
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the dynamic nature of Mo sites for methane dehydroaromatization†
Ina
Vollmer
a,
Bart
van der Linden
a,
Samy
Ould-Chikh
 b,
Antonio
Aguilar-Tapia
c,
Irina
Yarulina
ab,
Edy
Abou-Hamad
d,
Yuri G.
Sneider
e,
Alma I.
Olivos Suarez
a,
Jean-Louis
Hazemann
c,
Freek
Kapteijn
a and
Jorge
Gascon
*ab
b,
Antonio
Aguilar-Tapia
c,
Irina
Yarulina
ab,
Edy
Abou-Hamad
d,
Yuri G.
Sneider
e,
Alma I.
Olivos Suarez
a,
Jean-Louis
Hazemann
c,
Freek
Kapteijn
a and
Jorge
Gascon
*ab
aCatalysis Engineering, Chemical Engineering Department, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: jorge.gascon@kaust.edu.sa
bKing Abdullah University of Science and Technology, KAUST Catalysis Center, Advanced Catalytic Materials, Thuwal 23955, Saudi Arabia
cInst. Néel, UPR 2940 CNRS – Univ. Grenoble Alpes, F-38000 Grenoble, France
dKing Abdullah University of Science and Technology, Core Labs, Thuwal 23955, Saudi Arabia
eDipartimento di Ingegneria Chimica Materiali Ambiente, Sapienza Universitá di Roma, Via Eudossiana 18, 00184 Roma, Italy
First published on 30th April 2018
Abstract
The mechanism of methane activation on Mo/HZSM-5 is not yet fully understood, despite the great interest in methane dehydroaromatization (MDA) to replace aromatics production in oil refineries. It is difficult to assess the exact nature of the active site due to fast coking. By pre-carburizing Mo/HZSM-5 with carbon monoxide (CO), the MDA active site formation was isolated from coke formation. With this a clear 13C NMR signal solely from the active site and not obscured by coke was obtained, and it revealed two types of likely molecular Mo (oxy-)carbidic species in addition to the β-Mo2C nanoparticles often mentioned in the literature. Furthermore, separating the active site formation from coking by pre-carburization helped us examine how methane is activated on the catalytic site by carrying out MDA using isotopically labelled methane (13CH4). Carbon originating from the pre-formed carbide was incorporated into the main products of the reaction, ethylene and benzene, demonstrating the dynamic behavior of the (oxy-)carbidic active sites.
Introduction
Due to depletion of oil reserves and the increasing availability of cheap methane as a major component of shale gas as well as in methane hydrates, processes to valorize methane are in high demand.1 To date, some indirect routes of methane conversion based on syngas from methane reforming have been commercialized. Requiring few process steps, direct conversion of methane is a desired alternative. Among them, non-oxidative methods have attracted interest since the pioneering work of Bragin and later Wang et al.2,3 A great deal of research effort has focused on the most promising systems Mo/HZSM-5 and Mo/HMCM-22, typically achieving 10 to 12% conversion with a benzene selectivity of 60 to 70% at 700 °C.4,5 Other products include, in the order of decreasing selectivity, naphthalene, ethylene, ethane, xylene and toluene. The reaction is hampered by thermodynamics with ΔGor = +433 kJ mol−1 and ΔH0r = +531 kJ mol−1, and coke formation is very favorable at high temperatures, leading to fast deactivation.6–8 In Mo/HZSM-5, both the Brønsted acid site (BAS) and Mo make up the bifunctional catalytic nature of the system: CH4 is activated on the exchanged Mo active site, forming C2Hx.9 Then, the intermediate C2Hx moieties react on the BAS and are further converted into aromatic compounds.4,10–13There is a delay in the onset of the reaction, in which mono-14 and dimeric15 MoOx species from the as-synthesized Mo/HZSM-5 were shown to carburize, suggesting that reduced Mo is the active phase for methane dehydroaromatization.12,15–19 This delay is usually coined as ‘induction period’. A wide range of species have been reported to be active for MDA: MoC, Mo2C, coke modified Mo2C,20 Mo2C18,21 on the outside surface and reduced oxides in pores, any kind of Mo6+ and partially reduced Mo6+ as MoO(3 − x).11,22 However, thus far the exact nature of these reduced Mo species and which one of them dominates activity in the reaction is unknown. The observation of the active phase is difficult. 13C MAS NMR16,23–25 and UV Raman spectroscopy26,27 can be used to look at the carbon in the Mo carbide, but due to the dominant signal coming from carbonaceous species, it is hard to observe. This leaves only quite expensive or less available options, like synchrotron techniques,15,28 EPR or 95Mo MAS NMR,29,30 which, regrettably, are not conclusive either. Since the active phase forms under reaction conditions, operando techniques are necessary to get more insight into the structure of the active site. Operando X-ray Absorption Spectroscopy (XAS) was used to show the gradual carburization of the molybdenum sites during the early stage of the reaction.17 While this yields a lot of insight into the oxidation state of Mo, structural information to be gained is limited, because XAS represents a bulk technique, making it difficult to distinguish between the plethora of active sites present on the zeolite. The signal is dominated by the bigger clusters of Mo, which are always present on the catalyst. Information on mono- or dimeric species, believed to be responsible for catalysis, is mostly lost.31
In another economically very interesting reaction, Fischer–Tropsch, it is proposed that the carbide phase of iron is active for the reaction and is involved in initiating the chain-growth of hydrocarbons.32,33 Both DFT studies as well as experiments with labelled reactants suggest that the carbide is easily hydrogenated to CH2 species, which then further react with gas-phase species to form hydrocarbons.34 Afterwards the carbide is replenished again in a way similar to the oxides in the Mars–van-Krevelen mechanism.35 Although the Fe-carbide is present as nanoparticles in the Fischer–Tropsch catalyst and the initial active phase for Mo/HZSM-5 is expected to present itself either as molecular MoOxCy or cluster species, it is of high interest to consider a similar mechanism for this system. Especially, since the catalyst considered here, Mo/HZSM-5, was also found to be active for the Fischer–Tropsch reaction.36 In a few publications, understanding the activation of CH4 on the carbidic form of Mo/HZSM-5 is only theoretically approached.37–39
In this work, we experimentally investigate the interaction of methane with the relevant Mo active sites during the MDA reaction. Because of the aforementioned induction period of MDA, it is not possible to separate in time the formation of the Mo active phase via CH4 carburization and the MDA reaction itself. Furthermore, the MDA reaction produces coke surrounding the active site and prevents the possibility to probe the interaction of CH4 solely with the active Mo-carbide phase.40 To circumvent this issue, our study involves the prior synthesis of the Mo carbide by contacting the Mo/HZSM-5 pre-catalyst with carbon monoxide (CO) at a high temperature (780 °C). Indeed, carbon monoxide has been reported to carburize Mo oxides to Mo2C.41–43 We will demonstrate by a combination of online mass spectrometry (MS) and operando X-ray absorption spectroscopy that the Mo (oxy-)carbide produced via CO carburization and CH4 activation is equivalent. We report more direct structural characterization of the active site using 13C NMR. Then, the use of isotope labelling experiments and online-MS will highlight the dynamic behaviour of the Mo active phase when contacted with methane.
