
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect heterobenzylic fluorination, difluorination and trifluoromethylthiolation with dibenzenesulfonamide derivatives†
Michael
Meanwell
a,
Bharani Shashank
Adluri
a,
Zheliang
Yuan
ab,
Josiah
Newton
 ad,
Philippe
Prevost
a,
Matthew B.
Nodwell
a,
Chadron M.
Friesen
d,
Paul
Schaffer
b,
Rainer E.
Martin
c and
Robert
Britton
*a
ad,
Philippe
Prevost
a,
Matthew B.
Nodwell
a,
Chadron M.
Friesen
d,
Paul
Schaffer
b,
Rainer E.
Martin
c and
Robert
Britton
*a
aDepartment of Chemistry, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada. E-mail: rbritton@sfu.ca
bLife Science Division, TRIUMF, Vancouver, BC V6T 2A3, Canada
cMedicinal Chemistry, Roche Pharma Research and Early Development (pRED), Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd, Grenzacherstrasse 124, CH-4070 Basel, Switzerland
dDepartment of Chemistry, Trinity Western University, Langley, British Columbia V2Y 1Y1, Canada
First published on 7th June 2018
Abstract
Functionalization of heterocyclic scaffolds with mono- or difluoroalkyl groups provides unique opportunities to modulate drug pKa, influence potency and membrane permeability, and attenuate metabolism. While advances in the addition of fluoroalkyl radicals to heterocycles have been made, direct C(sp3)–H heterobenzylic fluorination is comparatively unexplored. Here we demonstrate both mono- and difluorination of a range of alkyl heterocycles using a convenient process that relies on transient sulfonylation by the electrophilic fluorinating agent N-fluorobenzenesulfonimide. We also report heterobenzylic trifluoromethylthiolation and 18F-fluorination, providing a suite of reactions for late-stage C(sp3)–H functionalization of drug leads and radiotracer discovery.
Introduction
In recent years considerable effort has been directed towards the development of new methods to selectively fluorinate C(sp2)–H or C(sp3)–H bonds in structurally complex molecules.1 These efforts have been stimulated by the profound effect that fluorination can have on biological activity2 and strategic advantages manifest by late-stage C–H functionalization in medicinal and agrochemistry.3 For example, fluorination can significantly impact potency, selectivity, lipophilicity and membrane permeability of drug leads,2b and modulate the pKa of proximal heterocycles (e.g., 1,42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 and 3,6Fig. 1). The characteristically strong C–F bond is also routinely exploited in medicinal chemistry as a replacement for C–H bonds and, in particular, a means to block oxidative metabolism (e.g., 3).2 Furthermore, fluorinated alkyl groups can serve as bioisosteres for more polar or less stable functionalities, and the replacement of a hydroxyl group with a fluorine atom is a common tactic.2 Likewise, the CF2H group (H-bond donor) is a lipophilic bioisostere for alcohols or thiols and the CF2R group can serve as a carbonyl or alkoxy group mimic.2c Considering that roughly 60% of FDA approved drugs include a nitrogen-containing heterocycle,7 the development of synthetic strategies that provide access to heterobenzylic fluorides is of particular interest and much success has been realized in trifluoromethylation of heterocycles.8 However, introduction of heterobenzylic monofluoroalkyl or difluoroalkyl groups remains largely reliant on cross coupling reactions9 or deoxyfluorination of heterobenzylic alcohols10 and carbonyls,11 processes that require prior functionalization. As a notable exception, Baran has reported innate C(sp2)–H functionalization of heterocycles as a means to add each of the CHF2,12a CH2F12b and CF2CH312c groups (e.g., 5–7) by employing the corresponding zinc sulfinate salts in Minisci-like radical addition processes. Likewise, the introduction of difluoroacetates 8,13 difluoroacetamides 914 and difluorophosphonates 1015 has been accomplished via transition metal catalysis or radical processes.16 Unfortunately, despite considerable advances in C(sp3)–H benzylic mono- and difluorination,17 heterobenzylic C(sp3)–H fluorination10,18 or difluorination are largely unexplored owing to fundamental incompatibilities between common fluorine transfer reagents19 (e.g., N-fluorobenzenesulfonimide (NFSI)) and nucleophilic heterocycles.20 Towards this goal, Van Humbeck has very recently described the fluorination of several alkylheterocycles induced by single electron transfer to Selectfluor.18e Previously, we reported the serendipitous finding that 2- and 4-alkylpyridines (e.g., 11) undergo pyridylic fluorination by reaction with the electrophilic fluorination agent NFSI, a process that involves the transient formation of a sulfonylpyridinium intermediate.21 Here, we demonstrate that activation by transient sulfonylation is general for a range of alkylheterocycles and can be extended to heterobenzylic difluorination and trifluoromethylthiolation. Collectively, these convenient processes provide a platform for late-stage functionalization of drug leads and enable direct 18F-fluorination of alkylheterocycles for the purpose of radiotracer synthesis for positron emission tomography (PET) imaging.
