
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExceptionally rapid oxime and hydrazone formation promoted by catalytic amine buffers with low toxicity†
Dennis
Larsen‡
 ,
Anna M.
Kietrys
,
Spencer A.
Clark
,
Hyun Shin
Park
,
Andreas
Ekebergh§
and
Eric T.
Kool
*
,
Anna M.
Kietrys
,
Spencer A.
Clark
,
Hyun Shin
Park
,
Andreas
Ekebergh§
and
Eric T.
Kool
*
Department of Chemistry, Stanford University, Stanford, CA 94305, USA. E-mail: kool@stanford.edu
First published on 21st May 2018
Abstract
Hydrazone and oxime bond formation between α-nucleophiles (e.g. hydrazines, alkoxy-amines) and carbonyl compounds (aldehydes and ketones) is convenient and is widely applied in multiple fields of research. While the reactants are simple, a substantial drawback is the relatively slow reaction at neutral pH. Here we describe a novel molecular strategy for accelerating these reactions, using bifunctional buffer compounds that not only control pH but also catalyze the reaction. The buffers can be employed at pH 5–9 (5–50 mM) and accelerate reactions by several orders of magnitude, yielding second-order rate constants of >10 M−1 s−1. Effective bifunctional amines include 2-(aminomethyl)imidazoles and N,N-dimethylethylenediamine. Unlike previous diaminobenzene catalysts, the new buffer amines are found to have low toxicity to human cells, and can be used to promote reactions in cellular applications.
Introduction
Bioconjugation techniques are a cornerstone of the field of chemical biology, and much scientific interest has focused on developing new bioorthogonal chemical transformations that can be performed in water at neutral pH.1 Particular attention has been given to developing reactions with very high second-order rate constants in order to promote rapid bond formation even when the reactants are present at low concentrations.2In addition to development of new conjugation reactions, substantial research efforts have gone toward accelerating existing reactions that have broad application but are limited by slow rates. In this light, we have recently focused on developing catalysts for the well-established hydrazone and oxime conjugation reactions.3 Hydrazones and oximes, once formed, can be hydrolytically stable species4 that have been employed extensively for bioconjugations and polymer functionalization.5 The reactions remain appealing because of the simplicity of the reacting groups and the ease of introducing the reactants into biomolecules and probes. However, the relatively slow rate of product formation under neutral conditions (ca. 0.01–0.1 M−1 s−1)1d has limited the utility of these reactions.
To overcome this, researchers have developed strategies to speed up the hydrazone and oxime formation reactions. Gillingham and others have developed specialized aromatic aldehydes or ketones with a boronic acid group in the o-position that result in extremely rapid bond formation with aminooxy groups.6 Another approach to speed up this reaction without requirement of specialized reactant structures is the development of catalysts for hydrazone and oxime formations. Building on seminal work by Cordes and Jencks,7 Dawson introduced the use of aniline as a nucleophilic amine catalyst for hydrazone and oxime bioconjugations,4d,8 and subsequent work in several groups has identified simple aniline derivatives that exceed aniline's catalytic ability.9
We have focused on developing bifunctional catalysts that employ intramolecular proton transfer from acid/base groups in proximity to a nucleophilic amino-group to facilitate rapid hydrazone and oxime formation,3 building on the early work of Hine.10 A substantial advantage of the use of catalysis in this reaction (rather than specialized substrates) is that the reaction can be reversed or equilibrated under the influence of a catalyst, while the products remain relatively stable in its absence. This enables diverse applications such as exchange reactions for cellular imaging,11 dynamic combinatorial selections,8a,12 and reversal of formalin crosslinks in fixed tissue specimens.13
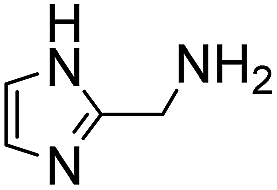
