97D/6-31G(d) level.30,31 Gibbs free energies in the solution phase were calculated using single-point energy calculations at the same level according to the PCM solvation model (chloroform: ε = 4.9) for the optimized structures.32
 |
| | Scheme 5 Model system for calculation. | |
First, the energy profile of the sequential SN2/syn-SN2′ (net anti-SN2′) pathway was identified to confirm whether intermediate Bmodel undergoes further syn-SN2′ reaction or not (Fig. 3), because this sequential pathway was experimentally proven to be negative. In accordance with the experimental confirmation of the structure of B,24 the formation of the allylic phosphate ester was considered as the first SN2 step. Therefore, as shown in CP-1, the substitution reaction was initiated through the protonation of imidate nitrogen NLG of the leaving group by the H–N moiety of 3e. The following SN2 reaction proceeded via transition state TS-1 in which the phosphoryl oxygen attacked the carbon having the leaving group to generate phosphate ester intermediate INT-1. Transition state analysis of the subsequent syn-SN2′ step was thoroughly conducted using the phosphate ester INT-2 (=Bmodel).26 As a result, in contrast to the relatively small energy barrier of TS-1 (the first SN2 step), TS-2 (the second syn-SN2′ step) exhibited a larger energy barrier than TS-1. Based on the present energy profile of the sequential SN2/syn-SN2′ pathway, the initial SN2 step was considered to be a catalyst deactivation process. Indeed, as shown in Table 2, entry 9, in the reaction of substrate 1j having an ortho-methylphenyl substituent, a significant amount of phosphate ester was detected by NMR monitoring21 and a trace amount of the desired product was formed.
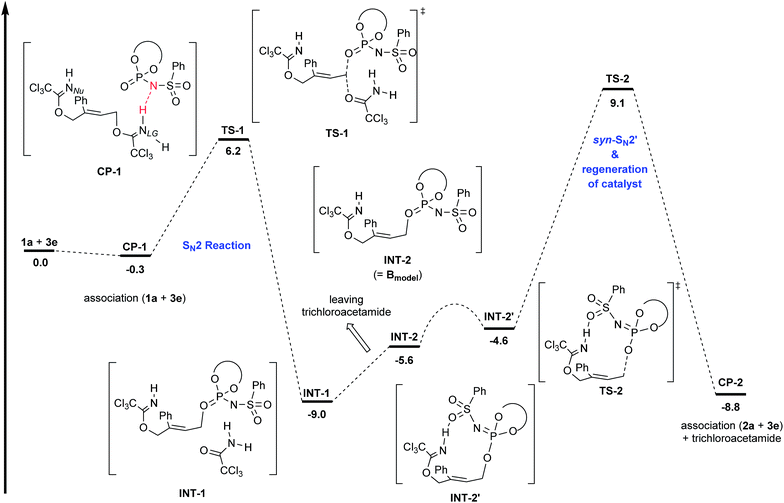 |
| | Fig. 3 Energy profile of the sequential SN2/syn-SN2′ (net anti-SN2′) pathway for the model reaction. The potential energy for the sum of 1a and 3e was set to zero. Geometries were optimized and characterized using frequency calculations at the B97D/6-31G(d) level. Gibbs free energies (kcal mol−1) in solution phase were calculated using single-point energy calculations at the same level as those for the optimized structures according to the SCRF method based on PCM (CHCl3). | |
The energy profile of the concerted anti-SN2′ pathway for the model system is shown in Fig. 4. In order to identify the transition state of the concerted anti-SN2′ pathway, a variety of initial structures were thoroughly generated, in which catalyst H–N protonated the imidate nitrogen NLG of the leaving group, analogous to CP-1 (Fig. 3). However, all attempts to find the transition states of the concerted anti-SN2′ pathway were unsuccessful and resulted in the transition states of the SN2 reaction of the phosphoryl oxygen at the carbon atom having the leaving group, analogous