Results and discussion
Catalyst preparation
Catalysts were based on a commercial HZSM-5 zeolite (Südchemie) having Si/Al = 13 (HZ-13) and were prepared with 1, 2 and 5 wt% loading (x) of Mo denoted as xMoHZ-13 using (NH4)6Mo7O24 as a precursor.44Temperature programmed carburization
To understand the conditions necessary to prepare the active Mo site by CO carburization, CO consumption and simultaneous CO2 production were monitored by MS during Temperature Programmed Carburization (TPC) with 13CO. Fig. 1 shows that carburization starts at 450 °C and continues until the maximum temperature is reached. After Mo carburization is complete, CO consumption and CO2 production continue at a lower level, which can be seen as a sign of complete carburization. From this we concluded that 1 h of carburization at 780 °C is sufficient to complete catalyst activation. The reproducibility of this experiment is demonstrated in Fig. S2.†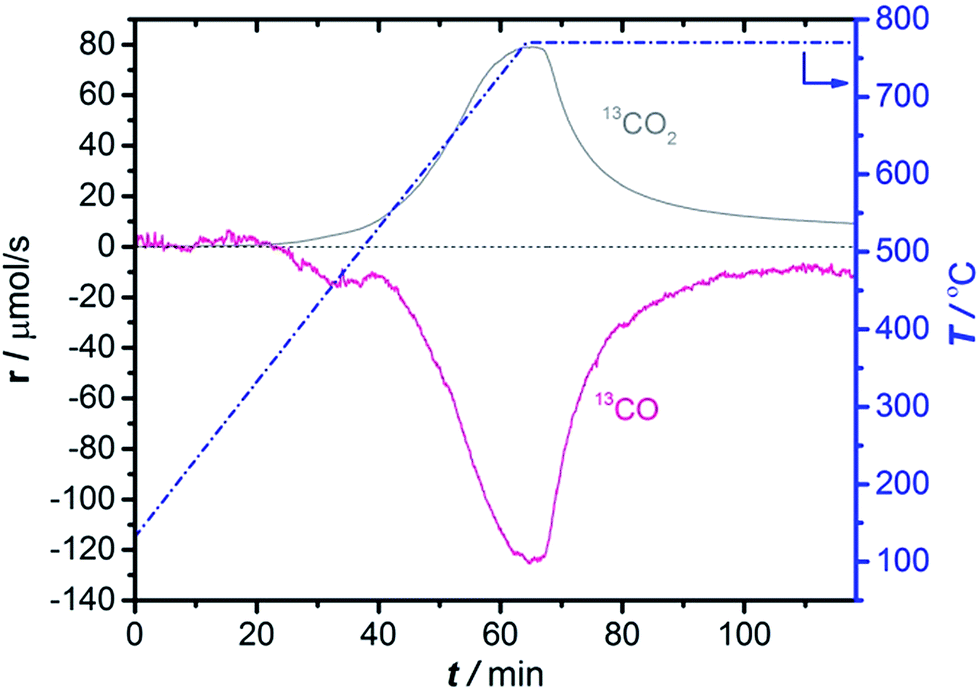 | ||
| Fig. 1 13CO consumption and simultaneous 13CO2 production in TPC of 2MoHZ-13 with 30 ml min−1, 2.5% 13CO in He. The temperature was increased to 780 °C at a rate of 10 °C min−1 (right axis). (cf. Fig. S4† for TPC for all Mo loadings). | ||
Probing the induction period – CH4 pulsing
Pulsing CH4 to the catalyst is a quantitative way to look at the length of the activation period.16 We performed pulsing experiments both on the untreated as well as the same catalyst carburized in CO for 1 h at 780 °C (Fig. 2). The untreated Mo/HZSM-5 started to produce benzene after 2–15 pulses of methane depending on the Mo loading (Fig. 2A and S5†), while benzene was formed immediately on the CO-carburized catalyst (Fig. 2B and S5†). Similar conclusions are made for ethane and ethylene formation. These observations attest to the immediate activity of the CO carburized catalyst. In another experiment, a H2 treatment at 700 °C was used to verify if carbidic carbon needs to be present at the active site for it to be able to activate CH4 or whether a merely reduced species is sufficient (see Fig. S6†). This pretreatment did not lead to instantaneous activity in the same manner as the CO treatment did. This was also observed previously12,16 and leads us to conclude that the carbon atoms incorporated into the active Mo phase play an essential role in activating the C–H bond of methane.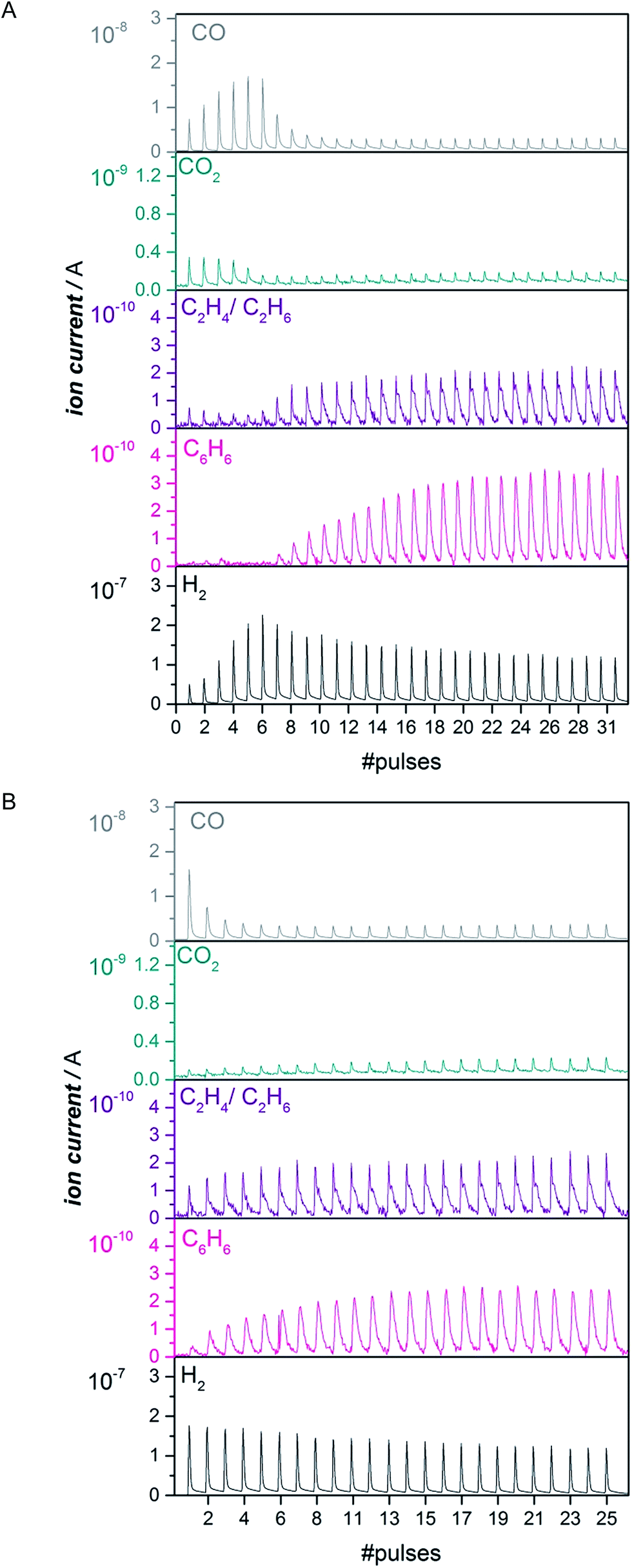 | ||
| Fig. 2 Evolution of CO, C2H4/C2H6, CO2, C6H6 and H2 with a consecutive pulsing of 223 μmol CH4 at 700 °C to 300 mg of an untreated 2MoHZ-13 (Panel A) and the same catalyst pre-carburized with a 30 ml min−1 flow of 2.5% CO in He at 780 °C for 1 h (Panel B). | ||
Same reduction with CO as with CH4 – operando XANES