In a recent study, we identified 2-(aminomethyl)imidazoles as a new, highly active bifunctional catalyst scaffold for these transformations.3c Further work in our laboratory led us to realize that these and related compounds have pKa values near the neutral range, and that the best way to increase the catalyst concentration, and thus speed up the desired reaction even further, without exceeding the buffer capacity of the system, is to use the catalysts themselves as buffering agents. Note that these agents are used here in super-stoichiometric amounts, but still follow a catalytic mechanism (see below). Here we study this new strategy of using catalyzing amine buffers (Amine Buffer/Catalysts, ABCs) to dramatically enhance the rate of oxime and hydrazone formation at neutral pH (Fig. 1).
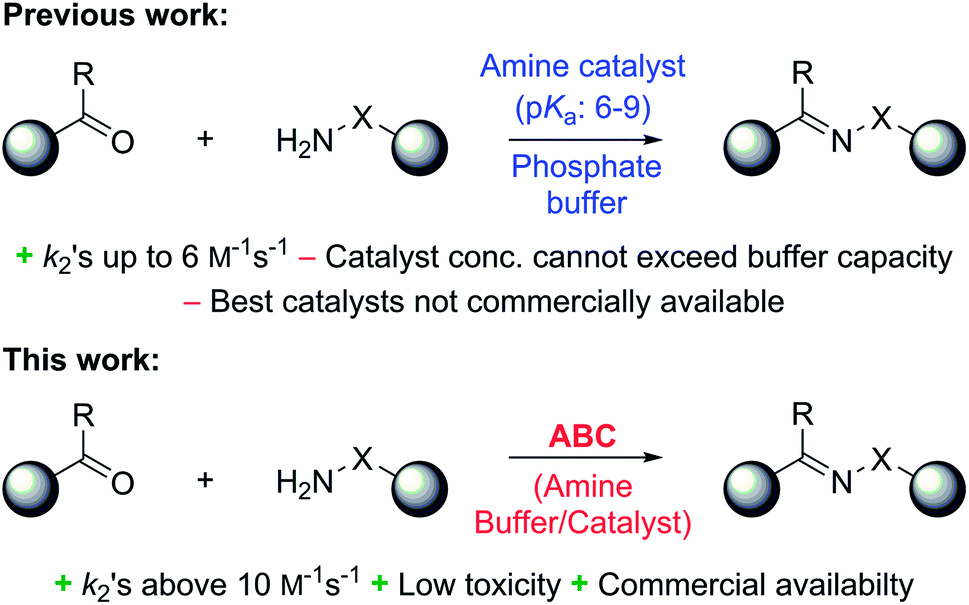 | ||
| Fig. 1 In previous studies, nucleophilic amines were used to catalyze hydrazone (X = NH) and oxime (X = O) formations in a phosphate buffer at pH 7.4. In the present study, nucleophilic amines with proximal acid/base groups with pKa values in the 6–9 range are used both as catalysts for the reactions as well as buffering agents for the aqueous reaction medium. | ||
Results and discussion
ABC screening
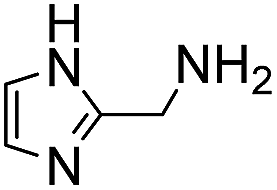
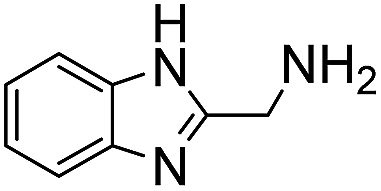
A range of ABCs were chosen for the initial screening (Table 1). To emulate the conditions found in buffered saline solutions often used in biological experiments, all the catalyzing buffers were screened in 150 mM NaCl (saline). Although recent studies have highlighted that high salt concentrations (>1 M) influence formation rates of oximes,14 at the NaCl concentrations applied here we did not see remarkable changes when compared to control experiments with no NaCl (see details in Table S1 in the ESI†). For the initial testing of buffers, we used a buffer concentration of 50 mM, but the best catalyzing buffers were also found to be effective at 10 mM (Table S1†). All the ABCs have an acid/base group with a pKa in the biologically relevant range, defined here as 6–9, ensuring that they can in fact function as buffering agents at biologically relevant pH levels. No pKa values have been reported for 2-(aminomethyl)pyrimidine 20, nor have they been determined for the two phosphonic acids 17 and 18, but they are estimated to fall within the 6–9 pKa range (see full list of known pKa values in Table S2†). A potentiometric titration was used to determine the pKa values of 2-(aminomethyl)imidazole 4 to be 4.38 and 8.13 in water (see ESI†).| No. | Buffer | k 2 (M−1 s−1) | k rel | No. | Buffer | k 2 (M−1 s−1) | k rel |
|---|---|---|---|---|---|---|---|
| a Second-order rate constants, k2, listed as mean values ± standard deviations based on triplicate measurements. Relative reaction rates, krel, in comparison to reaction in phosphate buffer (entry 1). Reactions were performed in saline solution (150 mM NaCl), and buffer pH was adjusted (using either 1 M NaOH or 1 M HCl) to pH 7.40 ± 0.04 prior to use. | |||||||
| 1 | Phosphate | 0.466 ± 0.007 |
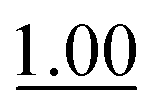
|
11 |
|
0.34 ± 0.01 | 0.72 |
| 2 | Tris | 0.481 ± 0.002 | 1.0 | 12 |
|
2.15 ± 0.02 | 4.6 |
| 3 | HEPES | 0.28 ± 0.02 | 0.60 | 13 |
|
2.77 ± 0.06 | 5.9 |
| 4 |
|
10.6 ± 0.6 | 23 | 14 |
|
2.26 ± 0.02 | 4.9 |
| 5 |
|
8.6 ± 0.9 | 18 | 15 |
|
0.34 ± 0.02 | 0.74 |
| 6 |
|
10.7 ± 0.5 | 23 | 16 |
|
(Not soluble at 50 mM) | |
| 7 |
|
8.0 ± 0.5 | 17 | 17 |
|
(Not soluble at 50 mM) | |
| 8 |
|
0.222 ± 0.004 | 0.48 | 18 |
|
2.9 ± 0.4 | 6.3 |
| 9 |
|
0.304 ± 0.004 | 0.65 | 19 |
|
6.2 ± 0.2 | 13 |
| 10 |
|
0.22 ± 0.01 | 0.46 | 20 |
|
1.46 ± 0.04 | 3.1 |
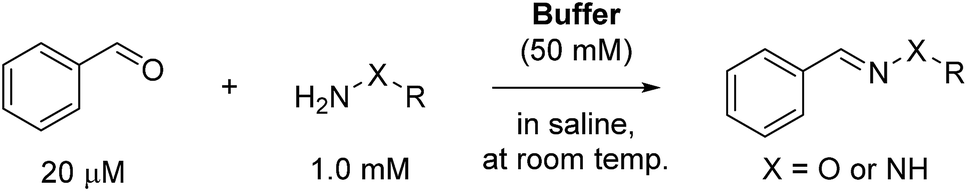
To initially evaluate the catalyzing buffers, we followed the reaction between benzaldehyde and phenylhydrazine (Scheme 1). The reaction is conveniently monitored by absorption at or near 340 nm (approx. λmax of the hydrazone product), and the reaction rates were determined by non-linear regression assuming full conversion of the limiting reagent into product.3c This is a reasonable assumption at a fifty-fold excess of one reagent, and was also confirmed by chromatographic analysis of reactions in ABC 4 (Fig. S9†), eliminating the concern that these buffers can form stable Schiff-bases or (hemi)aminals that could preclude full conversion.
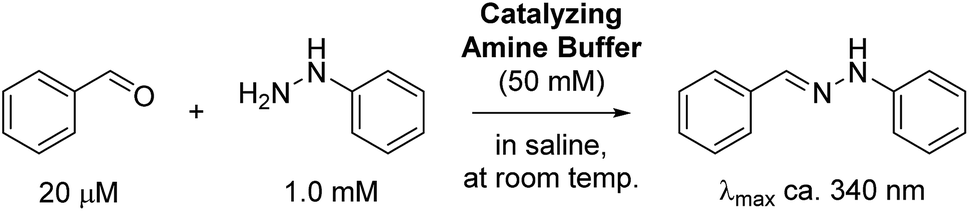 | ||
| Scheme 1 Model reaction between benzaldehyde and phenylhydrazine. | ||
The reaction rate determined in phosphate buffer was used as the point of reference (Table 1, entry 1). Two other common buffers (Tris and HEPES, entries 2 and 3) were also employed for comparison, but although Tris is a primary amine buffer, it did not afford any significant improvement compared to the phosphate buffer. HEPES buffered saline was found to give an even slower reaction rate.