to TS-1 (Fig. 3). Taking these unsuccessful results into consideration, we surmised that the hydrogen bond acceptor site generated on the catalyst conjugate base is too far to interact with the proton attached to nucleophilic nitrogen NNu. We therefore attempted to switch the acidic site, namely, the hydrogen bond donor site, of 3e. Proton shift from the initial H–N form to the phosphoryl oxygen generated H–O acid of the catalyst.33 As shown in CP-3, the generated H–O acid protonates nitrogen atom NLG to activate the leaving group, whereas the oxygen atom of the sulfonamide moiety interacts with the hydrogen atom to enhance the nucleophilicity of nitrogen atom NNu. Because the bond connection sequence was elongated, NNu–H⋯OS–NP–O–H⋯NLG, the concerted anti-SN2′ pathway became feasible as a rational reaction mechanism. In fact, after thorough screening for the transition states, corresponding TS-3 was successfully calculated. Careful analysis of TS-3 indicates that the reaction does not proceed in a completely concerted fashion because the leaving group has been cleaved off prior to the C3–NNu bond formation. In TS-3, the cleaving C1–O bond of the leaving group elongates to 1.97 Å, whereas the distance between C3 and NNu is 2.89 Å and still too large to form a C3–NNu bond. However, subsequent analysis of the intrinsic reaction coordinate (IRC) from TS-3 revealed the formation of ternary associated complex CP-4 consisting of product 2a, catalyst 3e, and trichloroacetamide. Consequently, the present reaction mechanism is considered to proceed in a synchronous anti-SN2′ fashion rather than the ideal concerted pathway. More importantly, the comparable energy levels between TS-3 (Fig. 4) and TS-1 (Fig. 3) imply that these pathways compete with each other. In fact, the formation of phosphate ester B was observed during the course of NMR monitoring, in which the leaving group was cleaved off from the reaction system prior to the C3–NNu bond formation step, whereas enantioselectivity was markedly dependent on the leaving group and hence the leaving group was considered to be involved in EDS, namely, the C3–NNu bond formation step. Although there seems to be a contradiction, these results are well rationalized if these pathways would take place in parallel, as evaluated in the calculation.
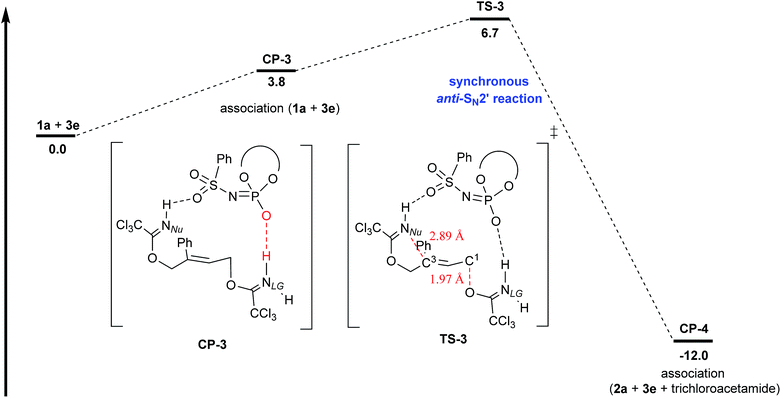 |
| | Fig. 4 Energy profile of the synchronous anti-SN2′ pathway for the model reaction. The potential energy for the sum of 1a and 3e was set to zero. Geometries were optimized and characterized using frequency calculations at the B97D/6-31G(d) level. Gibbs free energies (kcal mol−1) in solution phase were calculated using single-point energy calculations at the same level as those for the optimized structures according to the SCRF method based on PCM (CHCl3). | |
Real system.