5 and 3,6Fig. 1). The characteristically strong C–F bond is also routinely exploited in medicinal chemistry as a replacement for C–H bonds and, in particular, a means to block oxidative metabolism (e.g., 3).2 Furthermore, fluorinated alkyl groups can serve as bioisosteres for more polar or less stable functionalities, and the replacement of a hydroxyl group with a fluorine atom is a common tactic.2 Likewise, the CF2H group (H-bond donor) is a lipophilic bioisostere for alcohols or thiols and the CF2R group can serve as a carbonyl or alkoxy group mimic.2c Considering that roughly 60% of FDA approved drugs include a nitrogen-containing heterocycle,7 the development of synthetic strategies that provide access to heterobenzylic fluorides is of particular interest and much success has been realized in trifluoromethylation of heterocycles.8 However, introduction of heterobenzylic monofluoroalkyl or difluoroalkyl groups remains largely reliant on cross coupling reactions9 or deoxyfluorination of heterobenzylic alcohols10 and carbonyls,11 processes that require prior functionalization. As a notable exception, Baran has reported innate C(sp2)–H functionalization of heterocycles as a means to add each of the CHF2,12a CH2F12b and CF2CH312c groups (e.g., 5–7) by employing the corresponding zinc sulfinate salts in Minisci-like radical addition processes. Likewise, the introduction of difluoroacetates 8,13 difluoroacetamides 914 and difluorophosphonates 1015 has been accomplished via transition metal catalysis or radical processes.16 Unfortunately, despite considerable advances in C(sp3)–H benzylic mono- and difluorination,17 heterobenzylic C(sp3)–H fluorination10,18 or difluorination are largely unexplored owing to fundamental incompatibilities between common fluorine transfer reagents19 (e.g., N-fluorobenzenesulfonimide (NFSI)) and nucleophilic heterocycles.20 Towards this goal, Van Humbeck has very recently described the fluorination of several alkylheterocycles induced by single electron transfer to Selectfluor.18e Previously, we reported the serendipitous finding that 2- and 4-alkylpyridines (e.g., 11) undergo pyridylic fluorination by reaction with the electrophilic fluorination agent NFSI, a process that involves the transient formation of a sulfonylpyridinium intermediate.21 Here, we demonstrate that activation by transient sulfonylation is general for a range of alkylheterocycles and can be extended to heterobenzylic difluorination and trifluoromethylthiolation. Collectively, these convenient processes provide a platform for late-stage functionalization of drug leads and enable direct 18F-fluorination of alkylheterocycles for the purpose of radiotracer synthesis for positron emission tomography (PET) imaging.
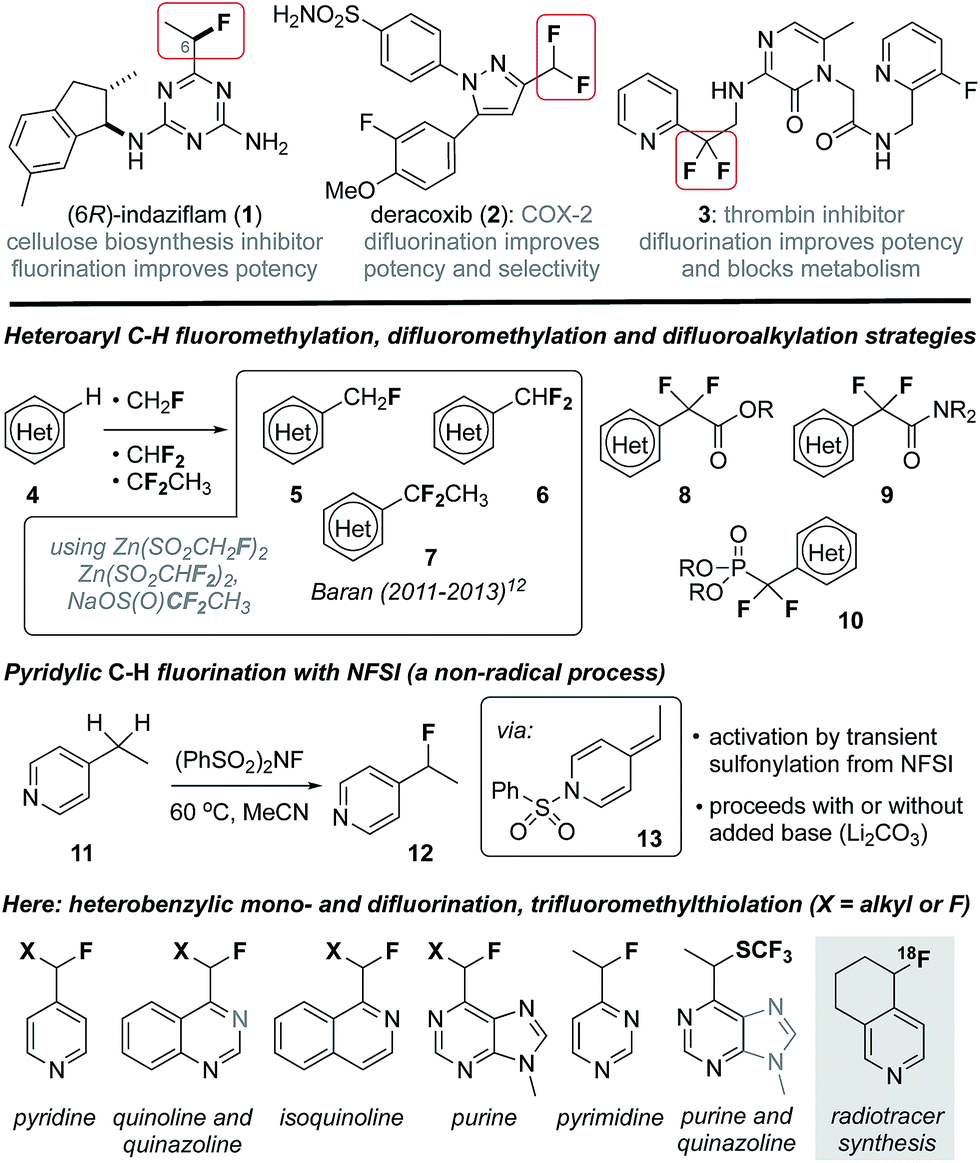 | ||
| Fig. 1 Heterobenzylic fluorides in drug discovery and strategies for their synthesis. | ||
Results and discussion
Mono- and difluorination of 4-ethylpyridine and 4-alkylquinolines