It is worth mentioning that the reaction rate in phosphate buffer reported here (ca. 0.47 M−1 s−1) is more than two-fold faster than the known rate in less concentrated phosphate buffered saline (0.21 M−1 s−1 in ca. 12 mM phosphate),3c illustrating that the reaction is dependent on buffer concentration. This is indicative of a reaction that follows a general acid/base mechanism, and supports Jencks' well-established conclusion that the reaction proceeds via a rate-determining acid-catalyzed dehydration of the intermediate hemiaminal.15
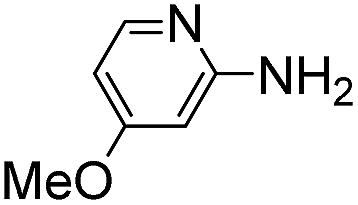
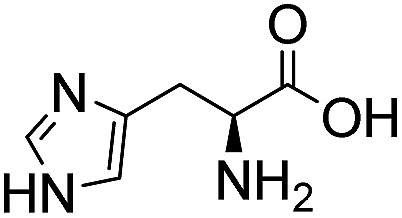
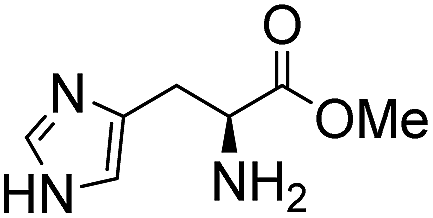
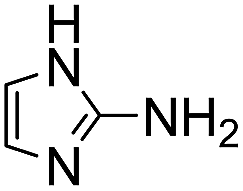
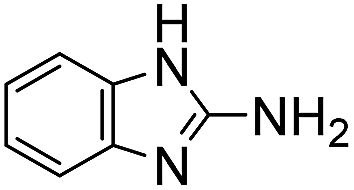
Comparative evaluation of twenty buffers revealed (Table 1) that aminopyridines and aminoimidazoles (compounds 8–11) were the least effective catalysts, giving rates equal to or below that of phosphate buffer. Histidine 12 and its simple derivatives 13 and 14 were moderately effective as catalytic buffers, showing improved reaction rates compared to both phosphate or the more structurally similar and well-known biological buffer imidazole (15), but still falling well below the best buffers in efficiency.
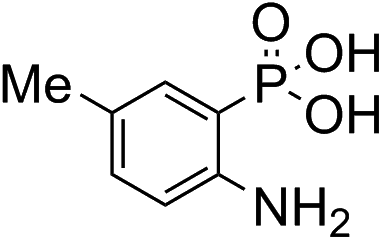
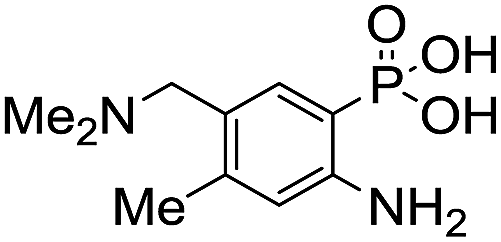
Anthranilic and phosphanilic acids are among the most effective catalysts known for this reaction.3a,3b We found that the poor solubility of anthranilic acids prevent their use at 50 mM, and addition of a ring nitrogen (16) still did not confer solubility at 50 mM. In tests at lower buffer concentrations of 10 mM, 16 was found to give a rate constant almost 10-fold slower than with 4 (Table S1†). Catalyst 17 is the most efficient of previously identified catalysts in hydrazone formation when employed at lower concentrations in phosphate buffered saline.3a It is hampered in this catalyzing buffer context by relatively low solubility at neutral pH (estimated to be in the 15–20 mM range at room temperature, pH 7).13 In fact, when employed as a catalyzing buffer at 10 mM, 17 fared quite well, giving a relatively high second-order rate constant of 3.3 ± 0.2 M−1 s−1, though it was not as high as the 4.6 ± 0.2 M−1 s−1 achieved with ABC 4 at 10 mM concentration (see further examples in Table S1†). Therefore, we synthesized the more water-soluble 18, hoping it would retain the catalytic effect of 17. While 18 was soluble enough to be used at 50 mM, it was also found that the rate enhancements achieved (ca. 6-fold compared to phosphate) were not as high as for the best ABCs 4–7. We attribute the poorer effectiveness of 18 relative to 17 to the undesired inductive effect of the dimethylammonium group on the aromatic ring.
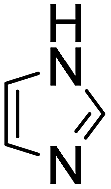
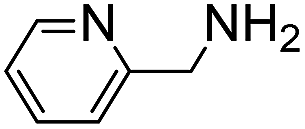
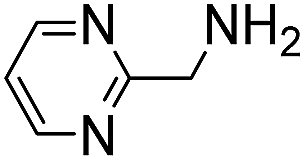
(Aminomethyl)imidazoles (4, 5) proved to be stable under all conditions tested here, and provided among the highest rate enhancements (18- to 23-fold over phosphate buffer) for the test reaction. For comparison, we examined other aminomethyl-substituted heterocycles (19, 20). 2-(Aminomethyl)pyridine 19 gives a high rate enhancement of 13-fold. Structurally similar to imidazoles 4 and 5, the relatively high pKa value of 8.6 for 19 may explain why it does not perform as well as 4 and 5, whose pKa values (8.13 and 7.83 respectively)16,17 are closer to the pH level used in these screening reactions. Pyrimidine compound 20 is less effective, likely due to its significantly lower pKa.
Next, we turned our attention to the most effective catalytic buffers, 4–7. As we found in a recent study, 2-(aminomethyl)(benz)imidazoles 4 and 5 catalyze hydrazone and oxime formations when used at only 1 mM concentration in phosphate buffer,3c and when employed as catalyzing buffers here, we find that they achieve remarkably high second-order reaction rates of greater than 10 M−1 s−1. This represents a roughly 20-fold enhancement compared to phosphate at the same buffer concentration, and the rates are higher than could be achieved with the best previously identified catalyst, phosphonic acid 17, on the model reaction between benzaldehyde and phenylhydrazine in purely aqueous media using our previous conditions.3a Thus, using 17 as a catalyst at 1.0 mM in a 12 mM phosphate buffered saline at pH 7.4, the rate constant was found to be 6.1 ± 0.3 M−1 s−1.
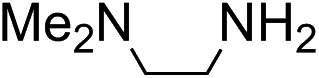
Hine showed that N,N-dimethylethylenediamine 6 forms imines much faster than other primary amines, due to intramolecular acid-catalyzed dehydration of the intermediate hemi-aminal.10b Here, we find that it is highly effective as a catalytic buffer for hydrazone formation at 50 mM. Due to the proximal location of the two amino groups in 6, the primary amine has a pKa of 6.93, while the tertiary amine has a pKa of 9.53,18 leaving the primary amine mostly in its nucleophilic state at biological pH levels. Although 6 was found to be a relatively poor catalyst under previous conditions,3c in this catalyzing buffer context it achieves a second-order rate constant in excess of 10 M−1 s−1, a >20-fold rate enhancement compared to phosphate. As was later discovered (see below), the stability of 6 in the presence of phenylhydrazine is limited, especially at pH levels of 5.5–6.5, somewhat limiting its applicability in the lower part of the biological pH range.