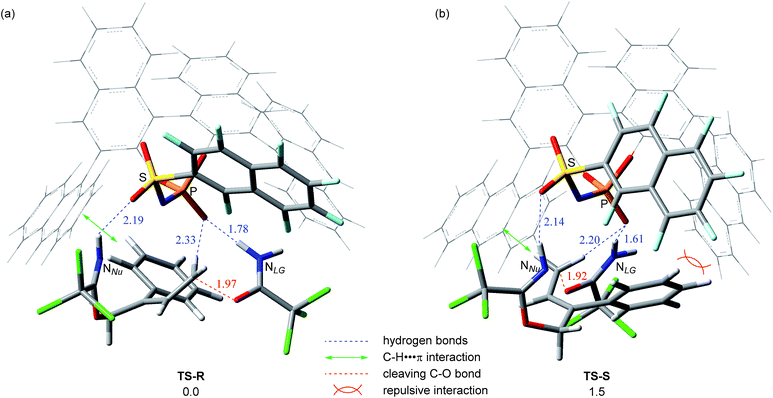
The product formation pathway was clarified on the basis of the experimental studies and the energy profiles of the model calculations. We next turned our attention to the elucidation of the origin of the stereochemical outcome at the EDS, in particular, the intriguing substituent effect at the catalyst sulfonamide unit. As shown in Table 1, entries 8–11, the substituent at the sulfonamide unit influenced the enantioselectivity. In order to gain an insight into the origin of the stereochemical outcome as well as the substituent effect at the catalyst sulfonamide unit, we calculated the transition state of the EDS, namely, the synchronous anti-SN2′ step, using substrate 1a and (R)-3d, instead of simplified 3e. The calculated transition states for both major and minor pathways of the reaction using 1a are shown in Fig. 5.34 The energy difference between these two transition states is 1.5 kcal mol−1 with the one resulting in (R)-2a through TS-R being the energetically favoured one (Fig. 5a). The result of the calculations is consistent with the experimental finding that (R)-2a is the major enantiomer in the reaction of 1a catalyzed by (R)-3d.
 |
| | Fig. 5 Transition states of synchronous anti-SN2′ reaction of 1a catalyzed by (R)-3d. Geometries were optimized and characterized using frequency calculations at the B97D/6-31G(d) level. Relative Gibbs free energies (kcal mol−1) obtained by single-point energy calculations at the same level as those for the optimized structures with the SCRF method based on PCM (CHCl3) are shown. (a) Transition state for the formation of (R)-2a. (b) Transition state for the formation of (S)-2a. | |
In both transition states, the relative location of substrate 1a and catalyst (R)-3d is defined by three-point hydrogen bonds. The H–O acid moiety of 3d protonates nitrogen atom NLG of the leaving group, whereas the oxygen atom of the sulfonamide moiety interacts with the hydrogen atom attached to nucleophilic nitrogen atom NNu. These two hydrogen bonds are essential to accelerate the anti-SN2′ reaction. Meanwhile, an additional non-classical C–H⋯O hydrogen bond between the allylic hydrogen and the phosphoryl oxygen is formed in both transition structures.35 These three-point hydrogen bonding interactions, coupled with the use of a geometrically defined substrate, are the key to achieving the high enantioselectivity because the distinct discrimination of the substituents attached to the double bond is achieved by the anthryl and sulfonamide substituents of catalyst (R)-3d.36 Indeed, when the location of the nucleophilic NNu is fixed at the left front side, as shown in Fig. 5, the positions of the phenyl substituent and the leaving group are switched in these two transition states. In transition state TS-R (Fig. 5a) affording the major enantiomer, the phenyl substituent is oriented to occupy the sterically less hindered back side and further stabilizes this transition state through the T-shaped C–H⋯π interaction37,38 with the left side anthryl substituent of the catalyst. In transition state TS-S, the T-shaped C–H⋯π interaction also takes place between the allylic hydrogen of the substrate and the left side anthryl substituent of the catalyst (Fig. 5b). However, the phenyl substituent that locates to the right front side induces repulsive interaction with the right side anthryl and perfluoronaphthyl substituents of the catalyst. The substituent effect of the sulfonamide unit of the catalyst, as shown in Table 1, entries 8–11, is also well rationalized on the basis of these transition structures. This is because in TS-S, the phenyl substituent locates closely to the sulfonamide unit of the catalyst, inducing a repulsive interaction between them. The significant effect of the leaving group, as shown in Table 3, entry 3 in particular (introduction of the phenethyl substituent at the imidate nitrogen atom), is also rationalized on the basis of these transition states in which energetically favourable TS-R is destabilized by the steric congestion induced between the right side anthryl substituent of the catalyst and the modified leaving group.