The CH4 pulsing experiments show that the CO-treatment can effectively eliminate the induction period and should lead to a reduced Mo species equivalent to the one formed during the initial pulses of CH4 where no benzene is observed. To confirm this spectroscopically, the CH4 pulsing experiments were reproduced at the BM16 beamline (ESRF) and the evolution of Mo species was followed by operando X-ray absorption spectroscopy (cf. section “X-ray Absorption Spectroscopy” in the ESI for experimental details and Fig. S8–S10†). Fig. S10B† shows a similar pulsing experiment to what is depicted in Fig. 2A. Benzene is only detected at the 6th pulse of methane. It is therefore interesting to investigate what happens during the first five pulses of methane spectroscopically. This is depicted in the pink XANES spectra in Fig. 3. The XANES spectra collected during the initial five pulses of methane show a strong pre-edge peak at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 008 eV attributable to a 1s–4d quadrupole/dipole transition, characteristic of distorted oxidic Mo species17 and a 1s–5p dipole transition at 20025 eV followed by a relatively flat post-edge region. Clear changes in the pre-edge peak were detected especially between the 4th and 5th pulses accompanied by a total shift of the rising absorption edge of about 4.2 eV. These spectral changes have been previously studied by HERFD-XANES and vtc-XES and correspond to a gradual carburization of the molybdenum sites during the early stage of the reaction.17 Since the detection of benzene happened just at the 6th CH4 pulse (Fig. S10B†), it is particularly interesting to compare the spectrum corresponding to the 5th CH4 pulses of methane (bold pink curve, Fig. 3) with the spectrum of the CO treated catalyst uncontacted with methane (black curve, Fig. 3). Both spectra present the same spectral features with comparable intensity. This is also confirmed by EXAFS and FT-EXAFS of these two samples and their results are presented in Fig. S13 and S14.† Thus, operando X-ray absorption spectroscopy provides convincing evidence of a similar Mo chemical environment when the catalyst is CO pretreated or activated over time by CH4.
008 eV attributable to a 1s–4d quadrupole/dipole transition, characteristic of distorted oxidic Mo species17 and a 1s–5p dipole transition at 20025 eV followed by a relatively flat post-edge region. Clear changes in the pre-edge peak were detected especially between the 4th and 5th pulses accompanied by a total shift of the rising absorption edge of about 4.2 eV. These spectral changes have been previously studied by HERFD-XANES and vtc-XES and correspond to a gradual carburization of the molybdenum sites during the early stage of the reaction.17 Since the detection of benzene happened just at the 6th CH4 pulse (Fig. S10B†), it is particularly interesting to compare the spectrum corresponding to the 5th CH4 pulses of methane (bold pink curve, Fig. 3) with the spectrum of the CO treated catalyst uncontacted with methane (black curve, Fig. 3). Both spectra present the same spectral features with comparable intensity. This is also confirmed by EXAFS and FT-EXAFS of these two samples and their results are presented in Fig. S13 and S14.† Thus, operando X-ray absorption spectroscopy provides convincing evidence of a similar Mo chemical environment when the catalyst is CO pretreated or activated over time by CH4.