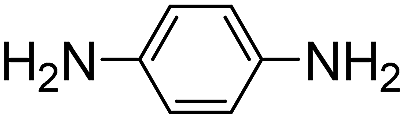
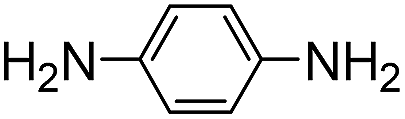
Both meta- and para-diaminobenzene have been identified as catalysts that are slightly superior to aniline for hydrazone and oxime ligation, and due in part to a much higher solubility in water, the meta derivative has been praised as a ‘highly efficient’ catalyst when employed at concentrations as high as 0.9 M.9bpara-Diaminobenzene 7 is the only isomer of the diaminobenzenes with a pKa value in the desired range (pKa 6.46),19 and thus the only derivative to be potentially useful as a catalyzing buffer at a biologically relevant pH. As is evident from Table 1, ABC 7 achieves a reasonably high rate constant of ca. 8 M−1 s−1, meaning it belongs in the ranks of the four standout catalyzing buffers discovered in this screening, ABCs 4–7. The oxidative instability of 7, however, leads to buffered solutions turning colored within minutes. This severely limits the applicability of the buffer for most UV/vis based assays, except where those assays are performed in less than ca. 30 min. Due to the large absorbance by the breakdown products of 7 (a broad, strongly absorbing band with a λmax at ca. 450–500 nm appears over the course of an hour), it was not possible to evaluate whether use of a more than one-hour old buffer solution significantly altered the rate of hydrazone formation.
All four ABCs 4–7 exhibited a linear relationship between reaction rate and buffer concentration in experiments from 10–50 mM in saline at pH 7.4 (see ESI† for full details), although for ABC 7 the linearity was not very good, likely due to optical interference from oxidation byproducts. Using the highly soluble 4 as a buffer at 500 mM in water, a second-order rate constant of 33.6 ± 1.0 M−1 s−1 was achieved for the model reaction between benzaldehyde and phenylhydrazine.
Applicability of ABCs at pH 4.5–9.0
Building on these results, we proceeded to evaluate the applicability of ABCs 4–7 at a range of pH levels from pH 4.5 to pH 9.0. All the catalyzing buffers improved the rate of reaction compared to phosphate buffer throughout the range, except for ABC 6, which proved to be unstable at pH 5.5 and pH 6.5 in the presence of phenylhydrazine (Fig. 2). The reaction rates were found to be higher below neutral pH, with a maximum near pH 5.5 in most cases, including with the use of phosphate buffer. This is in accord with the extensive research on imine formation reaction mechanisms by Cordes and Jencks.20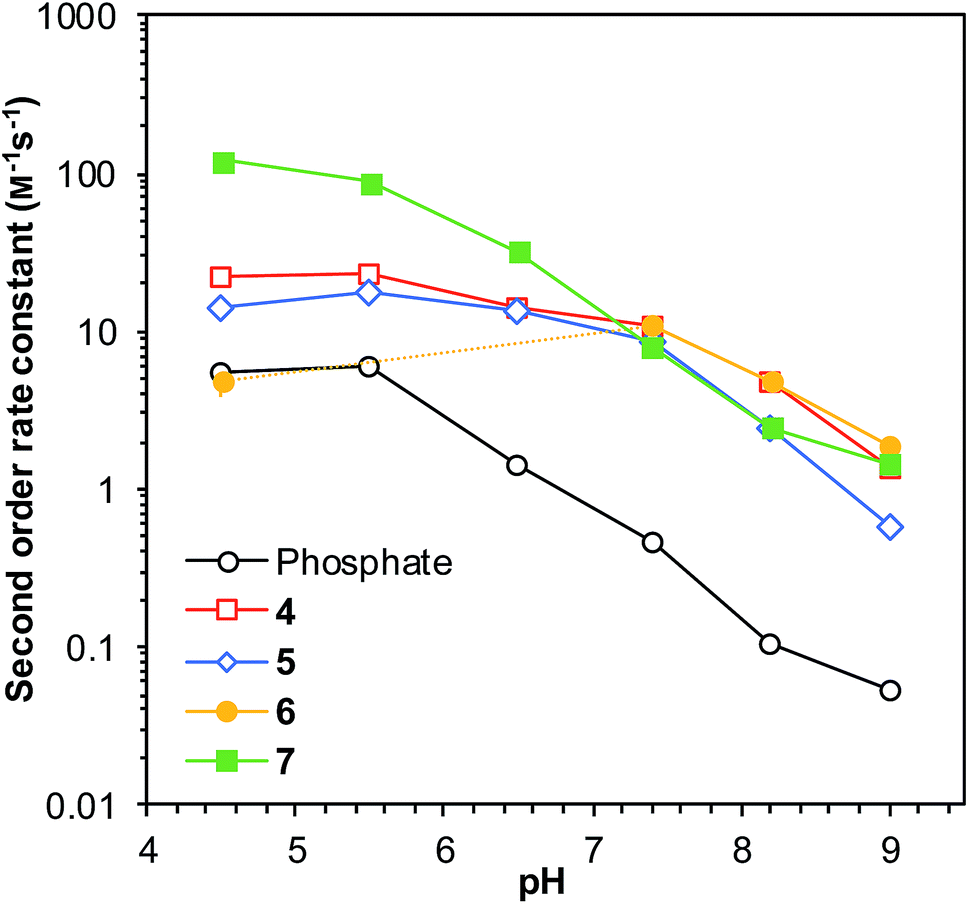 | ||
| Fig. 2 Second-order rate constants for the test reaction (Scheme 1) as a function of solution pH in a selection of ABCs and in phosphate buffer (50 mM buffer concentration). All reactions performed in saline (150 mM NaCl). Error bars show standard deviations based on triplicate measurements (see full data in Table S3†); where no error bars are visible, they are smaller than or equal to the size of the data point. The dashed part of the yellow line illustrates the pH levels where ABC 6 was found to be unstable in presence of phenylhydrazine. | ||
When comparing the reaction rates with ABCs 4–7 to that in phosphate buffer at different pH levels, an interesting pattern emerges. The rate enhancements relative to phosphate buffer go from below 5-fold (with exception of 7) at pH 4.5 to more than 20- or even 40-fold rate enhancements at pH 8.2 (Fig. 3). This strongly suggests that the catalysts function in a bifunctional manner by way of an intramolecular general acid/base mechanism, and it also demonstrates that these catalyzing buffers are highly effective in improving the reaction rate specifically in the biologically relevant part of the pH scale.