Conclusions
We have accomplished an enantioselective intramolecular anti-SN2′ cyclization reaction for the construction of a quaternary stereogenic center through the activation of the leaving group using a chiral Brønsted acid catalyst. Taking advantage of the present allylic substitution protocol not only for regioselective bond formation at the multi-substituted site but also for controlling the stereochemical outcome under acidic conditions, the reaction affords synthetically useful amino alcohol derivatives having a tetra-substituted carbon center in a highly enantioselective manner in most cases. To improve the enantioselectivity, modification of the sulfonamide unit of the phosphoramide catalyst was found to be an efficient approach. Experimental and theoretical studies elucidated the reaction pathway and the origin of the stereochemical outcomes at the generated quaternary stereogenic center. Experimental elucidation of the reaction mechanism suggests that the reaction proceeds through a concerted anti-SN2′ pathway, although the formation of the phosphorimidate ester, which is considered to deactivate the catalyst, was observed during 31P NMR monitoring of the reaction. The Gibbs free energy profile formulated by the theoretical calculations indicates that the reaction proceeds through a synchronous anti-SN2′ pathway rather than an ideal concerted pathway. Structural analysis of real model transition states at the enantio-determining step revealed that the distinct discrimination of the allyl substrate is achieved by the anthryl and sulfonamide substituents of the catalyst through the three-point hydrogen bonding interactions and the T-shaped C–H⋯π interactions, because of using a geometrically defined allylic substrate. Further studies of the development of enantioselective allylic substitution reactions using other types of nucleophiles are underway.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” from The MEXT, Japan (No. 23105002).
Notes and references
-
(a) C. J. Douglas and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5363–5367 CrossRef PubMed;
(b)
J. Christoffers and A. Baro, Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed;
(c) B. M. Trost and C. Jiang, Synthesis, 2006, 369–396 CrossRef;
(d) C. Hawner and A. Alexakis, Chem. Commun., 2010, 46, 7295–7306 RSC;
(e) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593–4623 RSC;
(f) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682–2694 CrossRef PubMed;
(g) K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef PubMed;
(h) Y. Liu, S.-J. Han, W.-B. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740–751 CrossRef PubMed;
(i) F. Vetica, R. M. de Figueiredo, M. Orsini, D. Tofani and T. Gasperi, Synthesis, 2015, 47, 2139–2184 CrossRef;
(j) X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef PubMed;
(k) J. Feng, M. Holmes and M. J. Krische, Chem. Rev., 2017, 117, 12564–12580 CrossRef PubMed.
-
(a)
J. G. de Vries, Science of Synthesis, Stereoselective Synthesis 1, Stereoselective Reactions of Carbon-Carbon Double Bonds, Georg Thieme Verlag KG, 2010 Search PubMed;
(b)
G. A. Molander, Science of Synthesis, Stereoselective Synthesis 2, Stereoselective Reactions of Carbonyl and Imino Groups, Georg Thieme Verlag KG, 2010 Search PubMed;
(c)
P. A. Evans, Science of Synthesis, Stereoselective Synthesis 3, Stereoselective Pericyclic Reactions, Cross Coupling, and C-H and C-X Activation, Georg Thieme Verlag KG, 2010 Search PubMed.
- For reviews on organocatalysts, see:
(a)
Enantioselective Organocatalysis: Reactions and Experimental Procedures, ed. P. I. Dalko, Wiley-VCH, New York, 2007 Search PubMed;
(b)
Science of Synthesis, Asymmetric Organocatalysis 1, Lewis Base and Acid Catalysts, ed. B. List, Georg Thieme Verlag KG, New York, 2012 Search PubMed;
(c)
Science of Synthesis, Asymmetric Organocatalysis 2, Brønsted Base and Acid Catalysts, and Additional Topics, ed. K. Maruoka, Georg Thieme Verlag KG, New York, 2012 Search PubMed.