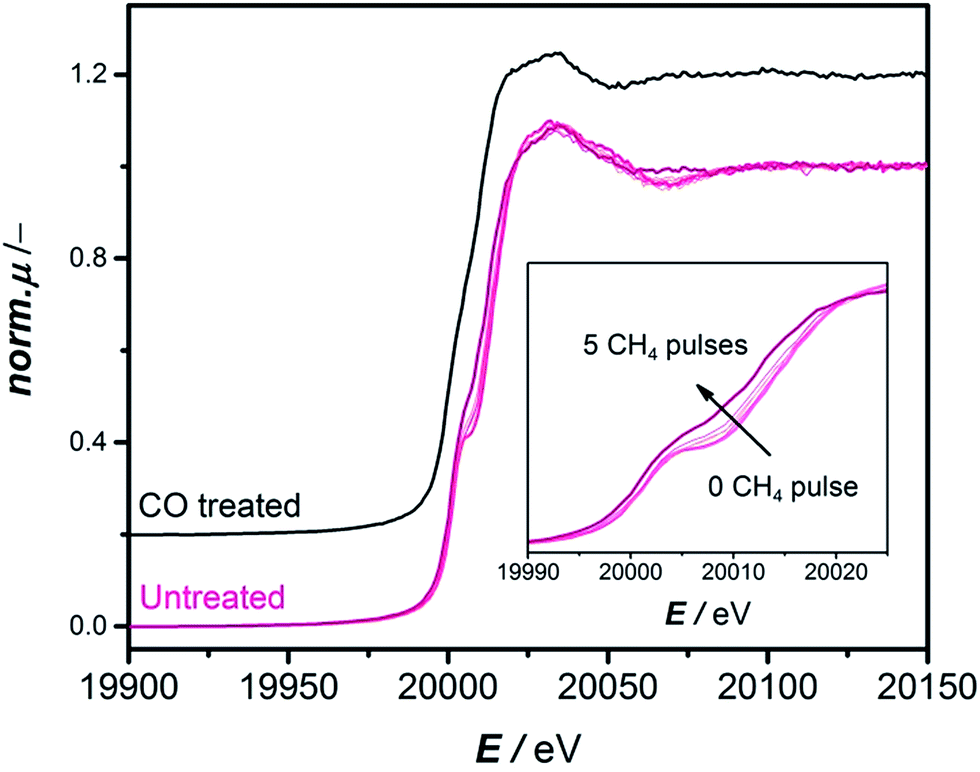 | ||
| Fig. 3 Comparison of operando XANES spectra recorded at Mo K-edge for a 2MoHZ-13 catalyst during consecutive pulsing of CH4 at 700 °C (pink curves) and a 2MoHZ-13 catalyst carburized at 780 °C with a 30 ml min−1 flow of 2.5% CO in He (black curve). | ||
If temperatures below 780 °C, applied for all experiments herein, are used for the CO-treatment, the induction period is not eliminated completely. This can be explained by insufficient reduction at these temperatures confirmed by the XANES spectra measured quasi-in-situ on samples carburized at 600 °C, 700 °C and 780 °C (Fig. S16 and S17†): only the spectra of the sample carburized at 780 °C are equivalent to the spectra of a sample activated in methane, while the spectra of the sample carburized at 600 °C still show a significant pre-edge feature representative of the 1s–5p transitions which allowed for distorted oxidic Mo species. This would explain that previous attempts at pre-carburizing Mo/HZSM-5 with CO at 700 °C12 did not lead to an elimination of the activation period.
Distinguishing among different Mo species – 13C NMR
The above presented TPC, operando XAS and pulsing experiments confirm that CO carburization can be used to produce an active Mo phase that is equivalent to the one formed during the activation period of the reaction with CH4. This opens up new possibilities for characterization, especially with 13C NMR, because the carbidic carbon from the active site can be probed directly without any interference from the signal of aromatic carbons. Indeed the 13C NMR results presented in Fig. 4 are devoid of non-carbidic carbon, since there is no resonance around 100 ppm, characteristic of aromatic carbon.24 In contrast to the XAS spectra, the NMR spectra can distinguish between different Mo carbide species present on the catalyst, showing distinct peaks for each species. Three main carbidic species can be detected, which corresponds well with Temperature Programmed Oxidation (TPO) experiments (Fig. S19†). A sharp contribution exists at a chemical shift of δ3 ∼ 270 ppm and two more broad resonances in the range of 400–250 ppm. The resonance of β-Mo2C at 270 ppm is attributed to hcp type β-Mo2C.23,24,45,46 Its rather sharp shape points towards nanoparticles. The fact that this resonance characteristic of bulk β-Mo2C is only significantly observed for the highest loadings of Mo, 5MoHZ-13, suggests an accumulation of the Mo at the outer surface of the zeolite crystal for the highest loading. An excess of Mo cannot effectively be anchored inside the pores of the zeolite. During synthesis Mo anchors to the Al of the framework through oxygen, replacing the proton of the active site, which leads to a high dispersion of Mo inside the pores of the zeolite.47 The importance of this anchoring was further demonstrated, by preparing a sample of Mo supported on the same zeolite, but containing no framework Al and therefore no anchoring sites. For this sample, (Mo on silicalite-1, 2MoS) only the resonance at ∼270 ppm can be observed.31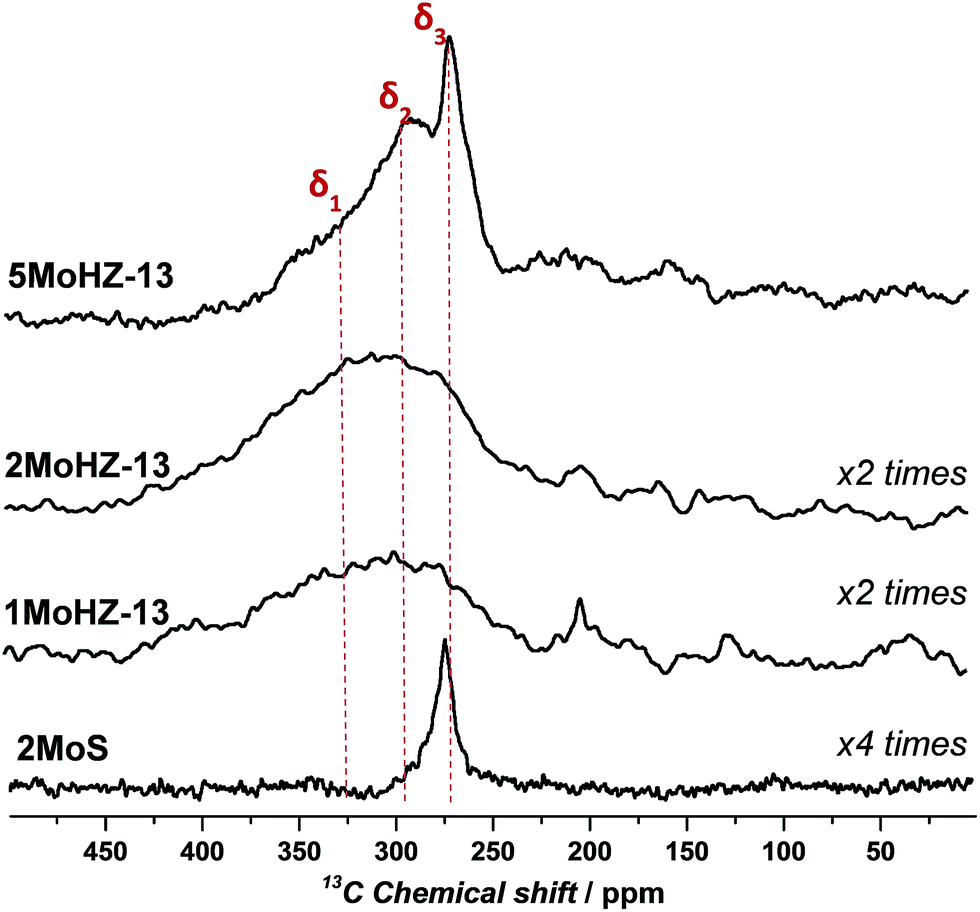 | ||
| Fig. 4 13C MAS NMR spectra of Mo loaded HZ-13 after 13CO carburization at 780 °C for 1 h using 30 ml min−1, 2.5% 13CO in He. Deconvolution results of the spectra can be found in Fig. S18.† | ||