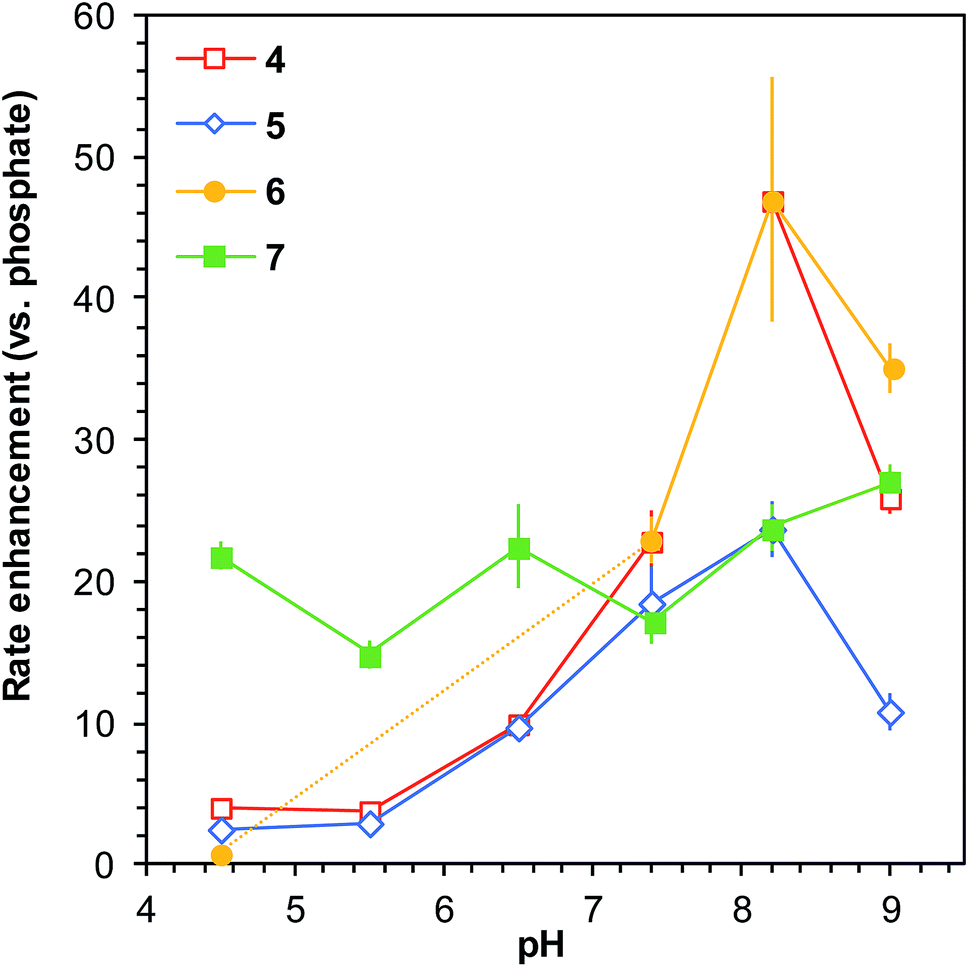 | ||
| Fig. 3 Comparison of rate enhancements of the model reaction (Scheme 1) for a selection of ABCs relative to phosphate buffer as a function of solution pH (buffer concentration 50 mM). All reactions performed in saline (150 mm NaCl). Error bars show compounded standard deviations based on triplicate measurements (see full data in Table S3†); where no error bar is visible, the standard deviation is smaller than or equal to the size of the data point. The greatest rate enhancements in comparison to phosphate buffer are achieved in the biologically relevant region of pH 6–9. The dashed part of the yellow line illustrates the pH levels where ABC 6 was found to be unstable in presence of phenylhydrazine. | ||
Scope investigations
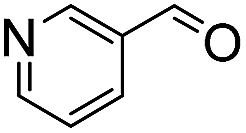
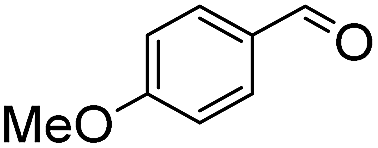
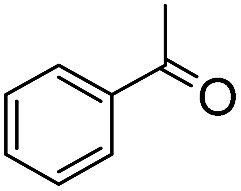
We next employed ABCs 4–7 in a range of experiments to investigate the scope of their abilities to catalyze hydrazone, oxime and acylhydrazone formations. First, phenylhydrazine was reacted with three different carbonyl compounds in both phosphate buffer and using ABCs 4–7 (Table 2). The results show that ABCs give higher rate enhancements with otherwise disfavored electron-rich carbonyl electrophiles like 4-methoxybenzaldehyde than with electron-poor 3-formylpyridine or neutral benzaldehyde (compare to values in Table 1). The data also show high rate enhancements for otherwise challenging ketone substrates such as acetophenone. Using ABC 6, a heretofore unseen rate of reaction between phenylhydrazine and acetophenone of almost 10 M−1 s−1 is achieved, representing a more than 60-fold rate enhancement in comparison to the phosphate buffer. This is a potentially useful finding for protein functionalization, as robust methods to incorporate 4-acetylphenylalanine into proteins have been developed.1a,1b,21|
|
|||||||
|---|---|---|---|---|---|---|---|
| Buffer | Carbonyl compound | ||||||
|
|
|
|
|||||
| k 2 | k rel | k 2 | k rel | k 2 | k rel | ||
| a Second-order rate constants (M−1 s−1), k2, listed as mean values ± standard deviations based on triplicate measurements. Conditions as per Table 1. Relative reaction rates, krel, is in comparison to reaction in phosphate buffer for each substrate respectively. n.d., no data could be obtained due to UV spectral overlap from the buffer. | |||||||
| Phosphate | 0.407 ± 0.007 |
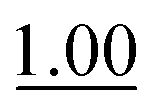
|
0.093 ± 0.007 |
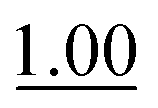
|
0.118 ± 0.015 |
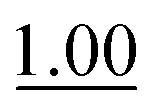
|
|
|
4 | 5.7 ± 0.6 | 14 | 2.8 ± 0.2 | 30 | 1.5 ± 0.4 | 13 |
|
5 | 5.4 ± 0.3 | 13 | 2.47 ± 0.12 | 27 | 0.55 ± 0.03 | 4.6 |
|
6 | 2.13 ± 0.04 | 5.2 | 3.77 ± 0.06 | 41 | 8.0 ± 1.2 | 67 |
|
7 | 11.9 ± 0.8 | 29 | 2.60 ± 0.16 | 28 | n.d. | |
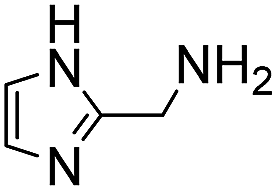
Next, we reacted O-benzylhydroxylamine and benzoylhydrazide with benzaldehyde to evaluate the catalyzing buffer method in oxime and acylhydrazone formations, respectively (Table 3). As is widely observed in the literature, oxime bond formations are roughly an order of magnitude slower than hydrazone formations, which is also reflected in the quite low rate constant seen here in phosphate buffer for this reaction (0.0213 M−1 s−1). However, using ABC buffers we were delighted to see that using either 4 or 6 we could achieve reaction rates in excess of 1.0 M−1 s−1. Although even higher oxime formation rates have been reported using specially activated aldehydes,6a,22 the rate enhancements reported here (75–140-fold in comparison to phosphate buffer), now makes the use of oxime ligations with standard, non-activated substrates a viable alternative without requiring the synthesis of specialized reactants.