- For selected recent examples on organocatalysis, see:
(a) M. Rueping, T. Theissmann, A. Kuenkel and R. M. Koenigs, Angew. Chem., Int. Ed., 2008, 47, 6798–6801 CrossRef PubMed;
(b) N. Hara, R. Tamura, Y. Funahashi and S. Nakamura, Org. Lett., 2011, 13, 1662–1665 CrossRef PubMed;
(c) J. Zhang, X. Liu, X. Ma and R. Wang, Chem. Commun., 2013, 49, 3300–3302 RSC;
(d) O. D. Engl, S. P. Fritz and H. Wennemers, Angew. Chem., Int. Ed., 2015, 54, 8193–8197 CrossRef PubMed;
(e) A. Kondoh, Y. Ota, T. Komuro, F. Egawa, K. Kanomata and M. Terada, Chem. Sci., 2016, 7, 1057–1062 RSC;
(f) T. Takeda, A. Kondoh and M. Terada, Angew. Chem., Int. Ed., 2016, 55, 4734–4737 CrossRef PubMed;
(g) S. Nakamura, N. Matsuda and M. Ohara, Chem.–Eur. J., 2016, 22, 9478–9482 CrossRef PubMed.
-
(a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef PubMed;
(b) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258–297 CrossRef PubMed;
(c) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427–440 RSC;
(d) N. A. Butt and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929–7967 RSC.
-
(a) A. Alexakis, J. E. Bäckvall, N. Krause, O. Pàmies and M. Diéguez, Chem. Rev., 2008, 108, 2796–2823 CrossRef PubMed;
(b) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef PubMed.
- For seminal works of chiral phosphoric acid catalysts, see:
(a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef PubMed;
(b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef PubMed; For selected reviews of chiral phosphoric acid, see:
(c) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef PubMed;
(d) G. Adair, S. Mukherjee and B. List, Aldrichimica Acta, 2008, 41, 31–39 Search PubMed;
(e) M. Terada, Chem. Commun., 2008, 4097–4112 RSC;
(f) M. Terada, Synthesis, 2010, 1929–1982 CrossRef;
(g) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef PubMed.
-
(a) M. Rueping, U. Uria, M.-Y. Lin and I. Atodiresei, J. Am. Chem. Soc., 2011, 133, 3732–3735 CrossRef PubMed;
(b) P.-S. Wang, X.-L. Zhou and L.-Z. Gong, Org. Lett., 2014, 16, 976–979 CrossRef PubMed;
(c) M. Zhuang and H. Du, Org. Biomol. Chem., 2014, 12, 4590–4593 RSC;
(d) Y. Kuroda, S. Harada, A. Oonishi, Y. Yamaoka, K. Yamada and K. Takasu, Angew. Chem., Int. Ed., 2015, 54, 8263–8266 CrossRef PubMed;
(e) J. Zhou and H. Xie, Org. Biomol. Chem., 2018, 16, 380–383 RSC.
- Y. Ishino, K. Wakamoto and T. Hirashima, Chem. Lett., 1984, 13, 765–768 CrossRef.
-
(a) M. Sabat and C. R. Johnson, Org. Lett., 2000, 2, 1089–1092 CrossRef PubMed;
(b) L. Grigorjeva and A. Jirgensons, Eur. J. Org. Chem., 2011, 2421–2425 CrossRef;
(c) K. Klimovica, L. Grigorjeva, A. Maleckis, J. Popelis and A. Jirgensons, Synlett, 2011, 2849–2851 Search PubMed;
(d) L. Grigorjeva, A. Kinens and A. Jirgensons, J. Org. Chem., 2015, 80, 920–927 CrossRef PubMed;
(e) V. Kumar, K. Klimovica, D. Rasina and A. Jirgensons, J. Org. Chem., 2015, 80, 5934–5943 CrossRef PubMed.