The other two broad resonances are centered at δ1 ∼ 338 ppm and δ2 ∼ 290 ppm and show a large anisotropy, probably arising from several similar species with slightly varying geometry and orientation. In addition, these two resonances, δ1 and δ2, show a strong up-field shift compared to those of Mo2C species. This could be due to deshielding by an electronegative atom (oxygen) and could be a further indication that oxygen is present at the Mo active site, as was previously claimed.17,18,48 The extent of the chemical shift (20 ppm and 68 ppm) makes quantum size effects an unlikely cause for this shift, as the shifts arising from quantum size effects are smaller, i.e. 12 ppm for a difference of 24 Å.49 We propose an oxycarbidic structure rather than a carbonylic one, considering that carbon in Mo(CO)6 grafted on zeolites resonates at ∼200 ppm while we observe resonances that are much more upfield.50 The presence of oxygen at the active site is also suggested by the limited amount of oxygen removed during CO carburization based on quantification of MS signals (cf. section “Quantification” in the ESI†). According to this analysis, only one oxygen is removed per Mo site, while depending on the geometry of the initially present Mo oxide two or three oxygens have to be removed in order to fully reduce the active site.44 While both quantification of the MS signals during CO carburization as well as 13C NMR suggest an oxycarbide, the XAS spectra show high similarity to a fully carbidic β-Mo2C. Comparing the XANES spectra of a sample carburized in CO with the reference spectra measured for β-Mo2C in Fig. S12† it can be found that they look similar, although the post-edge features differ. Also the FT-EXAFS spectra show that Mo–Mo distances on the samples activated in CO as well as in CH4 match the one for β-Mo2C, while being much less intense (Fig. S14†). We speculate that this is due to the presence of some bigger Mo clusters formed under reaction conditions, which are fully reduced, while a majority of species still retain their oxycarbidic form leading to a flattening of the post-edge features but not giving rise to significant additional signals due to the many configurations these molecular species can take inside the pores of the zeolite. Importantly, the resonance representing bulk β-Mo2C (δ3 ∼ 270 ppm) is missing in the 13C NMR spectra for both 1MoHZ-13 and 2MoHZ-13. These two catalysts are immediately active to form benzene after the CO-treatment (Fig. S5†). We therefore conclude that these broader species are active sites. Furthermore, there is a linear relationship between the total carbon content determined by deconvolution of the 13C NMR spectra and the initial conversion of methane in the reaction (Fig. S20†), while the initial conversion does not linearly correlate with the Mo content (Fig. S21†). This indicates that Mo does not constitute an active site unless carbon is present at the active site. This is similar to what was found for Ru/SiO2 in Fischer–Tropsch.51 Note that the amount of carbon increases with Mo loading, but the C/Mo ratio decreases (see Tables S2 and S3†).
