
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single and double activation of acetone by isolobal B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) N and BB triple bonds†
N and BB triple bonds†
Julian
Böhnke‡
ab,
Tobias
Brückner‡
ab,
Alexander
Hermann‡
ab,
Oscar F.
González-Belman‡
c,
Merle
Arrowsmith
 ab,
J. Oscar C.
Jiménez-Halla
*c and
Holger
Braunschweig
*ab
ab,
J. Oscar C.
Jiménez-Halla
*c and
Holger
Braunschweig
*ab
aInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
cDepartamento de Química, Universidad de Guanajuato, Noria Alta S/N, 36050 Guanajuato, México. E-mail: jjimenez@ugto.mx
First published on 30th April 2018
Abstract
B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and BB triple bonds induce C–H activation of acetone to yield a (2-propenyloxy)aminoborane and an unsymmetrical 1-(2-propenyloxy)-2-hydrodiborene, respectively. DFT calculations showed that, despite their stark electronic differences, both the BN and BB triple bonds activate acetone via a similar coordination-deprotonation mechanism. In contrast, the reaction of acetone with a cAAC-supported diboracumulene yielded a unique 1,2,3-oxadiborole, which according to DFT calculations also proceeds via an unsymmetrical diborene, followed by intramolecular hydride migration and a second C–H activation of the enolate ligand.
N and BB triple bonds induce C–H activation of acetone to yield a (2-propenyloxy)aminoborane and an unsymmetrical 1-(2-propenyloxy)-2-hydrodiborene, respectively. DFT calculations showed that, despite their stark electronic differences, both the BN and BB triple bonds activate acetone via a similar coordination-deprotonation mechanism. In contrast, the reaction of acetone with a cAAC-supported diboracumulene yielded a unique 1,2,3-oxadiborole, which according to DFT calculations also proceeds via an unsymmetrical diborene, followed by intramolecular hydride migration and a second C–H activation of the enolate ligand.
Due to their intrinsic electron deficiency, linear compounds containing a multiply bonded, sp-hybridised boron atom are far more reactive and difficult to isolate than isolobal carbon-based compounds. Owing to their ease of derivatisation, monomeric iminoboranes of the form RB
NR′ (R, R′ = anionic substituents),1 which are formally isoelectronic to alkynes,2 have been the most widely studied class of two-coordinate boron compounds.3 The strong polarisation of the BN bond enables their participation in a vast array of spontaneous [2 + 2] cycloaddition4 and 1,2-addition reactions5 with polar substrates inaccessible to their alkyne counterparts. Only recently has our group shown that, with a suitable transition metal catalyst, iminoboranes can undergo [2 + 2] and [2 + 4] cycloaddition reactions with nonpolar alkynes.6
While linear RBNR′ compounds have been studied for over 30 years, isolobal LBBL compounds (L = neutral donor ligand) displaying two dicoordinate, zero-valent boron atoms long eluded isolation. Since our report of the first stable diboryne, (IDip)BB(IDip) (I, IDip = 1,3-bis(2,6-diisopropylphenyl)-imidazolidin-2-ylidene),7 we have shown that, by varying the π acceptor ability of L, the electronics and reactivity of these compounds can be fine-tuned.8,9 Thus, whereas unsaturated N-heterocyclic carbene (NHC)-supported diborynes such as I are inert towards H2,10 (SIDep)BB(SIDep) (II, SIDep = 1,3-bis(2,6-diethylphenyl)-4,5-(dihydro)imidazolidin-2-ylidene), which is supported by saturated NHCs of intermediate π acidity,11 adds H2 at 80 °C to yield a 1,2-dihydrodiborene.10 In turn, the use of even stronger π-accepting cyclic (alkyl)(amino)carbenes (cAACs)12 yields the cumulenic species (MecAAC)⋯B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B⋯(MecAAC) (III, MecAAC = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene),13 which, unlike I and II, activates H2 at room temperature10 and undergoes spontaneous [2 + 2] and [2 + 4] cycloadditions with acetylene.14
B⋯(MecAAC) (III, MecAAC = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene),13 which, unlike I and II, activates H2 at room temperature10 and undergoes spontaneous [2 + 2] and [2 + 4] cycloadditions with acetylene.14
Intrigued by the seeming lack of reactivity overlap between isolobal linear RBNR′ and LBBL species (Fig. 1), we were eager to investigate whether the diboron compounds undergo spontaneous polar cycloaddition reactions similar to those of iminoboranes. Herein we compare the reactivity of I–III and a highly sterically hindered iminoborane, (TMP)BNAr* (IV, Ar*= (2,6-(CHPh2)2-4-tBuC6H2); TMP = 2,6-tetramethylpiperidyl), towards acetone and show that, despite their marked electronic differences, compounds II and IV activate acetone following a similar mechanism, whereas cumulene II promotes an unprecedented spontaneous double activation of acetone.
 | ||
| Fig. 1 Side-by-side comparison of iminoboranes and diborynes. | ||
Heating a suspension of IV in hexanes with excess acetone overnight at 70 °C resulted in clean formation of the (2-propenyloxy)aminoborane 1 (Scheme 1A). 11B NMR data of 1 showed a resonance at 24.8 ppm, while the 1H NMR spectrum displayed a NH singlet at 3.49 ppm and two characteristic 1H resonances for the terminal methylidene protons of the enolate ligand at 4.36 and 4.11 ppm.