- A. Maleckis, K. Klimovica and A. Jirgensons, J. Org. Chem., 2010, 75, 7897–7900 CrossRef PubMed.
- For the formation of active intermediates through nucleophilic substitution reactions of phosphoric acid catalysts, see:
(a) N. D. Shapiro, V. Rauniyar, G. L. Hamilton, J. Wu and F. D. Toste, Nature, 2011, 470, 245–249 CrossRef PubMed;
(b) Z. Sun, G. A. Winschel, P. M. Zimmerman and P. Nagorny, Angew. Chem., Int. Ed., 2014, 53, 11194–11198 CrossRef PubMed;
(c) L. Liu, M. Leutzsch, Y. Zheng, M. W. Alachraf, W. Thiel and B. List, J. Am. Chem. Soc., 2015, 137, 13268–13271 CrossRef PubMed;
(d) Y. Kuroda, S. Harada, A. Oonishi, H. Kiyama, Y. Yamaoka, K. Yamada and K. Takasu, Angew. Chem., Int. Ed., 2016, 55, 13137–13141 CrossRef PubMed;
(e) J.-H. Tay, A. J. Argüelles, M. D. DeMars II, P. M. Zimmerman, D. H. Sherman and P. Nagorny, J. Am. Chem. Soc., 2017, 139, 8570–8578 CrossRef PubMed.
- The formation of the covalently bonded phosphate ester resembling phosphorimidate ester B (Scheme 1) from the SN2 reaction of parent phosphoric acid 4 with 1a was observed by NMR measurement. This implies that catalyst 4 was deactivated by the formation of this phosphate ester.
- D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626–9627 CrossRef PubMed.
- In contrast with the use of (E)-isomer as the substrate, the use of (Z)-1a resulted in the formation of product 2a in low yield with low enantioselectivity, see ESI† for details.
- Partial hydrolysis of bis-trichloroacetimidate 1a was observed under the reaction conditions. Ammonia was generated during the course of the hydrolysis, which deactivated the acid catalyst to prevent further transformation. Therefore, to remove moisture from the reaction system, MS 4A was added.
- Screening of the catalysts, see ESI† for details.
- For selected examples for modification of phosphoramide catalyst, see:
(a) C. H. Cheon and H. Yamamoto, J. Am. Chem. Soc., 2008, 130, 9246–9247 CrossRef PubMed;
(b) B. Guo, G. Schwarzwalder and J. T. Njardarson, Angew. Chem., Int. Ed., 2012, 51, 5675–5678 CrossRef PubMed;
(c) A. Borovika and P. Nagorny, Tetrahedron, 2013, 69, 5719–5725 CrossRef;
(d) M. Sai and H. Yamamoto, J. Am. Chem. Soc., 2015, 137, 7091–7094 CrossRef PubMed;
(e) P. S. J. Kaib and B. List, Synlett, 2016, 27, 156–158 Search PubMed;
(f) S. Lee, P. S. J. Kaib and B. List, Synlett, 2017, 28, 1478–1480 CrossRef; For selected papers on imidodiphosphates or imidodiphosphoramide catalyst, see:
(g) S. Vellalath, I. Čorić and B. List, Angew. Chem., Int. Ed., 2010, 49, 9749–9752 CrossRef PubMed;
(h) I. Čorić and B. List, Nature, 2012, 483, 315–319 CrossRef PubMed;
(i) S. Das, L. Liu, Y. Zheng, M. W. Alachraf, W. Thiel, C. K. De and B. List, J. Am. Chem. Soc., 2016, 138, 9429–9432 CrossRef PubMed;
(j) L. Liu, P. S. J. Kaib, A. Tap and B. List, J. Am. Chem. Soc., 2016, 138, 10822–10825 CrossRef PubMed;
(k) P. S. J. Kaib, L. Schreyer, S. Lee, R. Properzi and B. List, Angew. Chem., Int. Ed., 2016, 55, 13200–13203 CrossRef PubMed;
(l) N. Tsuji, J. L. Kennemur, T. Buyck, S. Lee, S. Prévost, P. S. J. Kaib, D. Bykov, C. Farès and B. List, Science, 2018, 359, 1501–1505 CrossRef PubMed.