|
|
|||||
|---|---|---|---|---|---|
| Buffer | α-nucleophile | ||||
|
|
|
||||
| k 2 | k rel | k 2 | k rel | ||
| a Second-order rate constants (M−1 s−1), k2, listed as mean values ± standard deviations based on triplicate measurements. Conditions as per Table 1. Relative reaction rates, krel, is in comparison to reaction in phosphate buffer for each substrate respectively. n.d., no data could be obtained due to UV spectral overlap from the buffer. | |||||
| Phosphate | 0.0213 ± 0.0013 |
|
0.00141 ± 0.00004 |
|
|
|
4 | 2.9 ± 0.2 | 140 | 0.59 ± 0.01 | 420 |
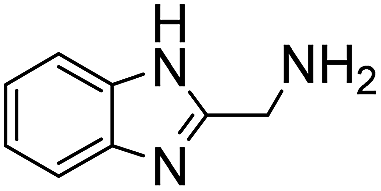
|
5 | n.d. | 0.32 ± 0.08 | 220 | |
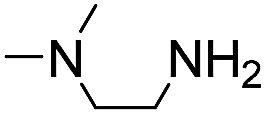
|
6 | 1.6 ± 0.2 | 75 | 0.31 ± 0.02 | 220 |
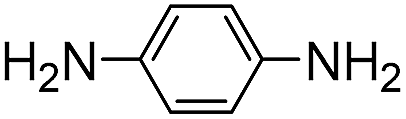
|
7 | n.d. | n.d. | ||
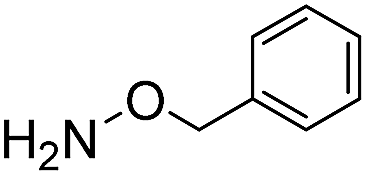
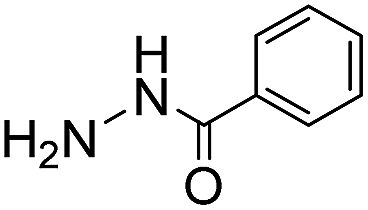
Due to the electron-withdrawing effect of the carbonyl, acylhydrazines (i.e. carbohydrazides) are also known to be significantly less nucleophilic than hydrazines, and therefore acylhydrazone formation is much slower than hydrazone formation. However, because of the ease of preparation and handling of hydrazides, many hydrazone conjugation procedures are performed with hydrazides as the nucleophile.23 In our tests, using benzoylhydrazide as the test nucleophile instead of phenylhydrazine leads in phosphate buffer to a dramatic loss of reactivity of more than two orders of magnitude (Table 3). Importantly, use of ABCs 4–6 makes up for this lost reactivity, with rate enhancements of more than 200-fold, and even more than 400-fold for ABC 4. The high rate enhancements achieved with benzoylhydrazide underline the broad applicability of the catalyzing buffer method, and makes the use of more easily-attained hydrazides as conjugation partners much more feasible.
Protein labelling studies
ABCs 4 and 6 were found to be useful for catalyzing hydrazone formation between phenylhydrazine and the aldehyde produced in situ by periodate oxidation of threonine (i.e. glyoxalic aldehyde), reaching second-order rate constants of 0.459 ± 0.005 and 0.318 ± 0.009 M−1 s−1, respectively (see ESI† for details). ABCs 5 and 7, on the other hand, were found to be unstable in solutions containing periodate, which we tentatively ascribe to oxidative polymerization processes.24To test a labelling application of the catalysis, we applied a periodate-based oxidation method to illustrate improved labelling of protein fragments using buffer 4 (compared to PBS) with a commercially available fluorescent hydrazide for visualisation of the peptides in a gel (Fig. S8†). This shows that the catalyzing buffer method is also applicable to systems relying on in situ formation of aldehydes by periodate oxidation of naturally occurring 1,2-diols, such as sialylated glycoproteins and nucleic acids that display 3′-ribonucleotides,11b,25 or 1,2-amino alcohols such as N-terminal serine or threonine residues in peptides and proteins.26
Toxicity studies
To evaluate whether ABCs 4–7 might be useful in biological studies, we carried out toxicity studies of the buffers with the HeLa cell line. Living cells were subjected to ABCs (0.2–20 mM) in the DMEM growth medium (supplemented with HEPES buffer to maintain pH) for 6 hours. The results show (Fig. 4 and S6†) that ABCs 4, 5, and 6 show generally low toxicities, allowing for use of these ABCs in cellular applications. Diaminobenzene 7, on the other hand, shows significant toxicity at concentrations of 5 mM and higher, suggesting that ABC 7 is not well suited for use in live cell applications.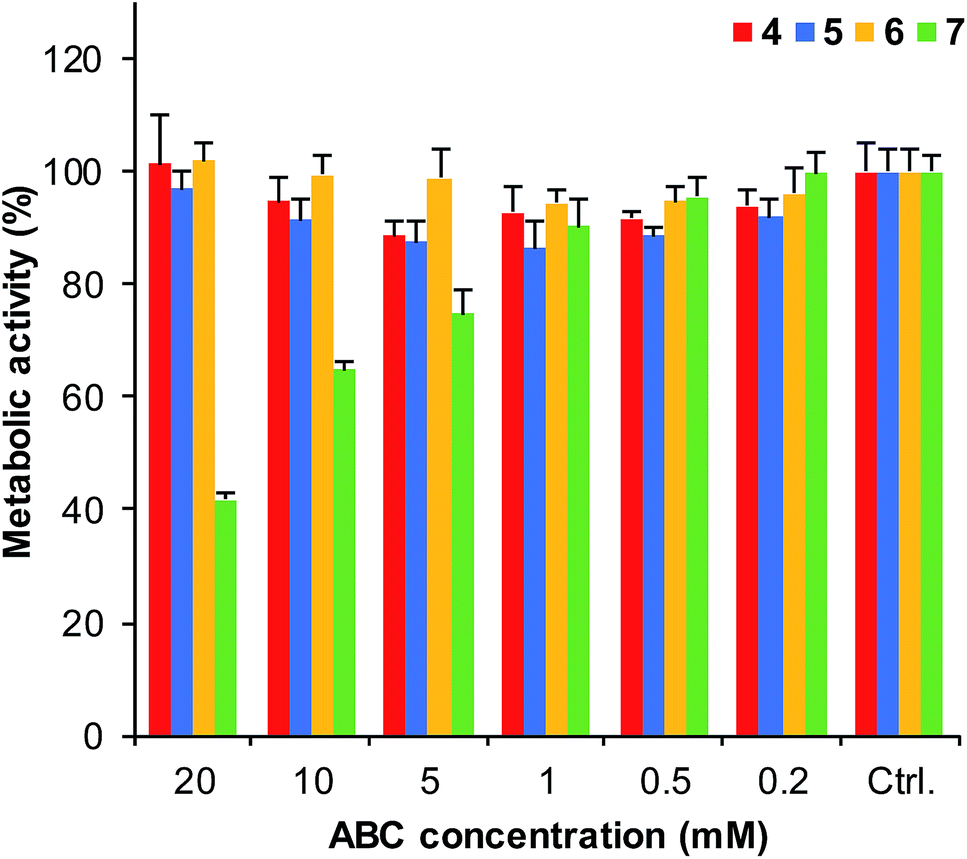 | ||