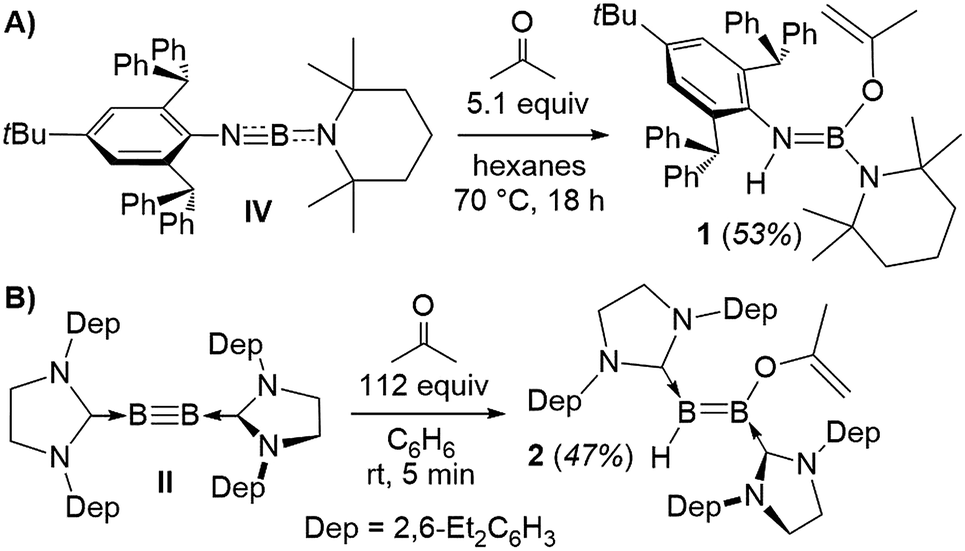 | ||
| Scheme 1 Enolic activation of acetone by (A) iminoborane IV and (B) diboryne III. Dep = 2,6-diethylphenyl. | ||
X-Ray crystallographic analysis of 1 (Fig. 2) confirms the sp2-hybridisation at B1 (Σ(∠B1) = 359.9(18)°) and N1 (B1–N1–C4 126.92(16)°) as well as the elongation of B1–N1 to a single bond (1.424(3) Å). The 2-propenyloxide ligand coordinated to B1 displays a C1–O1 single bond (1.343(2) Å) and a terminal C1C2 double bond (1.320(3) Å). Formally, compound 1 results from the addition of 2-propenol, the enol form of acetone, across the polar BN triple bond of iminoborane IV. While there is, to our knowledge, no literature precedent for the reactivity of monomeric iminoboranes with enolisable ketones, the dimeric iminoborane [BuBNtBu]2 has been shown to undergo 1,4-enol addition to the ring-opened iminoborane dimer with acetone, acetophenone and 3,3-dimethylbutan-2-one.15 This contrasts with the reactivity of iminoboranes towards aldehydes4d and CO2,4c which yields the 1,3,2-oxazaboretidine [2 + 2] cycloaddition products.
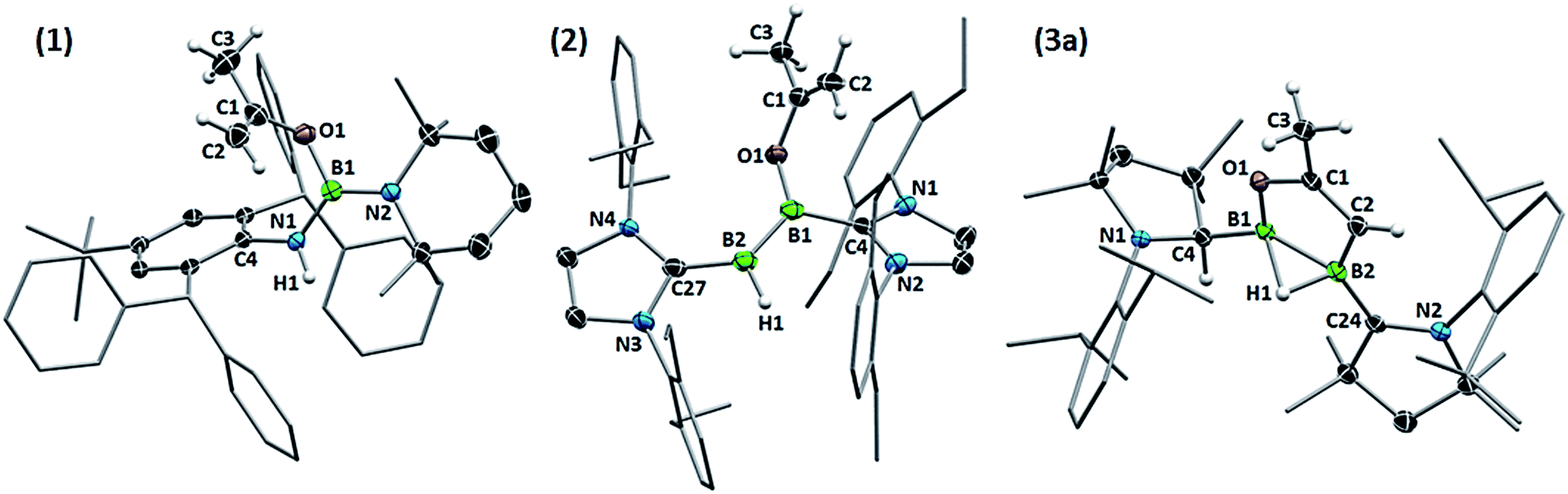 | ||
| Fig. 2 Crystallographically determined solid-state structures of 1, 2 and 3a. Atomic displacement ellipsoids depicted at the 50% probability level. Atomic displacement ellipsoids of peripheral substituents omitted for clarity. Hydrogen atoms omitted, except for those of the activated acetone moieties, the protonated cAAC carbon atom and those bound to boron. Selected bond lengths (Å): (1) B1–N1 1.424(3), B1–N2 1.429(3), B1–O1 1.418(3), O1–C1 1.343(2), C1–C2 1.320(3); (2) B1–C4 1.574(4), B2–C27 1.523(4), B1–B1 1.599(4), B2–H1 1.09(2), B1–O1 1.455(3), O1–C1 1.352(3), C1–C2 1.316(4); (3a) B1–C4 1.5295(19), B2–C24 1.5295(19), B1–B2 1.721(2), B1–H1 1.485(17), B2–H1 1.213(16), B1–O1 1.4064(17), O1–C1 1.3824(15), C1–C2 1.3301(18), C2–B2 1.5743(18). | ||
Whereas diboryne I proved unreactive towards acetone even under forcing conditions, diboryne II reacted rapidly with excess acetone in benzene at room temperature to yield the green-coloured 1,2-enol addition product 2 (Scheme 1). Compound 2 presents two broad 11B NMR resonances at 38.1 and 19.3 ppm in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, attributable to the BH and the BOC3H5 moieties of the unsymmetrical diborene, respectively. The 1H NMR spectrum displayed two inequivalent SIDep ligands, as well as the inequivalent terminal methylene protons of the 2-propenyloxide ligand at 3.93 and 3.47 ppm. X-Ray crystallographic analysis of 2 showed a trans-1-alkoxy-2-hydrodiborene with a B–B double bond of 1.599(4) Å similar to that of its dihydrodiborene relative, (SIDep)HBBH(SIDep) (1.589(4) Å).10 The SIDep ligand at the BH moiety is near coplanar with the diborene core (torsion (N4, C27, B2, B1) 12.1(5)°) and displays a short B2–C27 bond (1.523(4) Å), indicative of π backdonation. In contrast, the SIDep ligand supporting the BOC3H5 moiety is twisted ca. 35.5° out of the diborene plane and displays a pure σ-donor interaction (B1–C4 1.574(4) Å). The planar 2-propenyloxide ligand lies at a ca. 58° angle with respect to the diborene plane, and its bond lengths (O1–C1 1.352(3), C1C2 1.316(4) Å) are similar to those of 1. With Kinjo and co-workers recently reporting the first diborene with two different donor ligands16 and our group having just published the first fully unsymmetrical diborene,17 compound 2 is only the second unsymmetrical diborene with respect to the anionic substituents.