- Catalysts 3c and 3d were synthesized from 3,3′-disubsutituted BINOL and trichloroiminophosphorane, which was prepared from phosphorus pentachloride and the corresponding sulfonamide having a perfluoro substituent at the sulfur atom, see ESI† for details.
- See ESI† for details. Also see:
(a) A. Khan, J. Xing, J. Zhao, Y. Kan, W. Zhang and Y. J. Zhang, Chem.–Eur. J., 2015, 21, 120–124 CrossRef PubMed;
(b) L. Yang, A. Khan, R. Zheng, L. Y. Jin and Y. J. Zhang, Org. Lett., 2015, 17, 6230–6233 CrossRef PubMed.
-
31P NMR monitoring of the reaction mixture was conducted and most of catalyst 3d was
consumed and phosphorimidate ester B (R = 2-MeC6H4), as shown in Scheme 1, was detected as the major phosphorous component.
-
(a) J. S. Cannon, S. F. Kirsch, L. E. Overman and H. F. Sneddon, J. Am. Chem. Soc., 2010, 132, 15192–15203 CrossRef PubMed;
(b) J. S. Cannon, A. C. Olson, L. E. Overman and N. S. Solomon, J. Org. Chem., 2012, 77, 1961–1973 CrossRef PubMed.
- For 31P NMR monitoring of the reaction of 1a catalyzed by 3c (10 mol%), see ESI† for details.
- The structure of substitution product B was estimated by 31P NMR analysis and comparison with a structurally similar compound, because B was too labile to isolate. See ESI† for details.
- Further control experiments were conducted by changing the nucleophilic imidate to a less nucleophilic ester. See ESI† for details.
- Control experiments were carried out to confirm the influence of the cleaved leaving group, such as trichloroacetamide and trifluoroacetamide, on the enantioselectivity. The reaction of 1a in the presence of one equivalent of trifluoroacetamide was performed using catalyst (R)-3c. The observed enantioselectivity of 2a (78% ee) was slightly lower than that of the original result (83% ee). However, it was much higher than that of the reaction of 9a (60% ee). Likewise, the reaction of 9a in the presence of one equivalent of trichloroacetamide afforded 2a in 58% ee, which was comparable to the original result (60% ee) and much lower than that of the reaction of 2a (83% ee). These results indicate that the cleaved leaving group, trihaloacetamide, is not primarily involved in the enantio-determining step, namely, the SN2′ process. See ESI† for details.
- A linear relationship between catalyst % ee and product % ee was observed, strongly suggesting that one catalyst molecule is involved in the reaction. Therefore, a monomeric catalyst was considered in the DFT studies. See ESI† for details.
- For the theoretical calculation of the syn-SN2′ mechanism, see ESI† for details.
-
M. J. Frisch, et al., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMedsee ESI† for the full citation.
- For the B97D method, see: S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef PubMed.
- For Gaussian basis sets, see:
(a) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef;
(b)
W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, John Wiley, New York, USA, 1986, and references cited therein Search PubMed.
- For the PCM model, see:
(a) R. Cammi, B. Mennucci and J. Tomasi, J. Phys. Chem. A, 1999, 103, 9100–9108 CrossRef;
(b) R. Cammi, B. Mennucci and J. Tomasi, J. Phys. Chem. A, 2000, 104, 5631–5637 CrossRef;
(c) M. Cossi, N. Rega, M. Scalmani and V. Barone, J. Chem. Phys., 2001, 114, 5691–5701 CrossRef.
- J. P. Lovie-Toon, C. M. Tram, B. L. Flynn and E. H. Krenske, ACS Catal., 2017, 7, 3466–3476 CrossRef.
- For 3D representations of the transition states from different angle, see ESI† for details.