Interaction of CH4 with the active site – 13CH4 pulsing
Preparation of the active carbidic phase of Mo without any coke surrounding it further allows probing the interaction of CH4 solely with the active Mo-carbide phase by isolating this interaction from interactions with undefined (hydro-)carbonaceous species.31To understand how methane interacts with the formed active Mo species, we performed a series of pulsing experiments using labelled methane, 13CH4. Prior to this pulsing, the catalyst was carburized using 12CO, forming 12C based carbidic or oxycarbidic Mo structures. This way, it was possible to track the incorporation of 12C from the catalytic Mo site into the products. Firstly, masses 84 to 78 arising from fragmentation of labelled benzene, 13C6H6, as well as benzene where some 12C is incorporated, were recorded on the MS (Fig. 5A). Fig. 5B shows the development of the abundance of the masses normalized by the one with the highest abundance, m/z = 84, while Fig. S25† shows the control experiment where both CO pretreatment and methane pulsing were performed with the same isotope of carbon, 13C. The ratio 83/84 is most informative in assessing the incorporation of 12C into the observed benzene, because m/z = 83 is the most abundant mass for 12C13C5H6 and should lead to a higher 83/84 ratio than for the control experiment where m/z = 83 only represents the 13C6H5 fragment. When using 12C for carburization and 13C for methane pulsing (Fig. 5), the ratio of 83/84 reaches a value of 0.67 for the first pulse and decreases to 0.28 over the next 8 methane pulses. This value of 0.28 is the constant fragmentation ratio in the control experiment. The higher value of 83/84 during the initial pulses can clearly be attributed to the presence of 12C13C5H6. The increased abundance of masses 82 to 78 further supports the incorporation of 12C into benzene. Similarly, incorporation of 12C into ethylene and ethane was investigated by tracking masses 26 to 30. Fig. S27† demonstrates that mostly ethylene with one carbon from the active site, 12C13CH4, is produced, evidenced by the increased abundance of mass 29. These findings demonstrate that in both benzene and ethylene at least one carbon from the active site is incorporated as long as 12C is still available at this site. In analogy to what is proposed for Fischer–Tropsch, the carbon from the Mo-carbide could easily be hydrogenated by the abundant H2 in the reaction atmosphere arising from dehydrogenation of methane already during the induction period.35 The CHx formed in this way easily reacts with the gas-phase or nearby adsorbed CHx species to form ethylene, suggested as the main reaction intermediate in the literature.4,10–13 Because of the high reactivity of this intermediate under the reaction conditions, it quickly reacts with other ethylene in its proximity.52 In addition, the small extent of incorporation of carbon from the active site into benzene can be explained by the small amount of carbidic 12C present compared to that of the 13CH4 fed. According to quantification of the amount of carbon left on 2MoHZ-13 (Fig. 1), the number of moles of CH4 fed in the first 4 pulses, where significant amounts of 12C13C5H6 are observed, is ∼9.6 times the amount of carbidic carbon on the catalyst.
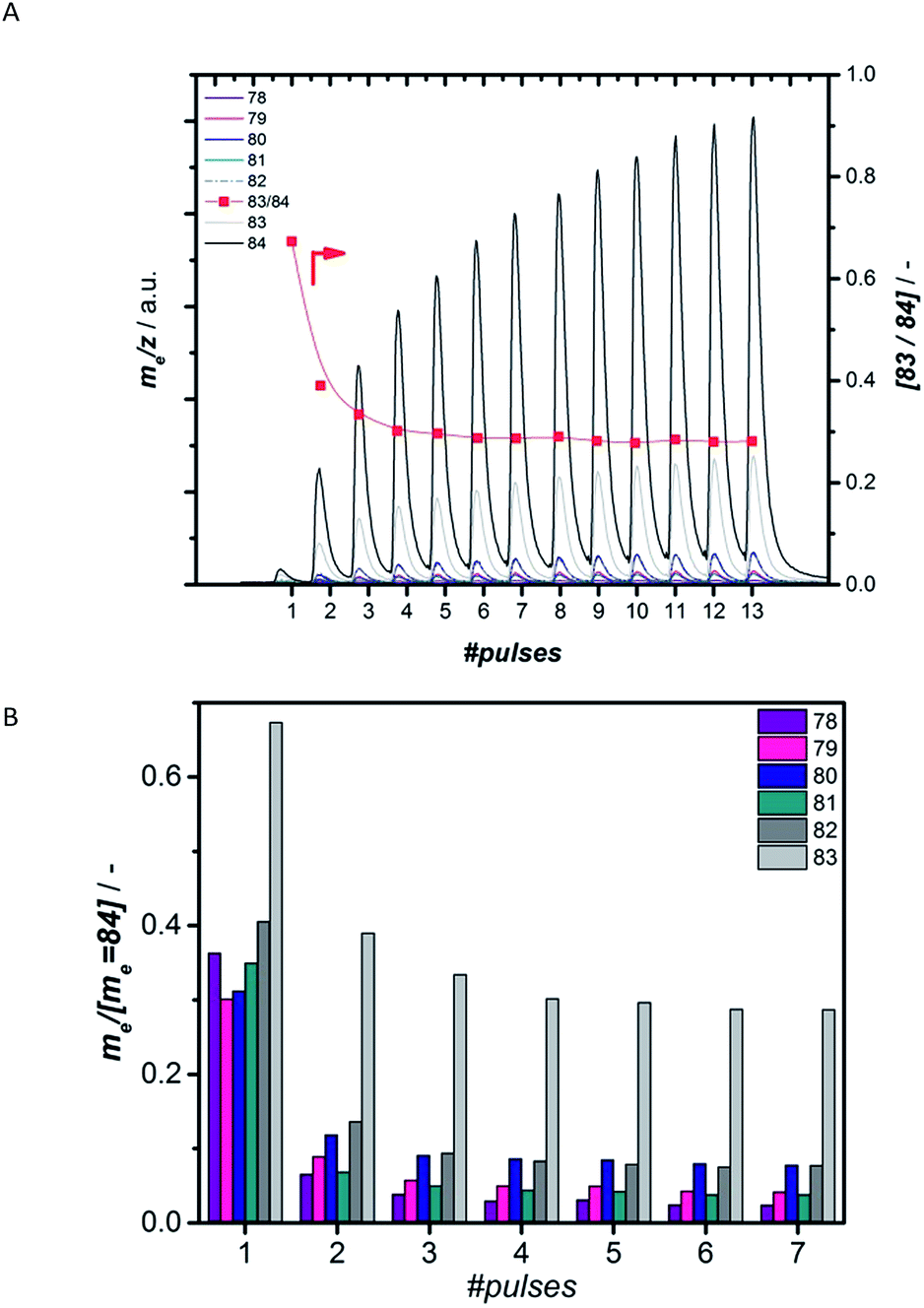 | ||
| Fig. 5 Evolution of masses typical for the fragmentation of benzene at a consecutive pulsing of 223 μmol 13CH4 (Panel A) to the 300 mg 2MoHZ-13 catalyst carburized at 780 °C with 30 ml min−1 2.5% 12CO in He. Panel B shows the area under each pulse for masses 78 to 83 normalized by me = 84. | ||
Through these labelling experiments the activation of methane on the catalytic Mo site could be studied at such a detailed level. Our work provides a good starting point for finding the precise molecular structure of the reduced Mo formed. The carbon in this structure is easily replaced by another carbon from methane, pointing to a dynamic active site. The high reactivity of the carbidic carbon is also evident from TPO experiments (Fig. S19†) showing that some of the carbidic species already oxidized at 50 °C.