:1 ratio, attributable to the BH and the BOC3H5 moieties of the unsymmetrical diborene, respectively. The 1H NMR spectrum displayed two inequivalent SIDep ligands, as well as the inequivalent terminal methylene protons of the 2-propenyloxide ligand at 3.93 and 3.47 ppm. X-Ray crystallographic analysis of 2 showed a trans-1-alkoxy-2-hydrodiborene with a B–B double bond of 1.599(4) Å similar to that of its dihydrodiborene relative, (SIDep)HBBH(SIDep) (1.589(4) Å).10 The SIDep ligand at the BH moiety is near coplanar with the diborene core (torsion (N4, C27, B2, B1) 12.1(5)°) and displays a short B2–C27 bond (1.523(4) Å), indicative of π backdonation. In contrast, the SIDep ligand supporting the BOC3H5 moiety is twisted ca. 35.5° out of the diborene plane and displays a pure σ-donor interaction (B1–C4 1.574(4) Å). The planar 2-propenyloxide ligand lies at a ca. 58° angle with respect to the diborene plane, and its bond lengths (O1–C1 1.352(3), C1C2 1.316(4) Å) are similar to those of 1. With Kinjo and co-workers recently reporting the first diborene with two different donor ligands16 and our group having just published the first fully unsymmetrical diborene,17 compound 2 is only the second unsymmetrical diborene with respect to the anionic substituents.
TDDFT calculations performed upon the optimised geometry of 2 at the (smd: n-pentane)lc-ωPBE/6-311+g(d) level of theory provided a maximum UV-vis absorbance at 592 nm (see Table S1 and Fig. S24 in the ESI†), which is in good agreement with the experimentally measured absorbance maximum in pentane at 605 nm (Fig. S16†). This corresponds to the HOMO–LUMO transition from the π-bonding orbital of the BB double bond into the empty pz orbital of the carbene carbon of the SIDep ligand supporting the BOC3H5 moiety, and is responsible for the blue-green color of the compound.
Surprisingly, the 1:1 reaction of diboracumulene III with acetone did not yield the expected cAAC analogue of 2. Instead, 11B NMR data revealed a 92:8 mixture of two sp2–sp3 diborane products, the major one (3a) showing two broad singlets at 42.8 (full width at half maximum: fwhm ≈ 370 Hz) and −1.9 ppm (fwhm ≈ 130 Hz), and the minor (3b) presenting a very broad resonance at 63.0 ppm (fwhm ≈ 630 Hz) and a broad BH doublet at −15.0 ppm (1JB–H = 50.8 Hz), suggesting a non-bridging hydride. The 1H NMR spectrum of the mixture showed very similar sets of resonances for 3a and 3b, which strongly suggests an isomeric relationship. Both compounds display one neutral cAAC ligand and one C1-protonated cAAC ligand (δ = 3a 4.02, 3b 4.24 ppm) as well as a single 1H alkene resonance (δ = 3a 3.50, 3b 3.78 ppm) (Scheme 2).