While examining the scope of the pyridylic fluorination reaction depicted in Fig. 1 (11 → 12),21 we found that at elevated temperatures (>65 °C) small amounts of the corresponding difluoroalkyl derivatives were formed and could be identified by a characteristic resonances at δ ∼ −95 ppm in 19F NMR spectra recorded on crude reaction mixtures. These observations prompted us to investigate the pyridylic difluorination reaction as a complimentary process. As summarized in Table 1, heating a solution of 4-ethylpyridine in MeCN with an excess of NFSI afforded exclusively the monofluorinated adduct 15 at 60 °C (entry 1). Increasing the reaction temperature above 80 °C (in a microwave) provided a complex mixture of products that included the corresponding acetamide derived from displacement of fluoride by solvent (MeCN).17d However, when the reaction was repeated at 75 °C with a further increase in equivalents of NFSI, a ∼1:1 mixture of the mono- and difluorinated ethylpyridines 15 and 16 were produced in good yield (74%, entry 2) and were readily separable by flash column chromatography. Notably, for difluorination, sequential activation by sulfonylation consumes 2 equivalents of NFSI and a further 2 equivalents are required for fluorination. The additional excess of NFSI is required to offset its slow decomposition over the course of the reaction (48 h). Several alternative solvents were evaluated and a modest increase in yield was realized in EtOAc (entry 3). The fluorination of 4-ethylquinoline (17) was also examined and we were pleased to find that heterobenzylic fluorination of this alkylquinoline provided the monofluoroethyl product 18 in good yield (entry 4). However, despite considerable effort, this substrate proved reluctant to undergo difluorination. Under more forcing conditions (e.g., >90 °C, microwave) decomposition occurred, and after 36 h at 75 °C with a large excess of NFSI only ∼7% of the difluoroethyl quinoline 19 was produced (entry 5). Considering the importance of both the mono- and difluoromethyl groups as bioisosteres,2c we also investigated the fluorination of 4-methyl quinoline (20) and were surprised to find that difluorination predominated even at low conversion, suggesting that here the second fluorination event is a more facile process (entry 6). Increasing the equivalents of NFSI and reaction temperature (entry 7) as well as concentration (entry 8) ultimately provided the difluoromethyl quinoline 22 in excellent yield while further increases in reaction temperature, time or equivalents of NFSI failed to promote trifluorination on this or any other substrate. In both the mono- and difluorination of alkylquinolines 17 and 20, phenylsulfonyl fluoride was observed as a by-product, suggesting that these reactions rely on activation of quinoline through transient sulfonylation by NFSI.21 It is notable that this approach to heterobenzylic fluorination is complimentary to the Minisci-like radical reactions described by Baran, which favour trifluoromethylation at C7 or difluoromethylation at C2 of quinolines.12
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Hetero aromatic | Solvent (conc. (M)) | NFSI (equiv.) | Temp (°C) | Producta (ratio) | % Yieldb |
| a Ratio of mono- and difluorinated products determined by analysis of crude 1H and 19F NMR spectra. b Combined isolated yield of mono- and difluorinated products. c 1.1 equiv. of Li2CO3. d 5 equiv. of Li2CO3. | ||||||