Conclusions
In summary, we demonstrate that CO carburization is a powerful approach to isolate the formation of the active site on Mo/HZSM-5 for non-oxidative methane conversion from the catalytic cycle without undesired catalyst coking. This strategy allowed us to distinguish between three different structures for the active site with 13C NMR, obtaining high resolution spectra of the (oxy-)carbidic species present that are usually obscured by aromatic carbon.16,23–25 This provides a useful structural characterization tool to further our understanding beyond bulk techniques like XAS. Further, it was possible to study the activation of methane on the Mo catalytic active site at the molecular level. Rapid exchange reactions with the active Mo-site result in the incorporation of carbidic carbon into the products ethylene and benzene. This demonstrates that the carbon at the metal site in this structure is easily replaced by another carbon from methane, pointing to a dynamic active site. There are three observations that provide hard evidence on the mechanistic role of carbidic Mo species in the Mo/HZSM-5 catalyst: (a) Carbon from the active site is incorporated into the final products. (b) There is a linear relationship between carbon present at the active site and initial activity of the catalyst. (c) H2 reduction treatment during which no carbon is deposited at the active site does not lead to an elimination of the induction period. These let us conclude that the catalytic Mo site actively takes part in the catalytic reaction rather than acting as an adsorption site to lower the activation barrier of CH4, similar to what was observed for the Mars–van-Krevelen mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Sabic-NWO CATC1CHEM CHIPP project. Thanks go to Dr Christoph Dittrich (SABIC), Dr Frank Mostert (SABIC) and Dr Xander Nijhuis (SABIC) as well as Prof. Emiel Hensen (TU Eindhoven) and Nikolay Kosinov (TU Eindhoven) for helpful discussion. We also acknowledge the BM26B DUBBLE beamline and the BM16 FAME-UHD beamline of the European Synchrotron Radiation Facility (ESRF), Grenoble, France for letting us use their facilities. Thanks also go to Maarten Gorseling (TU Delft) for performing GC-MS analysis. The FAME-UHD project is financially supported by the French "grand emprunt" EquipEx (EcoX, ANR-10-EQPX-27-01), the CEA-CNRS CRG consortium and the INSU CNRS institute.References
- B. L. Farrell, V. O. Igenegbai and S. Linic, ACS Catal., 2016, 6, 4340–4346 CrossRef CAS.
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35–41 CrossRef CAS.
- O. Bragin, T. Vasina, Y. I. Isakov, B. Nefedov, A. Preobrazhenskii, N. Palishkina and K. M. Minachev, Russ. Chem. Bull., 1982, 31, 847 CrossRef.
- J. J. Spivey and G. Hutchings, Chem. Soc. Rev., 2014, 43, 792–803 RSC.
- S. Ma, X. Guo, L. Zhao, S. Scott and X. Bao, J. Energy Chem., 2013, 22, 1–20 CrossRef CAS.
- B. S. Liu, L. Jiang, H. Sun and C. T. Au, Appl. Surf. Sci., 2007, 253, 5092–5100 CrossRef CAS.
- K. Honda, X. Chen and Z.-G. Zhang, Catal. Commun., 2004, 5, 557–561 CrossRef CAS.
- Y. Song, Y. Xu, Y. Suzuki, H. Nakagome and Z.-G. Zhang, Appl. Catal., A, 2014, 482, 387–396 CrossRef CAS.
- S. Liu, L. Wang, R. Ohnishi and M. Ichikawa, J. Catal., 1999, 181, 175–188 CrossRef CAS.
- V. T. T. Ha, L. V. Tiep, P. Meriaudeau and C. Naccache, J. Mol. Catal. A: Chem., 2002, 181, 283–290 CrossRef CAS.
- Y. Xu and L. Lin, Appl. Catal., A, 1999, 188, 53–67 CrossRef CAS.
- D. Wang, J. Lunsford and M. Rosynek, Top. Catal., 1996, 3, 289–297 CrossRef CAS.
- Y. Xu, X. Bao and L. Lin, J. Catal., 2003, 216, 386–395 CrossRef CAS.
- J. Gao, Y. Zheng, J.-M. Jehng, Y. Tang, I. E. Wachs and S. G. Podkolzin, Science, 2015, 348, 686–690 CrossRef CAS PubMed.
- W. Ding, S. Li, G. D Meitzner and E. Iglesia, J. Phys. Chem. B, 2001, 105, 506–513 CrossRef CAS.
- H. Jiang, L. Wang, W. Cui and Y. Xu, Catal. Lett., 1999, 57, 95–102 CrossRef CAS.
- I. Lezcano-González, R. Oord, M. Rovezzi, P. Glatzel, S. W. Botchway, B. M. Weckhuysen and A. M. Beale, Angew. Chem., Int. Ed., 2016, 55, 5215–5219 CrossRef PubMed.
- F. Solymosi, A. Szöke and J. Cserényi, Catal. Lett., 1996, 39, 157–161 CrossRef CAS.
- S. Liu, L. Wang, R. Ohnishi and M. Lchikawa, Kinet. Catal., 2000, 41, 132–144 CrossRef CAS.
- B. M. Weckhuysen, M. P. Rosynek and J. H. Lunsford, Catal. Lett., 1998, 52, 31–36 CrossRef CAS.
- F. Solymosi, A. Erdöhelyi and A. Szöke, Catal. Lett., 1995, 32, 43–53 CrossRef CAS.