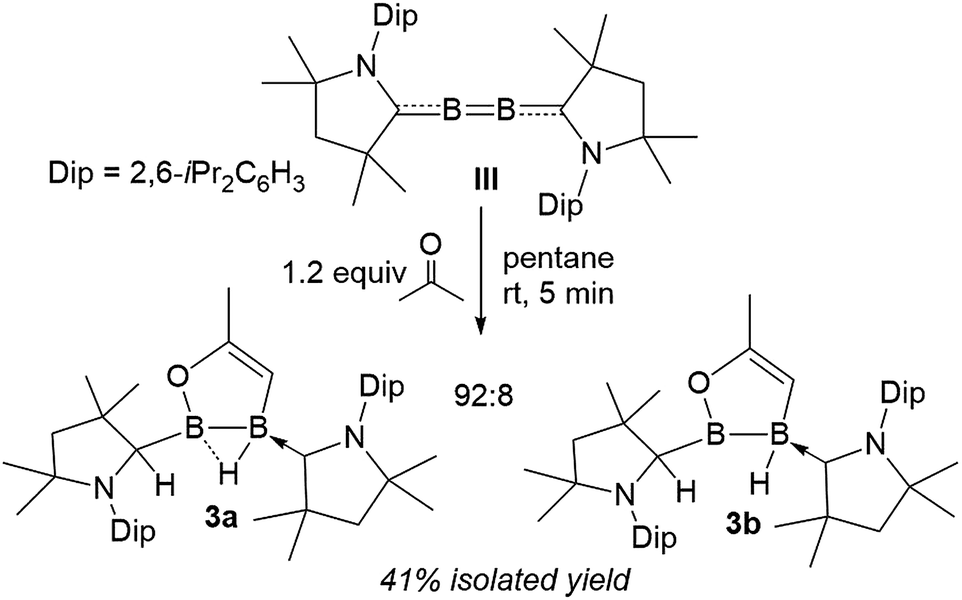 | ||
| Scheme 2 Double activation of acetone by diboracumulene III. | ||
Single-crystal X-ray crystallography revealed a unique planar 2,3-dihydro-5-methyl-1,2,3-oxadiborole heterocycle displaying an endocyclic C1C2 double bond (1.3301(18) Å, Fig. 2). The B–B bond is unsymmetrically μ2-bridged by a hydride (B1–B2 B1–H1 1.213(16), B2–H1 1.485(17) Å) positioned orthogonally to the B2C2O heterocycle (torsion (H1, B2, B1, C2) 105.7(9)°) and shows a bond length of 1.721(2) Å typical of a diborane (5). The alkenylborane moiety around B1 is supported by a neutral cAAC ligand with a relatively short B1–C4 bond (1.5295(19) Å) and forms an angle of only ca. 19° with the plane of the B2C2O heterocycle (torsion (N1, C4, B1, C2) 14.4(2)°), which is indicative of π conjugation. The enoxyborane moiety around B2 bears a protonated cAAC ligand displaying clear sp3-hybridisation at C24 (B2–C2 4 1.6045(18), C24–N2 1.4878(16) Å). The structure of 3a is reminiscent of the products obtained from the reduction of (SIMes)BBr2BAr2 diborane (5) precursors (SIMes = 1,3-Mes2-4,5-dihydroimidazol-2-ylidene, Mes = 2,4,6-trimethylphenyl; Ar = Mes, 9-anthryl). These display a central, μ2-hydride-bridged, planar B2C5 heterocycle, resulting from the C–H activation of one aryl substituent by an intermediate boraborylene, and coordinated on one side by a neutral SIMes ligand and on the other by the second aryl substituent.18
Although single crystals of the minor species in solution were never obtained, the propensity for cAAC-supported hydroboranes to undergo 1,2-hydrogen shifts from boron to an adjacent cAAC carbene centre, which has been demonstrated both experimentally and computationally,19 first prompted us to identify the second isomer as compound 3taut, a tautomeric form of 3a, in which the neutral cAAC ligand coordinates to the enoxyborane moiety, and the protonated cAAC ligand coordinates to the alkenylborane moiety (Fig. 3). DFT optimisations at the ONIOM(M06-2X/6-311+G(d):PM6) level (see ESI† for details) showed, however, that 3taut is 8.4 kcal mol−1 higher in energy than 3a and that its calculated 11B NMR shifts (δ = 45.1, 6.1 ppm) do not fit the experimental data (δ = 63.0, −15.0 ppm). Since 3a presents two stereocentres, one at B2, which is locked by the B2C2O ring and the asymmetrically bridging hydride, and one at the protonated cAAC carbon atom, the other possibility is that 3a and 3b could be diastereomers. This would also fit the observation that they do not exchange in solution even at high temperatures. To test this, the geometries and 11B NMR chemical shifts of the possible diastereomeric pairs derived from 3a were computed (Fig. 3).