| 1 | 14 | MeCN (0.1)c | 3 | 60 |
15:16 (>20:1) |
87 |
| 2 | 14 | MeCN (0.5)d | 10 | 75 |
15:16 (1:1) |
74 |
| 3 | 14 | EtOAc (0.5)d | 10 | 75 |
15:16 (2:3) |
82 |
| 4 | 17 | MeCN (0.1)c | 3 | 65 |
18:19 (>20:1) |
71 |
| 5 | 17 | EtOAc (0.5)d | 10 | 75 |
18:19 (10:1) |
81 |
| 6 | 20 | MeCN (0.1)c | 3 | 65 |
21:22 (1:3) |
30 |
| 7 | 20 | MeCN (0.3)d | 4 | 75 |
21:22 (1:8) |
61 |
| 8 | 20 | MeCN (0.5)d | 5 | 75 |
21:22 (1:10) |
74 |
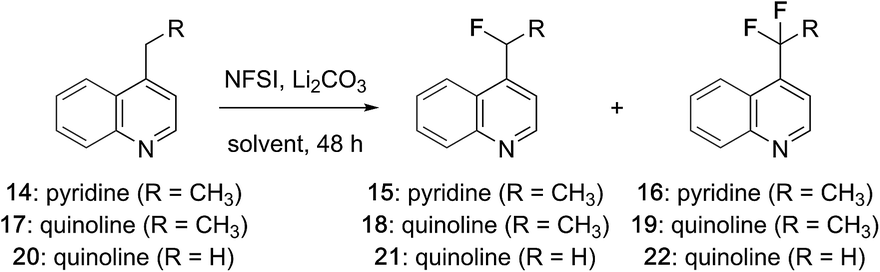
Scope of heterobenzylic mono- and difluorination
Encouraged by the susceptibility of 4-alkylquinolines 17 and 20 to undergo mono- or difluorination, we explored the scope of these reactions with a broader range of heterocycles including pyridines, isoquinolines, pyrimidines, quinazolines and purines. As summarized in Fig. 2, by simply modifying the equivalents of reagent and temperature, in several cases mono- or difluorination could be effected selectively. For example, both mono- and difluoroalkyl pyridines, quinazolines and purines could be produced in good yield following this straightforward procedure (e.g., 23/42, 30/44, 31/43 and 32/45). As noted above, alkylquinolines were reluctant to difluorinate but were monofluorinated in excellent yield providing 24 and 25. Conversely, a series of methylquinolines were transformed directly into the corresponding difluoromethylquinolines 33–39 in good yield. In addition to the obvious compatibility with azaheterocycles, substituted aromatics (e.g., 36–41), esters (e.g., 25) and amides (e.g., 24) were well tolerated. It is notable that both 2,4-dimethylquinoline and 2,4,6-trimethylpyridine failed to undergo fluorination (<5% yield) using our standard reaction conditions. Here, we postulate that steric hindrance from the adjacent alkyl group(s) impedes sulfonylation of the heterocycle by NFSI and thus prevents fluorination. In several cases, complete separation of mono- and difluorinated products by flash column chromatography proved challenging. Thus, while purified product could be isolated this way, yields for these reactions were determined by analysis of NMR spectroscopic data using an internal standard.21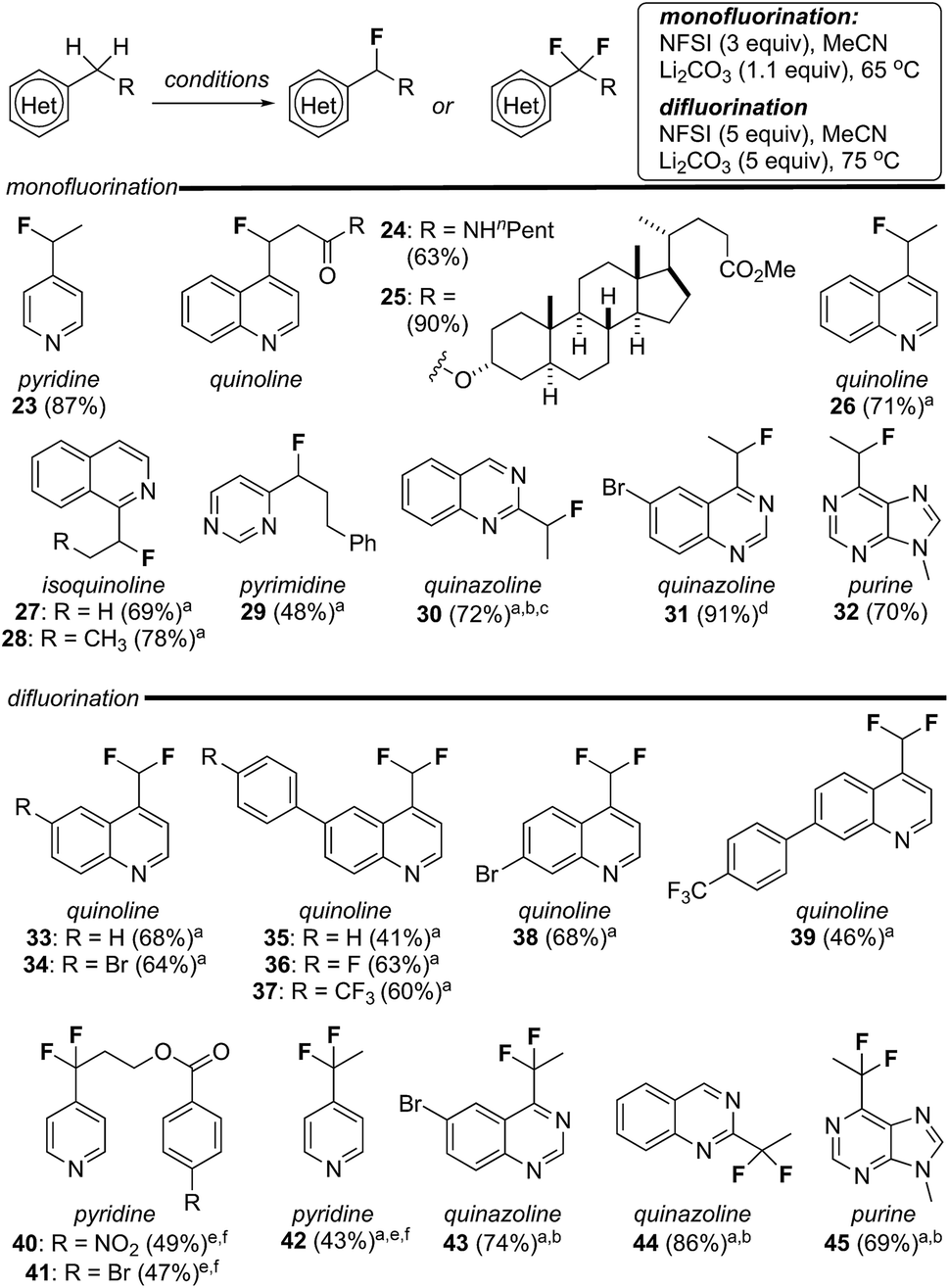 | ||
| Fig. 2 Mono- and difluorination of pyridines, quinolines, isoquinolines, quinazolines, pyrimidines and purines. aYield determined by analysis of NMR spectroscopic data using an internal standard; breaction at 125 °C in a microwave reactor; caccompanied by 25% of the difluorinated quinazoline 44; dreaction at 25 °C; eaccompanied by ∼40% of a monofluorinated product; f10 equiv. of NFSI in EtOAc. | ||
Sulfonyl transfer promotes heterobenzylic trifluoromethyl-thiolation