| Fig. 4 The viability of HeLa cells in presence of ABCs 4–7 were evaluated by subjecting the living cells to 0.2–20 mM concentration of the ABCs in DMEM medium supplemented with HEPES buffer for 6 h at 37 °C and determining the metabolic activity by measuring resorufin fluorescence (see details in ESI†); error bars show standard deviations based on measurements performed in multiple wells (n > 4). These results were supported by similar results using the MTT assay (Fig. S6†). | ||
Live cell experiments
Given the low toxicity of ABCs such as 4, we tested the application of this catalytic amine buffer to promote oxime and hydrazone formation in living cells. To provide optical evidence of oxime formation, we incubated the aldehyde-functionalized coumarin 21 with HeLa cells, visible by the coumarin's bright fluorescence, and then incubated the cells with the oxyamine-substituted dabcyl quencher 22 in phosphate-buffered saline (PBS) or in buffer 4 (10 mM) at pH 7.4 (Fig. 5). The results show that 21 was quenched relatively slowly in PBS, while in catalyzing buffer 4, the quenching was considerably more rapid, and was almost complete within one hour (compare images B and D in Fig. 5).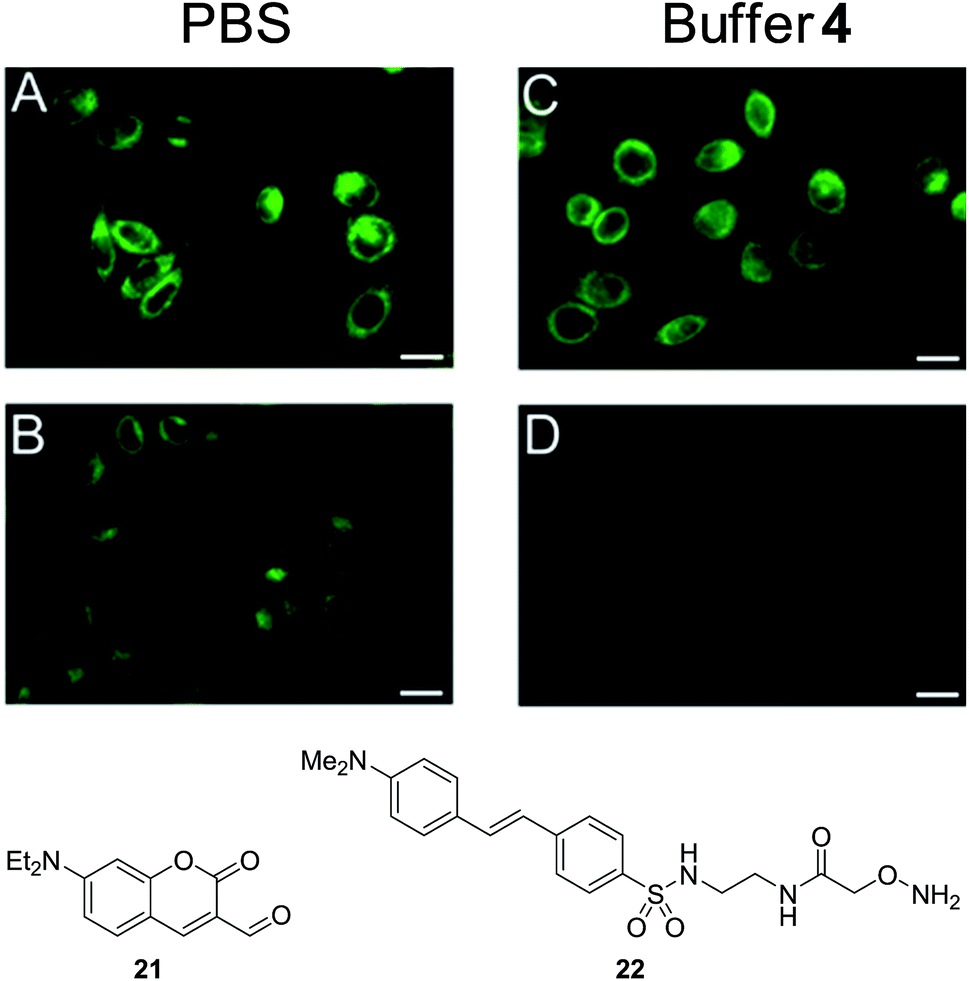 | ||
| Fig. 5 ABC-promoted oxime formation in HeLa cells. Epifluorescence images of cells treated with fluorescent coumarin 21 and incubated for 1 h. (A) In PBS, (B) in PBS with oxyamine quencher 22, (C) in buffer 4 (10 mM), (D) in buffer 4 (10 mM) with quencher 22. Excitation 400–440 nm; [21] = 10 μM and [22] = 50 μM; pH 7.4, 37 °C. The scale bar represents 10 μm. | ||
To further test the scope of hydrazone formation promoted by ABC 4 in living cells, we employed the DarkZone reagent developed recently for imaging of cellular aldehydic loads.11a In HeLa cells incubated in a medium supplied with formaldehyde (500 μM), a dramatic improvement in light-up of the DarkZone dye was seen in the presence of ABC 4 in comparison to medium in cell visualization studies (Fig. S7†). Taken together, these results provide evidence that an ABC can promote oxime and hydrazone formations, ordinarily slow reactions at physiological pH, at convenient time scales in living cells.
Conclusions
Our data show that catalytic amine buffers can provide a simple and low-toxicity strategy for speeding hydrazone and oxime conjugations. We show that reaction rate constants of ca. 20–30 M−1 s−1 can be achieved even at pH 7 and above, with rate enhancements of up to 400-fold possible. The buffer compounds are commercially available and serve a dual purpose, buffering solution pH and greatly accelerating these reactions both in vitro and in living cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the U.S. National Institutes of Health (GM110050 and CA217809) for support. D. L. acknowledges a postdoctoral fellowship from the Carlsberg Foundation, and A. E. acknowledges a postdoctoral stipend from the Foundation Blanceflor. We thank the Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University, for assistance with MS measurements.Notes and references
- (a) P. V. Chang and C. R. Bertozzi, Chem. Commun., 2012, 48, 8864 RSC; (b) K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764 CrossRef PubMed; (c) L. Carroll, H. L. Evans, E. O. Aboagye and A. C. Spivey, Org. Biomol. Chem., 2013, 11, 5772 RSC; (d) M. King and A. Wagner, Bioconjugate Chem., 2014, 25, 825 CrossRef PubMed.
- K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16 CrossRef PubMed.