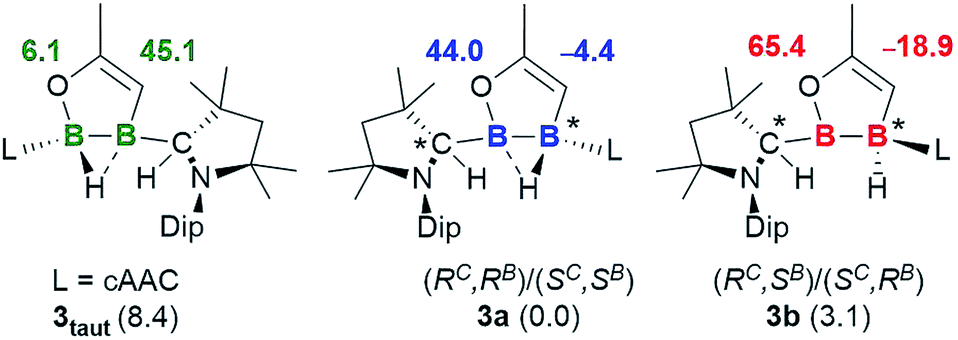 | ||
| Fig. 3 Calculated 11B NMR shifts (ppm, in bold) for the optimised structures of tautomer 3taut, and the diastereomeric pair 3a and 3b at the ONIOM(M06-2X/6-311+G(d):PM6) level. Experimental shifts for comparison: 3a: δ = 42.8 and −1.9 ppm; 3b: δ = 63.0 and −15.0 ppm. Relative energies (kcal mol−1) in brackets. | ||
The predicted 11B NMR chemical shifts for the (RC,RB)/(SC,SB)-3a pair (δcalc = 44.0, −4.4 ppm) adequately match the experimentally-observed shifts (δexp = 42.8, −1.9 ppm; Δ(δ) ≈ ±2 ppm). Calculations on the diastereomeric pair showed that a form with a non-bridging hydride is the most likely. This also correlates well with the observation that, unlike 3a, which shows two very broad 11B NMR resonances typical for a μ2-hydride-bridged diborane, 3b shows a doublet at −15.0 ppm (1J11B–1H = 50.8 Hz), indicating a terminal hydride rather than a bridging one. The predicted 11B NMR chemical shifts for the (RC,SB)/(SC,RB)-3b pair (δcalc = 65.4, −18.9 ppm) are comparable to the experimental ones (δexp = 63.0, −15.0 ppm; Δ(δ) ≈ ±3 ppm). The relative energy of (RC,SB)/(SC,RB)-3b, at 3.1 kcal mol−1 above (RC,RB)/(SC,SB)-3a, is consistent with the experimentally observed ratio of 92:8.
The spontaneous formation of 3a/b is particularly remarkable in view of the fact that there is seemingly no literature precedent for a one-step, uncatalysed, 100% atom-efficient double C–H activation of acetone or other enolisable ketones. We were therefore keen to investigate the mechanism of the formation of 3a/b and compare it to that of the boron enolates 1 and 2. While the reaction of dimeric iminoboranes with enolisable ketones always yielded the 1,4-enol addition products, Paetzold and co-workers showed that with acetophenone, which is less prone to enolisation, a [2 + 4] cycloaddition product can also be isolated.15 However, it remained unclear whether or not the latter is an intermediate to the former. For comparison, nonpolar disilenes are known to first undergo [2 + 2] cycloaddition with acetone and acetophenone to form the corresponding 1,2,3-oxadisiletane heterocycles, which then rearrange to the 1,2-enol addition products.20 In our case, however, careful monitoring of the reaction of iminoborane IV and diboryne II with acetone showed no evidence of [2 + 2] cycloaddition products or intermediates.
DFT calculations carried out at the D3-PBE0/6-31G(d) level for IV and at the ONIOM(M06-2X/6-311+G(d):PM6) level for II and III showed that acetone activation does not proceed via 1,2-enol addition, as the enol form of acetone lies 15.3 kcal mol−1 higher than the reactants, well above the activation energy for direct acetone addition (Fig. 4, see ESI† for details on the methodology and the optimised structures of all reactants, products, intermediates and transition states).
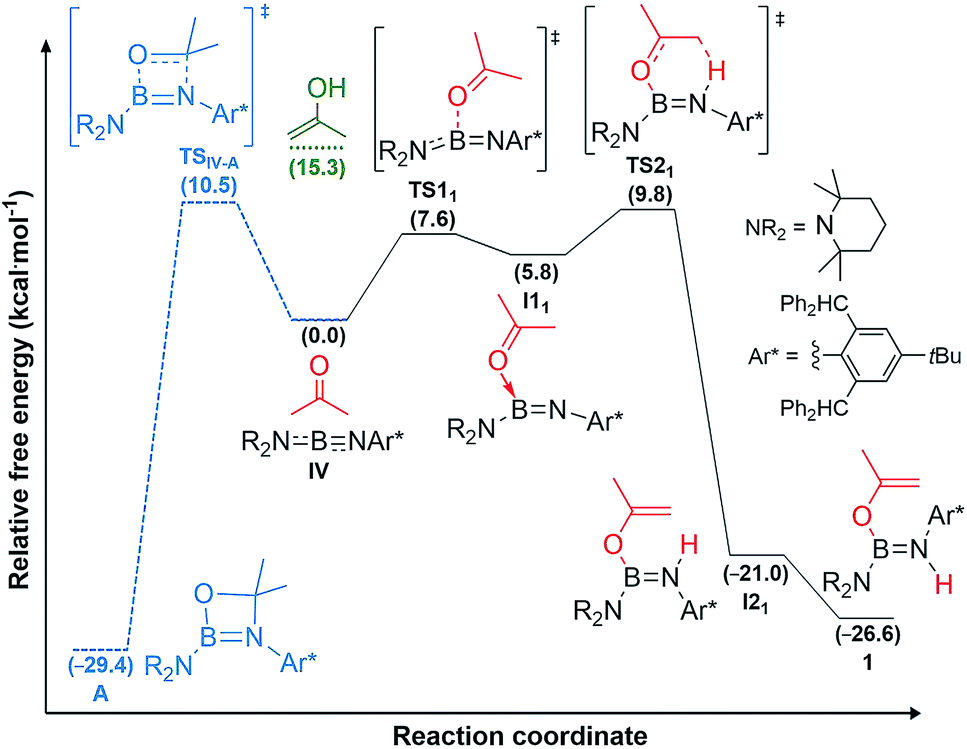 | ||
| Fig. 4 Mechanisms of acetone addition to iminoborane IV to yield aminoborane 1 (straight lines in black) and alternative [2 + 2] cycloaddition to yield A (dashed lines in blue), as well as energy level of the enol form of acetone (green) calculated at the D3-PBE0/6-31G(d) level of theory. Gibbs free energies (kcal mol−1) in brackets. | ||
For iminoborane IV two plausible mechanisms were investigated, the first via a 4,4-dimethyl-1,3,2-oxaboretidine [2 + 2] cycloaddition product (A), the second via concerted acetone coordination-deprotonation (Fig. 4). Although the cycloaddition product A is calculated to be more stable than 1 by 2.8 kcal mol−1, the energy barrier for the formation of A is slightly higher than for 1.§ Furthermore, as there is no thermodynamically viable reaction path from A to 1, a [2 + 2] cycloaddition mechanism followed by rearrangement to 1 can be ruled out.