- For representative and recent examples for C–H⋯O hydrogen bonds as the key interaction in asymmetric catalysis, see:
(a) E. J. Corey and J. J. Rohde, Tetrahedron Lett., 1997, 38, 37–40 CrossRef;
(b) M. N. Grayson and J. M. Goodman, J. Am. Chem. Soc., 2013, 135, 6142–6148 CrossRef PubMed;
(c) P. Maity, R. P. Pemberton, D. J. Tantillo and U. K. Tambar, J. Am. Chem. Soc., 2013, 135, 16380–16383 CrossRef PubMed;
(d) H. Wang, P. Jain, J. C. Antilla and K. N. Houk, J. Org. Chem., 2013, 78, 1208–1215 CrossRef PubMed;
(e) T. Ishii, R. Watanabe, T. Moriya, H. Ohmiya, S. Mori and M. Sawamura, Chem.–Eur. J., 2013, 19, 13547–13553 CrossRef PubMed;
(f) K. Kanomata, Y. Toda, Y. Shibata, M. Yamanaka, S. Tsuzuki, I. D. Gridnev and M. Terada, Chem. Sci., 2014, 5, 3515–3523 RSC;
(g) N. Grimblat, M. Sugiura and S. C. Pellegrinet, J. Org. Chem., 2014, 79, 6754–6758 CrossRef PubMed;
(h) Y. Xie, G.-J. Cheng, S. Lee, P. S. J. Kaib, W. Thiel and B. List, J. Am. Chem. Soc., 2016, 138, 14538–14541 CrossRef PubMed;
(i) M. N. Grayson, Z. Yang and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 7717–7720 CrossRef PubMed;
(j) Y. Takayama, T. Ishii, H. Ohmiya, T. Iwai, M. C. Schwarzer, S. Mori, T. Taniguchi, K. Monde and M. Sawamura, Chem.–Eur. J., 2017, 23, 8400–8404 CrossRef PubMed;
(k) F. Li, T. Korenaga, T. Nakanishi, J. Kikuchi and M. Terada, J. Am. Chem. Soc., 2018, 140, 2629–2642 CrossRef PubMed.
- R. C. Johnston and P. H.-Y. Cheong, Org. Biomol. Chem., 2013, 11, 5057–5064 Search PubMed.
- For C–H⋯π interaction, see:
(a) E. H. Krenske and K. N. Houk, Acc. Chem. Res., 2013, 46, 979–989 CrossRef PubMed;
(b) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef PubMed.
- For representative and recent examples for C–H⋯π as the key interaction in asymmetric catalysis, see:
(a) M. Yamakawa, I. Yamada and R. Noyori, Angew. Chem., Int. Ed., 2001, 40, 2818–2821 CrossRef;
(b) Y. Dang, S. Qu, Z.-X. Wang and X. Wang, J. Am. Chem. Soc., 2014, 136, 986–998 CrossRef PubMed;
(c) G. Jindal and R. B. Sunoj, Angew. Chem., Int. Ed., 2014, 53, 4432–4436 CrossRef PubMed;
(d) S.-S. Meng, Y. Liang, K.-S. Cao, L. Zou, X.-B. Lin, H. Yang, K. N. Houk and W.-H. Zheng, J. Am. Chem. Soc., 2014, 136, 12249–12252 CrossRef PubMed;
(e) B. Bhaskararao and R. B. Sunoj, J. Am. Chem. Soc., 2015, 137, 15712–15722 CrossRef PubMed;
(f) A. Changotra and R. B. Sunoj, Org. Lett., 2016, 18, 3730–3733 CrossRef PubMed;
(g) A. C. Doney, B. J. Rooks, T. Lu and S. E. Wheeler, ACS Catal., 2016, 6, 7948–7955 CrossRef;
(h) M. Pareek and R. B. Sunoj, ACS Catal., 2016, 6, 3118–3126 CrossRef;
(i) T. Korenaga, R. Sasaki, T. Takemoto, T. Yasuda and M. Watanabe, Adv. Synth. Catal., 2018, 360, 322–333 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01942h |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Azusa
Kondoh
a,
Azusa
Kondoh