- (a) P. Crisalli and E. T. Kool, Org. Lett., 2013, 15, 1646 CrossRef PubMed; (b) P. Crisalli and E. T. Kool, J. Org. Chem., 2013, 78, 1184 CrossRef PubMed; (c) D. Larsen, M. Pittelkow, S. Karmakar and E. T. Kool, Org. Lett., 2015, 17, 274 CrossRef PubMed.
- (a) J. Kalia and R. T. Raines, Angew. Chem., Int. Ed., 2008, 47, 7523 CrossRef PubMed; (b) V. Saggiomo and U. Lüning, Eur. J. Org. Chem., 2008, 2008, 4329 CrossRef; (c) V. Saggiomo and U. Lüning, Tetrahedron Lett., 2009, 50, 4663 CrossRef; (d) A. Dirksen and P. E. Dawson, Bioconjugate Chem., 2008, 19, 2543 CrossRef PubMed.
- (a) D. Rideout, Science, 1986, 233, 561 Search PubMed; (b) L. K. Mahal, K. J. Yarema and C. R. Bertozzi, Science, 1997, 276, 1125 CrossRef PubMed; (c) M. Rashidian, J. K. Dozier, S. Lenevich and M. D. Distefano, Chem. Commun., 2010, 46, 8998 RSC; (d) Z. Zhang, B. A. C. Smith, L. Wang, A. Brock, C. Cho and P. G. Schultz, Biochemistry, 2003, 42, 6735 CrossRef PubMed; (e) E. M. Brustad, E. A. Lemke, P. G. Schultz and A. A. Deniz, J. Am. Chem. Soc., 2008, 130, 17664 CrossRef PubMed; (f) J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13 CrossRef PubMed; (g) S. Ulrich, D. Boturyn, A. Marra, O. Renaudet and P. Dumy, Chem.–Eur. J., 2014, 20, 34 CrossRef PubMed; (h) D. K. Kölmel and E. T. Kool, Chem. Rev., 2017, 117, 10358 CrossRef PubMed.
- (a) P. Schmidt, C. Stress and D. Gillingham, Chem. Sci., 2015, 6, 3329 RSC; (b) A. Bandyopadhyay and J. Gao, Chem.–Eur. J., 2015, 21, 14748 CrossRef PubMed.
- E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 826 CrossRef.
- (a) A. Dirksen, S. Dirksen, T. M. Hackeng and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 15602 CrossRef PubMed; (b) A. Dirksen, T. M. Hackeng and P. E. Dawson, Angew. Chem., Int. Ed., 2006, 45, 7581 CrossRef PubMed.
- (a) M. Wendeler, L. Grinberg, X. Wang, P. E. Dawson and M. Baca, Bioconjugate Chem., 2014, 25, 93 CrossRef PubMed; (b) M. Rashidian, M. M. Mahmoodi, R. Shah, J. K. Dozier, C. R. Wagner and M. D. Distefano, Bioconjugate Chem., 2013, 24, 333 CrossRef PubMed.
- (a) J. Hine, F. E. Rogers and R. E. Notari, J. Am. Chem. Soc., 1968, 90, 3279 CrossRef; (b) J. Hine, M. S. Cholod and W. K. Chess, J. Am. Chem. Soc., 1973, 95, 4270 CrossRef; (c) J. Hine, Acc. Chem. Res., 1978, 11, 1 CrossRef.
- (a) L. H. Yuen, N. S. Saxena, H. S. Park, K. Weinberg and E. T. Kool, ACS Chem. Biol., 2016, 11, 2312 CrossRef PubMed; (b) Y. Zeng, T. N. C. Ramya, A. Dirksen, P. E. Dawson and J. C. Paulson, Nat. Methods, 2009, 6, 207 CrossRef PubMed.
- (a) A. Dirksen, S. Yegneswaran and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 2023 CrossRef PubMed; (b) S. R. Beeren, M. Pittelkow and J. K. M. Sanders, Chem. Commun., 2011, 47, 7359 RSC; (c) R. Nguyen and I. Huc, Chem. Commun., 2003, 942 RSC.
- S. Karmakar, E. M. Harcourt, D. S. Hewings, A. F. Lovejoy, D. M. Kurtz, T. Ehrenschwender, L. J. Barandun, C. Roost, A. A. Alizadeh and E. T. Kool, Nat. Chem., 2015, 7, 752 CrossRef PubMed.
- S. Wang, G. N. Nawale, S. Kadekar, O. P. Oommen, N. K. Jena, S. Chakraborty, J. Hilborn and O. P. Varghese, Sci. Rep., 2018, 8, 2193 CrossRef PubMed.
- W. P. Jencks, in Prog. Phys. Org. Chem., ed. S. G. Cohen, A. Streitwieser and R. W. Taft, John Wiley & Sons, Inc., 1964, vol. 2, pp. 63–128 Search PubMed.
- Based on potentiometric titration, see ESI.†.
- A. A. El-Sherif, J. Solution Chem., 2010, 39, 1562 CrossRef.
- H. Irving and J. M. M. Griffiths, J. Chem. Soc., 1954, 213 RSC.
- F. Mata, J. M. Leal and B. Garcia, Z. Phys. Chem., 1980, 261, 1059 Search PubMed.
- (a) E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1962, 84, 832 CrossRef; (b) E. H. Cordes and W. P. Jencks, J. Am. Chem. Soc., 1963, 85, 2843 CrossRef.
- (a) D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592 CrossRef PubMed; (b) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef PubMed; (c) D. Datta, P. Wang, I. S. Carrico, S. L. Mayo and D. A. Tirrell, J. Am. Chem. Soc., 2002, 124, 5652 CrossRef PubMed.
- P. Schmidt, L. Zhou, K. Tishinov, K. Zimmermann and D. Gillingham, Angew. Chem., Int. Ed. Engl., 2014, 53, 10928 CrossRef PubMed.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, Boston, 3rd edn, 2013 Search PubMed.
- (a) I. Amer, D. A. Young and H. C. M. Vosloo, Eur. Polym. J., 2013, 49, 3251 CrossRef; (b) M.-R. Huang, X.-G. Li and Y. Yang, Polym. Degrad. Stab., 2000, 71, 31 CrossRef.
- (a) F. Hansske, M. Sprinzl and F. Cramer, Bioorg. Chem., 1974, 3, 367 CrossRef; (b) J. A. Hunt, Biochem. J., 1973, 131, 315 CrossRef PubMed.
- D. Chelius and T. A. Shaler, Bioconjugate Chem., 2003, 14, 205 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary tables and figures, experimental details, and synthetic procedures. See DOI: 10.1039/c8sc01082j |
| ‡ Current address: Department of Chemistry, Technical University of Denmark, DK-2800 Lyngby, Denmark. |
| § Current address: Department of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-412 96 Gothenburg, Sweden. |
| This journal is © The Royal Society of Chemistry 2018 |