Instead, for compounds II–IV the first reaction step involves coordination of the carbonyl oxygen atom to one boron centre to form the acetone adducts I11, I12 and I13 (ΔG‡1 = 7.6 (IV), 20.6 (II), 10.1 (III) kcal mol−1), respectively (Fig. 4 and 5). This step is followed in all three cases by C–H activation of one of the pendant methyl groups of the coordinated acetone by either the nitrogen atom (for IV) or the electron-rich, second boron centre (for II and III), to yield the cis-aminoborane I21, and the SIDep- and cAAC-supported cis-diborenes I22 and I23, respectively (ΔG‡2 = 4.0 (IV), 14.1 (II), 14.9 (III) kcal mol−1). Finally, the trans-aminoborane 1 and the trans-diborenes 2 and I43 are obtained by rotation around the B–N and B–B bond, respectively. Overall, the formation of 2 presents the highest energy barrier and is also the most exergonic (ΔG = −43.2 kcal mol−1), followed by that of 1 (ΔG = −26.6 kcal mol−1) and I43 (ΔG = −27.4 kcal mol−1).
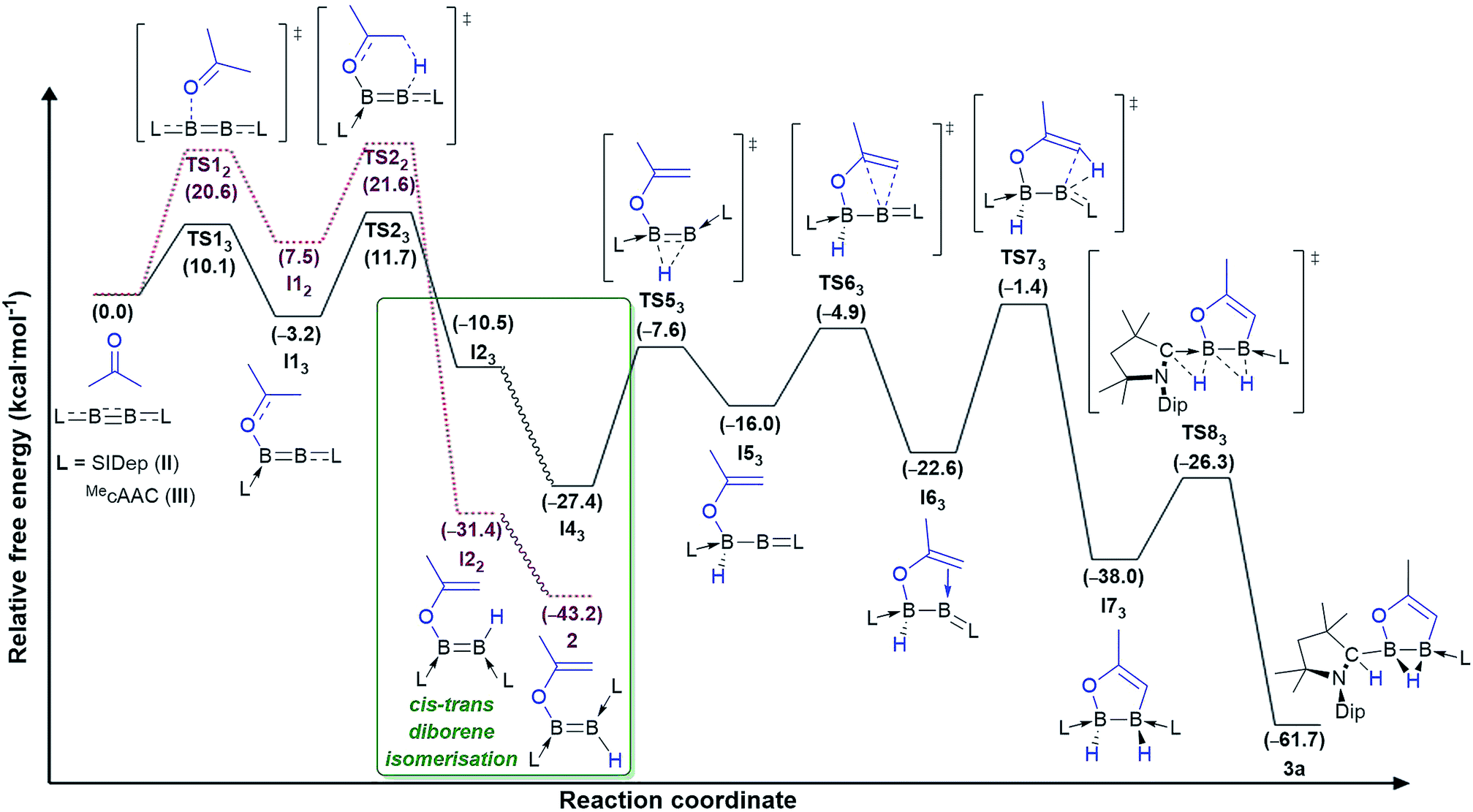 | ||
| Fig. 5 Mechanisms of double acetone activation by diboryne II (dotted lines in purple) and cumulene III (straight lines in black) calculated at the ONIOM(M06-2X/6-311+G(d):PM6) level of theory. Gibbs free energies (kcal mol−1) in brackets. Mechanistic detail of the cis–trans diborene isomerisation step provided in Fig. 6. | ||
The exergonic isomerisation step leading from the cis-diborenes I22 and I23 to the trans-diborenes 2 and I43, respectively, was further investigated to determine the rotation barrier in each case. Interestingly, DFT calculations showed two distinct mechanisms at work for the SIDep-stabilised and the cAAC-stabilised diborene, respectively (Fig. 6). For the SIDep analogue I22, rotation about the B–B bond is facilitated by shifting the π-electron density of the BB double bond into the π backbonding to the unsaturated carbene ligands. The resulting transition state TS32 now displays a B–B single bond, which allows facile rotation. The isomerisation process from I22 to 2 occurs with a low barrier of 9.7 kcal mol−1. In contrast, the lowest energy pathway for the cAAC analogue I23 proceeds via a 1,2-hydride shift from boron to the adjacent cAAC carbene carbon to yield the intermediate diborene I33 (ΔG‡3 = 8.9 kcal mol−1), in which the boron bearing the now protonated cAAC ligand is sp-hybridised. Rotation about this B–CcAACH single bond and a second 1,2-hydride shift back to the boron centre then yield the trans-diborene I43 with a low barrier of 9.3 kcal mol−1. This pathway is assisted on the one hand by the facile 1,2-hydride shuttling chemistry displayed by cAAC hydroboron compounds19 and on the other hand by the very strong π acceptor properties of cAAC,12 which enable the stabilisation of the coordinatively saturated intermediate I33.
 | ||
| Fig. 6 Divergent mechanisms of cis-to-trans isomerisation for diborene I22 (purple lines) and I32 (black lines) calculated at the ONIOM(M06-2X/6-311+G(d):PM6) level of theory. Gibbs free energies (kcal mol−1) in brackets. | ||