Considering that sulfonyl transfer from NFSI is a key feature of this process (e.g., 48, Fig. 3),21 we examined a small collection of dibenzenesulfonamide derivatives22 to explore their potential in the direct heterobenzylic functionalization of alkylquinazolines and purines. As depicted in Fig. 3, we found that both trifluoromethylthiolation (e.g., 49–51 and 53–55) and chlorination (e.g., 52) were facile processes. For example, 2- and 4-alkylquinazolines and 6-ethylpurine underwent heterobenzylic trifluoromethylthiolation using N-trifluoromethylthiodibenzenesulfonimide (N(SCF3)SI).22 Surprisingly, we observed no competing heteroaryl trifluoromethylthiolation22 of quinazolines and purines, and attempts to effect the equivalent transformation using trifluoromethylthiophthalimide, an electrophilic trifluoro-methylthiolation reagent,23 delivered none of the expected trifluoromethylthiolated products. This later result provides support for a mechanism involving activation by transient sulfonylation with dibenzenesulfonamide derivatives. Again, 2,4-disubstituted quinazolines failed to provide any trifluoromethylthiolated product (e.g., 56 or 57) presumably due to steric hindrance impeding sulfonylation of the heterocycle by N(SCF3)SI. Notably, this heterobenzylic trifluoromethylthiolation24 reaction offers a unique opportunity to significantly alter lipophilicity (Hansch hydrophobicity parameter π = 1.44)23 and pKa of a drug lead.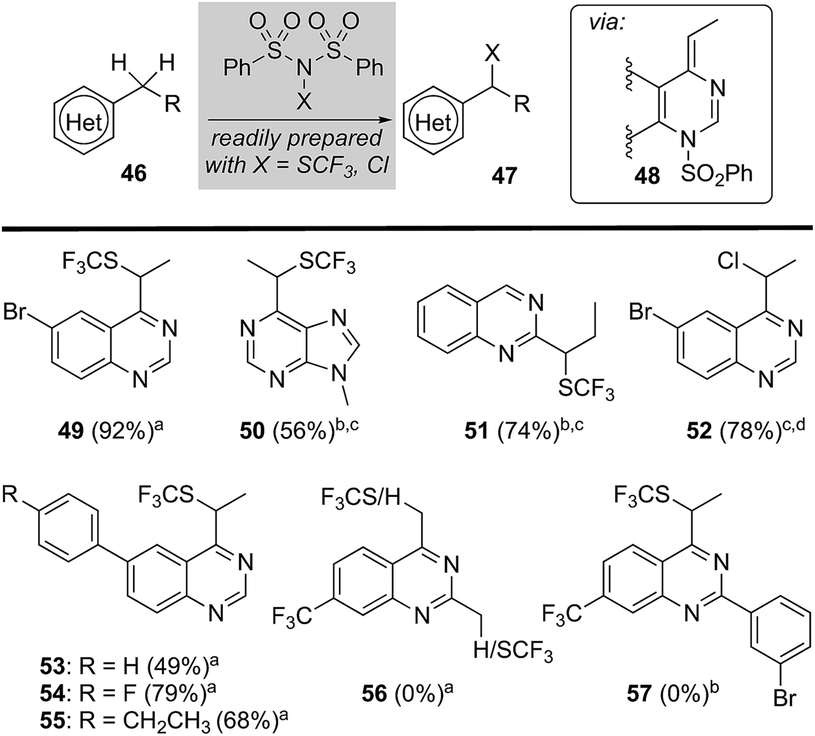 | ||
| Fig. 3 Trifluoromethylthiolation and chlorination of purine and quinazolines. aConditions: N(SCF3)SI (2.4 equiv.), Li2CO3 (1.1 equiv.), MeCN, 75 °C, 48 h; bN(SCF3)SI (2.4 equiv.), Li2CO3 (1.1 equiv.), MeCN, 125 °C (microwave), 50 min; cyield determined by analysis of NMR spectroscopic data using an internal standard; dN(Cl)SI (1.2 equiv.), Li2CO3 (1.1 equiv.), MeCN, 75 °C, 48 h. | ||
Heterobenzylic functionalization and 18F-fluorination of drug leads
In an effort to further demonstrate the utility of this suite of transformations, we explored the monofluorination, difluorination and trifluoromethylthiolation of quazodine (58),25 a cardiac stimulant. As depicted in Scheme 1, each of these transformations proceeded smoothly and provided access to the unique quazodine derivatives 59–61 in good to excellent yield. To gauge the impact of heterobenzylic functionalization on relevant physiochemical properties, the pKa, distribution coefficient (logD) at pH 7.4 and aqueous solubility of each compound was measured. As summarized in Scheme 1, these transformations significantly affected each property and provide a straightforward means to modulate lipophilicity and basicity. Likewise, the peracetate 63 of the cytotoxic purine nucleoside analogue 6226 could be mono- or difluorinated, affording the analogues 64 or 65, respectively, in good yield. Finally, we explored the direct 18F-fluorination of the annulated pyridine 66 to demonstrate the additional utility of this transformation for rapidly generating radiotracers for positron emission tomography (PET) imaging. We have previously exploited [18F]NFSI27 in the direct radiofluorination of branched aliphatic amino acids28 and were pleased to find that simply heating a solution of the annulated pyridine 66 and [18F]NFSI in MeCN at 75 °C for 40 min provided the 18F-labelled derivative 67 in good radiochemical conversion (RCC) and yield (RCY). This streamlined heterobenzylic 18F-fluorination does not rely on prior functionalization or sensitive reagents and thus offers certain advantages for the rapid generation of radiotracers for PET imaging.