- H. Liu, W. Shen, X. Bao and Y. Xu, J. Mol. Catal. A: Chem., 2006, 244, 229–236 CrossRef CAS.
- J. Yang, D. Ma, F. Deng, Q. Luo, M. Zhang, X. Bao and C. Ye, Chem. Commun., 2002, 3046–3047 RSC.
- N. Kosinov, F. J. A. G. Coumans, E. A. Uslamin, A. S. G. Wijpkema, B. Mezari and E. J. M. Hensen, ACS Catal., 2016, 7, 520–529 CrossRef.
- H. Zheng, D. Ma, X. Liu, W. Zhang, X. Han, Y. Xu and X. Bao, Catal. Lett., 2006, 111, 111–114 CrossRef CAS.
- Y. Song, Y. Xu, Y. Suzuki, H. Nakagome, X. Ma and Z.-G. Zhang, J. Catal., 2015, 330, 261–272 CrossRef CAS.
- D. Ma, Y. Shu, X. Han, X. Liu, Y. Xu and X. Bao, J. Phys. Chem. B, 2001, 105, 1786–1793 CrossRef CAS.
- D. Ma, W. Zhang, Y. Shu, X. Liu, Y. Xu and X. Bao, Catal. Lett., 2000, 66, 155–160 CrossRef CAS.
- H. Zheng, D. Ma, X. Bao, J. Z. Hu, J. H. Kwak, Y. Wang and C. H. F. Peden, J. Am. Chem. Soc., 2008, 130, 3722–3723 CrossRef CAS PubMed.
- J. Z. Hu, J. H. Kwak, Y. Wang, C. H. F. Peden, H. Zheng, D. Ma and X. Bao, J. Phys. Chem. C, 2009, 113, 2936–2942 CAS.
- N. Kosinov, A. S. G. Wijpkema, E. Uslamin, R. Rohling, F. J. A. G. Coumans, B. Mezari, A. Parastaev, A. S. Poryvaev, M. V. Fedin, E. A. Pidko and E. J. M. Hensen, Angew. Chem., 2018, 57, 1016–1020 CrossRef CAS PubMed.
- V. P. Santos, T. A. Wezendonk, J. J. D. Jaén, A. I. Dugulan, M. A. Nasalevich, H.-U. Islam, A. Chojecki, S. Sartipi, X. Sun, A. A. Hakeem, A. C. J. Koeken, M. Ruitenbeek, T. Davidian, G. R. Meima, G. Sankar, F. Kapteijn, M. Makkee and J. Gascon, Nat. Commun., 2015, 6, 6451 CrossRef CAS PubMed.
- K. Xu, B. Sun, J. Lin, W. Wen, Y. Pei, S. Yan, M. Qiao, X. Zhang and B. Zong, Nat. Commun., 2014, 5, 5783 CrossRef CAS PubMed.
- V. V. Ordomsky, B. Legras, K. Cheng, S. Paul and A. Y. Khodakov, Catal. Sci. Technol., 2015, 5, 1433–1437 CAS.
- J. M. Gracia, F. F. Prinsloo and J. W. Niemantsverdriet, Catal. Lett., 2009, 133, 257 CrossRef CAS.
- S. Liu, A. C. Gujar, P. Thomas, H. Toghiani and M. G. White, Appl. Catal., A, 2009, 357, 18–25 CrossRef CAS.
- S. Xing, D. Zhou, L. Cao and X. Li, Chin. J. Catal., 2010, 31, 415–422 CrossRef CAS.
- D. Zhou, S. Zuo and S. Xing, J. Phys. Chem. C, 2012, 116, 4060–4070 CAS.
- H. Zhu, Y. Zhang, D. Zhou, J. Guan and X. Bao, Chin. J. Catal., 2007, 28, 180–186 CrossRef CAS.
- D. Ma, Y. Shu, W. Zhang, X. Han, Y. Xu and X. Bao, Angew. Chem., Int. Ed., 2000, 39, 2928–2931 CrossRef CAS PubMed.
- J. Dang, G. Zhang, L. Wang, K. Chou and P. C. Pistorius, J. Am. Ceram. Soc., 2016, 99, 819–824 CrossRef CAS.
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1998, 175, 347–351 CrossRef CAS.
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1998, 175, 338–346 CrossRef CAS.
- I. Vollmer, G. Li, I. Yarulina, N. Kosinov, E. J. Hensen, K. Houben, D. Mance, M. Baldus, J. Gascon and F. Kapteijn, Catal. Sci. Technol., 2018, 8, 916–922 CAS.
- T.-c. Xiao, A. P. E. York, V. C. Williams, H. Al-Megren, A. Hanif, X.-y. Zhou and M. L. H. Green, Chem. Mater., 2000, 12, 3896–3905 CrossRef CAS.
- T. Xiao, A. P. E. York, K. S. Coleman, J. B. Claridge, J. Sloan, J. Charnock and M. L. H. Green, J. Mater. Chem., 2001, 11, 3094–3098 RSC.
- D. Ma, X. Han, D. Zhou, Z. Yan, R. Fu, Y. Xu, X. Bao, H. Hu and S. C. F. Au-Yeung, Chem.–Eur. J., 2002, 8, 4557–4561 CrossRef CAS.
- D. Ma, Y. Shu, X. Bao and Y. Xu, J. Catal., 2000, 189, 314–325 CrossRef CAS.
- B. S. Zelakiewicz and A. C. de Dios Tong, J. Am. Chem. Soc., 2003, 125, 18–19 CrossRef CAS PubMed.
- H. Koller, A. R. Overweg, R. A. van Santen and J. W. de Haan, J. Phys. Chem. B, 1997, 101, 1754–1761 CrossRef CAS.
- T. M. Duncan, P. Winslow and A. T. Bell, J. Catal., 1985, 93, 1–22 CrossRef CAS.
- A. Mehdad and R. F. Lobo, Catal. Sci. Technol., 2017, 7, 3562–3572 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01263f |
| This journal is © The Royal Society of Chemistry 2018 |