For cumulene III, however, the reaction does not stop at trans-diborene I43 (Fig. 3). The latter undergoes hydride migration from B1 to B2 to form the (alkoxy)hydroboryl-(alkylidene)borane I43 (ΔG‡3 = 19.8 kcal mol−1). Coordination of the pendant terminal alkene to the two-coordinate boron yields adduct I53, which is 6.6 kcal mol−1 more stable. Subsequent C–H activation of the methylidene moiety yields the bis(cAAC)-stabilised 1,2,3-oxadiborole I63 (ΔG‡5 = 21.2 kcal mol−1). This is the highest energy barrier in the entire reaction mechanism. I63 then tautomerises to compound 3a by concomitant migration of the hydride on B1 to the adjacent cAAC carbene centre and bridging of the hydride on B2 (ΔG‡6 = 11.7 kcal mol−1). Overall the formation of 3a from III and acetone is exergonic by 61.7 kcal mol−1, which explains why the intermediate diborene cannot be isolated.
To conclude, we have shown that three linear, isolobal, multiply bonded boron compounds, iminoborane IV, diboryne II and cumulene III, all activate acetone via a similar acetone coordination-deprotonation mechanism, regardless of their polar or nonpolar nature. For the iminoborane-based reaction, an enol addition mechanism and a mechanism proceeding via a [2 + 2] cycloaddition intermediate, as would normally be expected for such a polar compound, were both ruled out. For diboron compounds II and III the addition of acetone first yields a cis-diborene intermediate which isomerises to the thermodynamic trans-diborene product through a low energy barrier. Calculations showed that this isomerisation process heavily relies on the π-accepting nature of the carbene ligands, coupled, in the case of the cAAC-supported diborene, with a hydride shuttling mechanism from boron to the carbene carbon and back. These cAAC-specific properties also enable an unprecedented second C–H activation of the enolate ligand to yield a novel 1,2,3-oxadiborole heterocycle, demonstrating once again the unique reactivity of cAAC-supported low-valent boron compounds.
Overall this study should act as a reminder that the parallels all too eagerly drawn between organic compounds and their isoelectronic/isolobal inorganic p-block counterparts only rarely translate into actual organomimetic behaviour when it comes to reactivity or reaction mechanisms. Furthermore, this first example of reactivity overlap between polar and nonpolar boron-based triple bonds opens up new avenues for attempting reactions that may have been previously disregarded, such as the addition of nonpolar small molecules to iminoboranes or, alternatively, of polar molecules to diborynes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the European Research Council (ERC) under the European Union Horizon 2020 Research and Innovation Program (grant agreement no. 669054). O. F. G.-B. and J. O. C. J.-H. thank CONACyT (Mexico) for a MSc fellowship and project CB2014-241803 for a research stay at Julius-Maximilians-Universität Würzburg and the use of their supercomputing facilities. A. H. thanks the Chemical Industry Fund (FCI) for his PhD fellowship.Notes and references
- P. Paetzold, A. Richter, T. Thijssen and S. Würtenberg, Chem. Ber., 1979, 112, 3811 CrossRef.