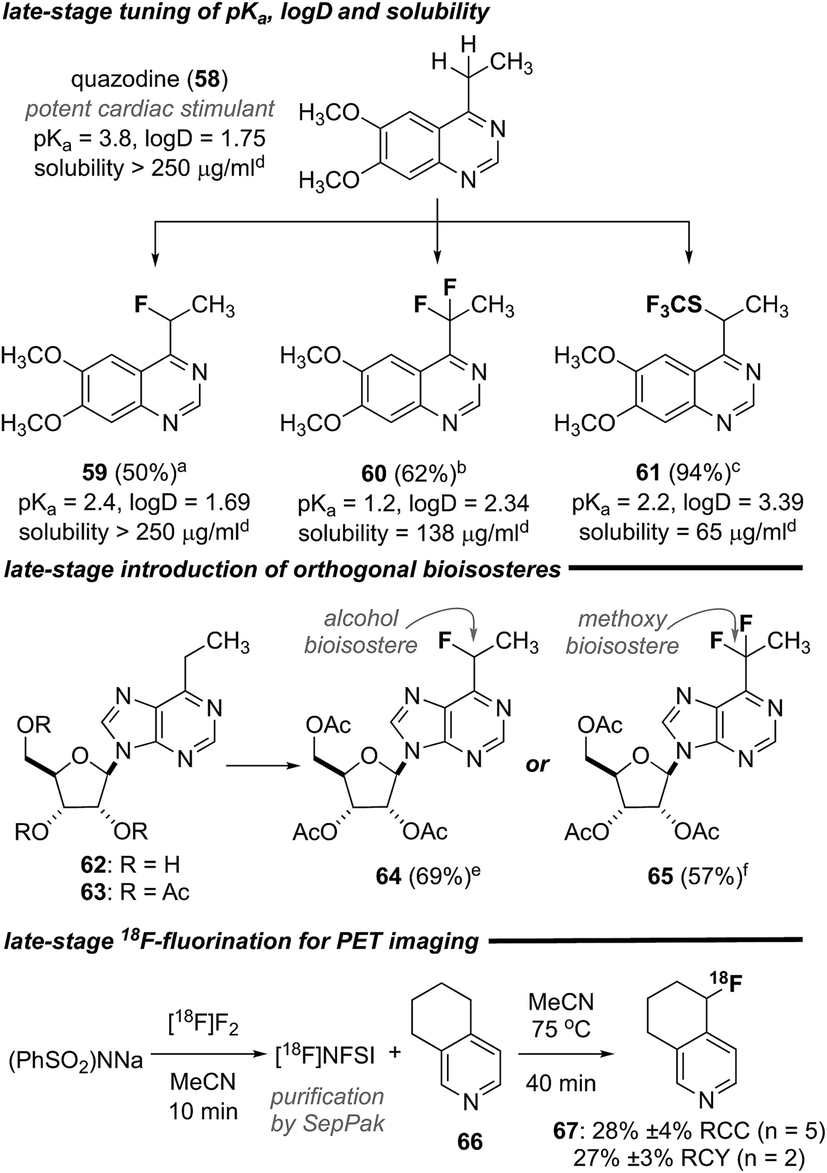 | ||
| Scheme 1 Late-stage mono- and difluorination, trifluoromethylthiolation and 18F-fluorination of heterocycles. aNFSI (1.2 equiv.), Li2CO3 (1.1 equiv.), MeCN, rt, 96 h; bNFSI (3.0 equiv.), Li2CO3 (1.1 equiv.), MeCN, 125 °C (microwave), 50 min; cN(SCF3)SI (2.4 equiv.), Li2CO3 (1.1 equiv.), MeCN, 75 °C, 48 h; dsolubility measured by lyophilisation solubility assay (LYSA) at pH 6.5 in 0.05 M phosphate buffer; eNFSI (3.0 equiv.), Li2CO3 (1.1 equiv.), MeCN, 75 °C, 48 h; fNFSI (5.0 equiv.), Li2CO3 (5.0 equiv.), MeCN, 75 °C, 48 h. | ||
Conclusions
In summary, we demonstrate that transient sulfonylation of a range of nitrogen-containing heterocycles enables direct heterobenzylic mono or difluorination using the bench stable electrophilic fluorinating agent NFSI or radiofluorination with [18F]NFSI. Taking advantage of this heterocycle activation process, both trifluoromethylthiolation and chlorination could also be achieved using the corresponding dibenzenesulfonamide derivatives. This collection of late-stage transformations should enable the rapid tuning of pKa and lipophilicity of heterocycle-containing drug leads and provides a complimentary means to incorporate pharmaceutically relevant bioisosteres (e.g., –CHF2, –CF2R and –CH(SCF3)R) as well as a method to rapidly generate 18F-labelled imaging agents for PET imaging.Experimental
General procedure for heterobenzylic monofluorination
To a solution of substrate in CH3CN (0.1–0.25 M substrate) was added N-fluorobenzenesulfonimide (NFSI) (3.0 equiv.) and Li2CO3 (1.1 equiv.). The resulting reaction mixture was then heated to 65 °C and maintained at this temperature for 18–24 h. The reaction mixture was cooled, diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The organic layer was dried (MgSO4), concentrated and the crude reaction product was purified by column chromatography on silica gel.General procedure for heterobenzylic difluorination
To a solution of substrate in CH3CN (0.25–0.50 M substrate) was added N-fluorobenzenesulfonimide (NFSI) (5.0 equiv.) and Li2CO3 (5.0 equiv.). The resulting reaction mixture was then either heated to 75 °C and maintained at this temperature for 48 h or heated to 125 °C and maintained at this temperature for 1 h in a microwave reactor. The reaction mixture was then cooled, diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The organic layer was dried (MgSO4), concentrated and the crude reaction product was purified by column chromatography on silica gel.General procedure for heterobenzylic trifluoromethyl-thiolation
To a solution of substrate in CH3CN (0.25–0.50 M substrate) was added N-trifluoromethylthiobenzenesulfonimide (2.4 equiv.) and Li2CO3 (1.1 equiv.). The resulting reaction mixture was then either heated to 75 °C and maintained at this temperature for 48 h or heated to 125 °C and maintained at this temperature for 1 h in a microwave reactor. The reaction mixture was cooled, diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The organic layer was dried (MgSO4), concentrated and the crude reaction product was purified by column chromatography on silica gel.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an NSERC Discovery Grants to R. B., a MSFHR Career Investigator Award to R. B., a Hoffmann-La Roche Fellowship (RPF) for M. B. N. an NSERC PGS and a SFU MYF for M. M. We would like to thank Pascal Hasenfratz, Laura Martin, Simona Capomolla, Maria Giraldo, Silvio Binkert and Christelle Jablonski for the preparation of starting materials, Catherine Karrer for LYSA, Aynur Ekiciler for logD and Bjoern Wagner for pKa measurements.