- (a) F. Zhang, P. Maksyutenko, R. I. Kaiser, A. M. Mebel, A. Gregušová, S. A. Perera and R. J. Bartlett, J. Phys. Chem. A, 2010, 114, 12148 CrossRef PubMed; (b) E. R. Lory and R. F. Porter, J. Am. Chem. Soc., 1973, 95, 1766 CrossRef; (c) C. N. Baird and R. K. Datta, Inorg. Chem., 1972, 11, 17 CrossRef.
- (a) P. Paetzold, Adv. Inorg. Chem., 1987, 31, 123 CrossRef; (b) H. Nöth, Angew. Chem., Int. Ed. Engl., 1988, 27, 1603 CrossRef.
- (a) B. Glaser, E. P. Mayer and H. Nöth, Z. Naturforsch., 1988, 43b, 449 Search PubMed; (b) C. Klofkorn, M. Schmidt, T. Spaniol, T. Wagner, O. Costisor and P. Paetzold, Chem. Ber., 1995, 128, 1037 CrossRef; (c) D. Männig, C. K. Narula, H. Nöth and U. Wietelmann, Chem. Ber., 1985, 118, 3748 CrossRef; (d) P. Paetzold, E. Schröder, G. Schmid and R. Boese, Chem. Ber., 1985, 118, 3205 CrossRef.
- (a) U. Braun, B. Böck, H. Nöth, I. Schwab, M. Schwartz, S. Weber and U. Wietelmann, Eur. J. Inorg. Chem., 2004, 3612 Search PubMed; (b) B. Böck, H. Nöth and U. Wietelmann, Z. Naturforsch., 2001, 56b, 659 Search PubMed; (c) A. Brandl, P. Kolle and H. Nöth, Chem. Ber., 1989, 122, 419 CrossRef; (d) P. Paetzold, C. V. Plotho, H. Schwan and H.-U. Meier, Z. Naturforsch., 1984, 39b, 610 Search PubMed.
- (a) H. Braunschweig, K. Geetharani, J. O. C. Jiménez-Halla and M. Schäfer, Angew. Chem., Int. Ed., 2014, 53, 3500 CrossRef PubMed; (b) H. Braunschweig, A. Damme, J. O. C. Jiménez-Halla, B. Pfaffinger, K. Radacki and J. Wolf, Angew. Chem., Int. Ed., 2012, 51, 10034 CrossRef PubMed.
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420 CrossRef PubMed.
- J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, K. Hammond, J. O. C. Jiménez-Halla, T. Kramer and J. Mies, Angew. Chem., Int. Ed., 2015, 54, 13801 CrossRef PubMed.
- M. Arrowsmith, H. Braunschweig and T. E. Stennett, Angew. Chem., Int. Ed., 2017, 56, 96 CrossRef PubMed.
- M. Arrowsmith, J. Böhnke, H. Braunschweig, M. A. Celik, T. Dellermann and K. Hammond, Chem.–Eur. J., 2016, 22, 17169 CrossRef PubMed.
- O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 2939 CrossRef PubMed.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 CrossRef PubMed.
- J. Böhnke, H. Braunschweig, W. C. Ewing, C. Hörl, T. Kramer, I. Krummenacher, J. Mies and A. Vargas, Angew. Chem., Int. Ed., 2014, 53, 9082 CrossRef PubMed.
- M. Arrowsmith, J. Böhnke, H. Braunschweig, M. A. Celik and C. Claes, Angew. Chem., Int. Ed., 2016, 55, 11271 CrossRef PubMed.
- P. Schreyer, P. Paetzold and R. Boese, Chem. Ber., 1988, 121, 195 CrossRef.
- W. Lu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2017, 139, 5047 CrossRef PubMed.
- T. E. Stennett, J. D. Mattock, I. Vollert, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 4098 CrossRef PubMed.
- (a) P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044 CrossRef PubMed; (b) S. R. Wang, D. Prieschl, J. D. Mattock, M. Arrowsmith, C. Pranckevicius, T. E. Stennett, R. D. Dewhurst, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 6347 CrossRef PubMed.
- (a) D. Auerhammer, M. Arrowsmith, H. Braunschweig, R. D. Dewhurst, J. O. C. Jiménez-Halla and T. Kupfer, Chem. Sci., 2017, 8, 7066 RSC; (b) M. R. Momeni, E. Rivard and A. Brown, Organometallics, 2013, 32, 6201 CrossRef.
- (a) R. West, Angew. Chem., Int. Ed., 1987, 26, 1201 CrossRef; (b) A. Schäfer and M. Weidenbruch, J. Organomet. Chem., 1985, 282, 305 CrossRef; (c) M. J. Fink, D. J. DeYoung and R. West, J. Am. Chem. Soc., 1983, 105, 1070 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental details, characterization data for all reported compounds and details of the DFT calculations. CCDC 1830168–1830171 and 1830420. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01249k |
| ‡ These authors contributed in equal parts to the paper. |
| § Since the calculated difference in energy barrier for products 1 and A is so low, the reaction of iminoborane IV with acetone was also carried out at higher temperatures to see if A could be obtained instead. However, this only led, beside the formation of 1, to an accumulation of intractable decomposition products. |
| This journal is © The Royal Society of Chemistry 2018 |