References
- For recent reviews, see: (a) C. Chatalova-Sazepin, R. Hemelaere, J.-F. Paquin and G. M. Sammis, Synthesis, 2015, 47, 2554 CrossRef; (b) D. E. Yerien, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 8398 RSC; (c) B. Lantaño and A. Postigo, Org. Biomol. Chem., 2017, 15, 9954 RSC; (d) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef PubMed; (e) W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727 CrossRef PubMed; (f) Y. Li, Y. Wu, G.-S. Li and X.-S. Wang, Adv. Synth. Catal., 2014, 356, 1412 CrossRef; (g) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073 CrossRef PubMed.
- (a) N. A. Meanwell, J. Med. Chem., 2018 DOI:10.1021/acs.jmedchem.7b01788; (b) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef PubMed; (c) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef PubMed; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (e) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- (a) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Acc. Chem. Res., 2016, 49, 1911 CrossRef PubMed; (b) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC; (c) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef PubMed.
- P. Jeschke, Pest Manage. Sci., 2017, 73, 1053 CrossRef PubMed.
- T. D. Penning, et al. , J. Med. Chem., 1997, 40, 1347 CrossRef PubMed.
- C. S. Burgey, et al. , J. Med. Chem., 2003, 46, 461 CrossRef PubMed.
- E. Vitaku, D. J. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef PubMed.
- For reviews on trifluoromethylation of (hetero)aromatics, see: (a) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef PubMed; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef PubMed; (c) A. A. Gakh and Y. Shermolovich, Curr. Top. Med. Chem., 2014, 14, 952 CrossRef PubMed; (d) X. F. Wu, H. Neumann and M. Beller, Chem.–Asian J., 2012, 7, 1744 CrossRef PubMed.
- For transition metal catalyzed difluoromethylation, see: (a) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524 CrossRef PubMed; (b) G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., Int. Ed., 2012, 51, 12090 CrossRef PubMed; (c) Y. Gu, X. Leng and Q. Shen, Nat. Commun., 2014, 5, 5405 CrossRef PubMed; (d) X.-L. Jiang, Z.-H. Chen, X.-H. Xu and F.-L. Qing, Org. Chem. Front., 2014, 1, 774 RSC; (e) C. Matheis, K. Jouvin and L. J. Goossen, Org. Lett., 2014, 16, 5984 CrossRef PubMed; (f) Z. Feng, Q.-Q. Min and X. Zhang, Org. Lett., 2016, 18, 44 CrossRef PubMed; (g) K. Aikawa, H. Serizawa, K. Ishii and K. Mikami, Org. Lett., 2015, 18, 3690 CrossRef PubMed; (h) L. Xu and D. A. Vicic, J. Am. Chem. Soc., 2016, 138, 2536 CrossRef PubMed; (i) J. R. Bour, S. K. Kariofillis and M. S. Sanford, Organometallics, 2017, 36, 1220 CrossRef; (j) Z. Feng, Q.-Q. Min, X.-P. Fu, L. An and X. Zhang, Nat. Chem., 2017, 9, 918 CrossRef PubMed.
- M. Meanwell and R. Britton, Synthesis, 2018, 50, A-I Search PubMed.
- For example, see: (a) W.-P. Deng, G. Nam, J. Fan and K. L. Kirk, J. Org. Chem., 2003, 68, 2798 CrossRef PubMed; (b) W. J. Middleton and E. M. Bingham, J. Org. Chem., 1980, 45, 2883 CrossRef.
- (a) Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2011, 134, 1494 CrossRef PubMed; (b) Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95 CrossRef PubMed; (c) Q. Zhou, A. Ruffoni, R. Gianatassio, Y. Fujiwara, E. Sella, D. Shabat and P. S. Baran, Angew. Chem., Int. Ed., 2013, 52, 3949 CrossRef PubMed.
- For examples, see: (a) T. Taguchi, O. Kitagawa, T. Morikawa, T. Nishiwaki, H. Uehara, H. Endo and Y. Kobayashi, Tetrahedron Lett., 1986, 27, 6103 CrossRef; (b) S. Murakami, H. Ishii, T. Tajima and T. Fuchigami, Tetrahedron, 2006, 62, 3761 CrossRef; (c) K. Fujikawa, Y. Fujioka, A. Kobayashi and H. Amii, Org. Lett., 2011, 13, 5560 CrossRef PubMed; (d) Y. Ohtsuka and T. Yamakawa, Tetrahedron, 2011, 67, 2323 CrossRef; (e) J. Jung, E. Kim, Y. You and E. J. Cho, Adv. Synth. Catal., 2014, 356, 2741 CrossRef; (f) C. Shao, G. Shi, Y. Zhang, S. Pan and X. Guan, Org. Lett., 2015, 17, 2652 CrossRef PubMed.
- For examples, see: (a) H. Chen, P. Li, M. Wang and L. Wang, Org. Lett., 2016, 18, 4794 CrossRef PubMed; (b) L. Wang, X.-J. Wei, W.-L. Jia, J.-J. Zhong, L.-Z. Wu and Q. Liu, Org. Lett., 2014, 16, 5842 CrossRef PubMed; (c) S. Ge, S. I. Arlow, M. G. Mormino and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 14401 CrossRef PubMed.
- For recent reviews, see: (a) M.-C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem.–Eur. J., 2015, 21, 12836 CrossRef PubMed; (b) D. E. Terien, S. Barata-Vallejo and A. Postigo, Chem.–Eur. J., 2017, 23, 14676 CrossRef PubMed.
- J. J. Douglas, H. Albright, M. J. Sevrin, K. P. Cole and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2015, 54, 14898 CrossRef PubMed.
- For examples, see: (a) W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024 CrossRef PubMed; (b) S. Bloom, M. McCann and T. Lectka, Org. Lett., 2014, 16, 6338 CrossRef PubMed; (c) S. Bloom, C. R. Pitts, R. Woltornist, A. Griswold, M. G. Holl and T. Lectka, Org. Lett., 2013, 15, 1722 CrossRef PubMed; (d) M. B. Nodwell, A. Bagai, S. D. Halperin, R. E. Martin, H. Knust and R. Britton, Chem. Commun., 2015, 51, 11783 RSC; (e) P. Xu, S. Guo, L. Wang and P. Tang, Angew. Chem., Int. Ed., 2014, 53, 5955 CrossRef PubMed; (f) J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494 CrossRef PubMed.
- For examples, see: (a) K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef PubMed; (b) K. B. McMurtrey, J. M. Racowski and M. S. Sanford, Org. Lett., 2012, 14, 4094 CrossRef PubMed; (c) B. S. Freeze, K. M. Gigstad, D. A. Janowick, H. M. Lee, Z. Shi, F. Soucy and S. Vyskocil, Millennium Pharmaceuticals Inc., WO2016118565 A1, 2016; (d) J.-J. Ma, W.-B. Yi, G.-P. Lu and C. Cai, Org. Biomol. Chem., 2015, 13, 2890 RSC; (e) K. E. Danahy, J. C. Cooper and J. F. Van Humbeck, Angew. Chem., Int. Ed., 2018, 57, 5134–5138 CrossRef PubMed.
- M. Rueda-Becerril, C. C. Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J.-F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026 CrossRef PubMed.
- J. M. Antelo, J. Crugeiras, J. Ramon Leis and A. Ríos, J. Chem. Soc., Perkin Trans. 2, 2000, 2071 RSC.
- M. Meanwell, M. B. Nodwell, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2016, 55, 13244 CrossRef PubMed.
- P. Zhang, M. Li, X.-S. Xue, C. Xu, Q. Zhao, Y. Liu, H. Wang, Y. Guo, L. Lu and Q. Shen, J. Org. Chem., 2016, 81, 7486 CrossRef PubMed.
- T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2013, 52, 12856 CrossRef PubMed.
- For the hydrotrifluoromethylthiolation of α-heteroaryl-α-diazo esters, see: Q. Lefebvre, E. Fava, P. Nikolaienko and M. Rueping, Chem. Commun., 2014, 50, 6617 RSC.
- J. R. Parratt and E. Winslow, Br. J. Pharmacol., 1971, 43, 612 CrossRef PubMed.
- A. E. A. Hassan, R. A. I. Abou-Elkhair, J. M. Riordan, P. W. Allan, W. B. Parker, R. Khare, W. R. Waud, J. A. Montgomery and J. A. Secrist III, Eur. J. Med. Chem., 2012, 47, 167 CrossRef PubMed.
- H. Teare, E. G. Robins, E. Arstad, S. K. Luthra and V. Gouverneur, Chem. Commun., 2007, 2330 RSC.
- M. B. Nodwell, H. Yang, M. Colovic, Z. Yuan, H. Merkens, R. E. Martin, F. Benard, P. Schaffer and R. Britton, J. Am. Chem. Soc., 2017, 139, 3595 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, purification and characterization data for all new compounds. See DOI: 10.1039/c8sc01221k |
| This journal is © The Royal Society of Chemistry 2018 |